

Abstract
Most solid tumours contain cancer-associated fibroblasts (CAFs) that support tumourigenesis and malignant progression. However the cellular origins of CAFs in epithelial ovarian cancers (EOCs) remain poorly understood, and their utility as a source of clinical biomarkers for cancer diagnosis has not been explored in great depth. Here, we report establishing in vitro and in vivo models of CAFs in ovarian cancer development. Normal ovarian fibroblasts and mesenchymal stem cells cultured in the presence of EOC cells acquired a CAF-like phenotype, and promoted EOC cell migration in vitro. CAFs also promoted ovarian cancer growth in vivo in both subcutaneous and intraperitoneal murine xenograft assays. Molecular profiling of CAFs identified gene expression signatures that were highly enriched for extracellular and secreted proteins. We identified novel candidate CAF specific biomarkers for ovarian cancer including NPPB, which was expressed in the stroma of 60% primary ovarian cancer tissues (n=145) but not in the stroma of normal ovaries (n=4). NPPB is a secreted protein that was also elevated in the blood of 50% of women with ovarian cancer (n=8). Taken together these data suggest that the tumor stroma is a novel source of biomarkers, including NPPB, that may be of clinical utility for detection of EOC.
Introduction
Identifying novel approaches to detect EOCs more effectively would have a substantial clinical impact in reducing ovarian cancer related mortality. Fewer than 30% of patients diagnosed with advanced epithelial ovarian cancer (EOC) survive more than 5 years after their initial diagnosis 1. One of the major reasons for the high rates of EOC-associated mortality is that more than 60% of patients are diagnosed with advanced stage disease and there are currently no biomarkers with sufficient specificity to detect new cases of early-stage EOC in the clinic. If more reliable blood-based biomarkers made it possible to detect ovarian cancers earlier, it is clear that may lives could be saved.
Historically, studies of the underlying biology and etiology of EOC have focused on the epithelial component of tumours 2. However, most tumour types, including ovarian cancers, also contain an abundant and complex stroma that supports tumour growth and is important for the maintenance of tumour tissue homeostasis. The stromal microenvironment is important throughout all stages of cancer development, co-evolving with the growing tumour epithelial cells, although in itself is not malignant. In EOCs up to 70% of the tumour is stroma, and in advanced stage EOCs a high stromal:epithelial ratio is associated with poorer prognosis 3. This suggests that stromal cells promote more aggressive disease phenotypes, which is supported by studies of models of ovarian and other cancer types and hematopoetic malignancies showing that cancer stroma promotes chemoresistance, evasion of apoptosis, invasion and metastasis 4-10. Stromal epithelial cross-talk is mediated by direct cell-cell contact as well as soluble signaling molecules, the release of stromal-epithelial signaling factors into the blood stream represents a potential source of biomarkers that has not yet been explored in great detail.
For some tumour types, and in particular for EOC, studying CAF biology has been hindered by a shortage of robust models of CAF development. Consequently, the origin of the cancer-associated fibroblasts (CAFs) present within ovarian cancers is poorly understood, although recent evidence suggests that CAFs derive from heterogenous origins in a mouse model of ovarian cancer 11. Genetic analyses suggest that the majority of ovarian CAFs arise from non-neoplastic cells 12, 13; but it is not currently known whether ovarian CAFs can originate from normal ovarian fibroblasts that have transdifferentiated into CAFs during neoplastic development. Alternatively ovarian CAFs can derive from mesenchymal stem cells (MSCs), which are multipotent cells produced in the bone marrow and have the ability both in vitro and in vivo to self-renew and differentiate into connective tissue cells including osteoblasts, adipocytes and chondrocytes. Naïve MSCs are also found in many normal tissues including fallopian tubes 14, and are recruited to wounds where they contribute to tissue repair. MSCs can differentiate into CAFs in vitro when exposed to cancer cells or transforming growth factor-beta, and can be recruited to tumours in in vivo models 11, 15, 16. Several reports have shown that MSCs can be tumour promoting in many solid tumours including breast, prostate and ovarian carcinoma 11, 17.
In this study we established heterotypic models of EOC, both in vitro and heterotypic in vivo, in order to directly compare NOFs and MSCs as potential sources of CAFs in EOCs. We found that both MSCs and NOFs have the potential to acquire molecular and phenotypic characteristics of CAFs when co-cultured with EOC cells. We also characterised the transcriptional reprogramming events that occur during the development of ovarian cancer-associated fibroblasts, and found that a plethora of secreted and extracellular molecules are produced early during ovarian CAF reprogramming. Such molecules represent novel candidate biomarkers for tumour detection.
Materials and Methods
Cell culture
All cell culture was performed within a certified BSL2+ facility. Culture media are listed in Supplementary Table 1. Primary mesenchymal stem cells (MSC) were purchased from Texas A&M Health Science Centre and were only used for assays within 5 passages of thawing. To test MSC multipotency, 50,000 cells were plated into 6-well plate and cultured until confluent. Cells were washed twice with PBS before adding 2ml of adipocyte/osteocyte differentiation medium (both Invitrogen) and incubated for 21 days. Media were replaced every 4 days. To fix and stain, cells were washed with PBS and fixed in buffered formalin for 1hr at room temperature. Adipocyte staining: Oil-Red-O working solution was prepared by mixing 3 parts of stock solution (1.25% Oil Red O (Sigma) w/v in isopropanol (VWR)) with 2 parts of PBS. After 10mins the solution was filtered through a 0.45μm filter. Fixed cells were washed with PBS, and 2mls Oil Red O working solution added for 20mins. After staining the cells were washed thoroughly with PBS. Osteocyte staining: Fixed cells were washed with deionised water and incubated in 2mls of Alizarin Red (Sigma) solution (1% Alizarin Red S in water, filtered through 0.45μm membrane) for 20mins. The cells were washed with deionised water until the background was clear. Stained cells were examined microscopically for adipocyte and osteocyte differentiation, both the MSC lines differentiated into adipocytes and osteocytes with efficiencies >75%.
TERT-immortalized normal ovarian fibroblasts and normal ovarian epithelial cells have been previously described 18, 19. Epithelial ovarian cancer cell lines (EOC) were a kind gift from Dr. G. Mills at the MD Anderson Cancer Center. All cells were maintained at 37°C and 5% CO2 and cultures were routinely screened for contaminating Mycoplasma infections and found to be negative. Epithelial line identities were verified by typing of short tandem repeats using the Promega PowerPlex16HS Assay (Promega). Typing was performed at the University of Arizona Genetics Core and profiles were compared to the ATCC and DSMZ databases, plus published EOC cell line profiles 20.
Cell Labeling and In vivo tumourigenicity assays
Hey.A8 cells were labeled with luciferase, using G418-luciferase lentiviral supernatants were purchased from the vector core at CHLA. Hey.A8luc cells were selected by the addition of 400μg/ml G418 to cell culture media (Sigma). All in vivo work was performed with permission from the University of Southern California Institutional Animal Care and Use Committee. For intraperitoneal injections, 1×106 Hey.A8 cells were injected alone or with 0.9×106 stromal cells. Cells were washed in PBS and resuspended in GFR phenol red free Matrigel (BD Biosciences) diluted 1:100 in ice cold PBS. Live animal imaging was performed at the USC Molecular Imaging Core. Animals were anesthetized by inhalation of 1-4% isoflurane and 50μg luciferin administered by tail vein injection. 1.5mins post-injection the animals were imaged using an IVIS® Imaging System 200 (Xenogen) and tumor foci measured by quantification of luciferase signal. For subcutaneous injections, 3×106 A2780 cells were injected alone (right flank) or with 1.5×106 stromal cells (left flank). Tumour growth was measured using digital calipers (VWR).
Conditioned media (CM) production
3×106 Hey.A8 cells were plated in complete growth media. After 24 hours the media was aspirated and the cells washed twice with PBS. 15mls of serum free medium was added to the cells. After 48hrs the CM was harvested, filtered through a 0.45μm filter and stored at -80°C.
Immunofluorescent staining and Western blotting
Cells were cultured on glass coverslips (VWR) and washed with PBS/1% bovine serum albumin (BSA, Sigma). Cells were fixed using ice-cold 3% formaldehyde (BDH) for 10mins, rinsed well in PBS, and permeabilised for 5mins with PBS/0.5%BSA/0.5%Triton-X-100 (Sigma). Cells were washed thoroughly with PBS and incubated with PBS/1%BSA for 30min. An anti-vimentin primary antibody (CRUK), diluted 1:500 in PBS/1%BSA, was applied for 1hour before were washing coverslips with PBS/1%BSA. An Alexa Fluor coupled secondary antibody (1:500 dilution, Invitrogen) was added for 30min. Coverslips were washed and transferred onto microscope slides with mounting medium containing DAPI (Vectashield). Images were captured using a fluorescence microscope (Olympus BX61) with Cytovision software.
For Western blotting: Cells were grown to 80% confluence and washed twice with ice-cold PBS. Cells were lysed and cell debris was removed by centrifugation at 13,000rpm for 30min at 4°C. For SDS-gel electrophoresis samples were denatured and reduced in Laemmli Buffer (Sigma) by boiling at 105°C for 5min. 10μg of each sample was run onto 8-16% precise protein gels (Pierce) at 150V for 45min. Proteins were electroblotted onto a polyvinylidene fluoride membrane and the membrane blocked in TBS-T/2% BSA overnight before incubation with an anti-smooth muscle actin (1:1000 dilution, Dako) or anti-actin (1:5000 dilution, Sigma) diluted in TBS-T for 1 hour. Unbounds antibody was removed by 3X TBS-T washes and an anti-mouse HRP coupled secondary antibody (Sigma) applied for 1 hour (1:5000 dilution). Membranes were washed again 3X in TBS-T and protein bands visualized by application of enhanced chemoluminescence liquid (Pierce) followed by exposure to chemiluminescence detection film. Quantification of protein bands was performed using ImageJ software 21.
In vitro co-culture assays
For transwell migration and invasion assays, stromal cells were starved for 24 hours, trypsinized and normalized to 0.12×106 or 0.5×106, for migration (Greiner Bio One) and invasion (Millipore) assays respectively. Transwell inserts with 8μm pores, coated or uncoated with matrix, were placed within a 24-well plate. Invasion membranes were rehydrated with 300μl prewarmed serum-free media for 30 minutes and 250μl removed. 250μl cell suspension was added to each membrane and cell incubated for 16-24 hours. Invasion assays were read using a luminescent dye, according to manufacturers instructions. Migration membranes were fixed in 100% methanol and stained with 2% crystal violet in 5% ethanol. Five fields of view were counted per membrane to determine the mean number of migrated cells per membrane. For testing migration of EOC cells towards stromal cells, 0.12×106 stromal cells were plated into 24 well plates and allowed to adhere for 24 hours before use as a chemoattractant. Stromal cells were washed thoroughly with PBS to remove all serum before use. Proliferation assays were performed by plating 5,000 cells into 96 well plates, and incubating with serum supplemented conditioned media for 48 hours. Proliferation was assayed using the Cell Titre Glo (Promega), according to manufacturers instructions. All assays were performed in triplicate.
For measuring gene expression, EOC cells were plated into 6 well plates and stromal cells plated into non-cell permeable transwell inserts (1μm pore size, Greiner Bio One). EOC:stromal cell ratio was 1:10. After 7 days RNA was extracted from EOC cells using the QIAgen RNeasy kit (QIAgen), according to manufacturers instructions. 2μg RNA was reverse transcribed into cDNA using the Promega MMLV reverse transcriptase enzyme and random hexamer priming (all Promega). The following TaqMan gene expression probes were used: beta-actin, Hs99999903_m1; GAPDH Hs02758991_g1; vimentin (Hs00185584_m1) and fibronectin, Hs00365052_m1, and PCRs were run and data recorded using the Applied Biosystems 7900HT Fast Real-Time PCR System. Expression of each gene of interest was calculated using the ΔΔCt method, with the Ct values for each gene normalized to the average Ct value for both control genes.
Gene expression microarray profiling and analysis
MSC1 and INOF cells were co-cultured with IOSE4 cells or Hey.A8 EOC cells. 1000 stromal cells were plated into 6 well plates and 10,000 epithelial cells plated into porous inserts (pore size 1μm, Greiner BioOne). After 24hrs, cells were refed and epithelial cell inserts added to the stromal cell cultures. Cells were grown in 50% INOF medium and 50% MSC medium for 7 days before harvesting RNA using the QIAgen RNeasy kit (QIAgen), according to manufacturers instructions. Samples were profiled using Illumina HumanHT-12 Expression BeadChip microarrays at the USC Epigenome Core facility. Array normalization was conducted with the Bioconductor ‘beadarray’ package 22 using Robust Multiarray Average (RMA). Bead-level data were log2 transformed and summarized after removing outliers using a 3 MAD (median absolute deviation) cutoff. Gene expression data were analysed using one-way blocked ANOVA, EOC vs. non-co-cultured and EOC vs IOSE, adjusted for MSC/NOF. To address potential departures from normality, a permutation-based FDR approach was applied 23 with 100 permutations and a threshold of 0.01. The final set of results was defined by the intersection of the two FDR = .01 sets, where probes were then ranked by max p-value, that is, each gene i was ranked by max(pi,non-cocultured vs. EOC, pi,IOSE vs. EOC). Gene ontology analyses were performed using the DAVID bioinformatics database (http://david.abcc.ncifcrf.gov) 24. GO terms were retrieved for the GOTERM_BP_FAT, GOTERM_CC_FAT and GOTERM_MF_FAT categories with Benjamini adjustments of p-values. KEGG pathway analyses were also performed using DAVID.
Validation of NPPB
Real-time PCR was performed as described above. Cells were cultured in 50% INOF medium and 50% MSC medium for 7 days before harvesting the RNA. Reverse transcription, Q-PCR and analysis were performed as described above, using a TaqMan probe: NPPB, Hs01057466_g1 (Life Technologies). Protein expression of NPPB in tumours was performed using an anti-human NPPB antibody (cat no: NBP1-47509, Novus Biologicals). Normal ovarian specimens, from women who underwent hysterectomy with bilateral salpingo-oophorectomy for non-malignant conditions such as fibroids, were retrieved from the archives. Additionally, a tissue microarray of 165 ovarian cancers was stained for NPPB expression. The development of this array has been previously described 25, patient characteristics can be found in Supplementary Table 2. Immunohistochemistry on paraffin-embedded tissue sections was performed by the USC Immunohistochemistry Laboratory within the USC Department of Pathology. NPPB expression in the tumor stroma was scored, by light microscopy, as negative (0), weak (1), moderate (2) or strong (3). Statistical analyses were performed to compare all expressing tumors (score 1-3) to negative tumors, using Fishers Exact and Chi Squared tests.
For measurement of NPPB in blood, patient sera were collected as part of the Gynecological Tissue and Fluid Repository at USC and stored at -80°C until use. Quantification of NT-proBNP was performed by Quest Diagnostics (test code 11188X). All human specimens used in this study were collected with informed consent, with the approval of the Institutional Review Boards at USC and OSHU.
Statistical Analysis
Unless otherwise stated, two-tailed paired Student's T-Tests were performed, assuming equal variance, using α=0.05.
Results
Stromal cells promote ovarian cancer tumourigenesis in vivo
For many tumour types it has been shown that co-injection of cancer-associated fibroblasts promotes cancer cell growth in vivo. Recent evidence suggests that bone-marrow derived mesenchymal stem cells (MSCs) make a substantial contribution to CAF population in a syngeneic ovarian cancer mouse model 11, 15. We addressed the hypothesis that normal human ovarian fibroblasts (INOFs) and/or bone-marrow derived human MSCs represent origins of ovarian cancer-associated fibroblasts by performing subcutaneous and intraperitoneal injections in immunocompromised mice of EOC cells alone and in the presence of INOFs/MSCs. Subcutaneous co-injection of human EOC cells with MSC and INOF cells was associated with a significantly more rapid tumour formation than for EOC lines alone; cells from the A2780 EOC line formed tumours in 19±0 days compared to 9.33±1.53 days when co-injected with MSCGFP and 4±0 when co-injected INOFs (P=0.009 and P=0.001 respectively. Fig. 1a). Co-injection of A2780 with MSC/INOF cells also resulted in a higher tumour take rate (Fig. 1a), a 3-7 day reduction in animal survival, and more aggressive tumours (Fig. 1b). We also evaluated in vivo tumour growth following intraperitoneal injection of Hey.A8 EOC cells with and without MSC/INOF cells; the results were consistent with those for subcutanous injection. Hey.A8 EOC cells were labeled with luciferase (Hey.A8luc) to enable live animal imaging. Ten days after injection, 2/5 animals had detectable tumours. By contrast, 5/5 animals co-injected with Hey.A8 and MSC/INOF cells had detectable tumours over the same time period (Fig. 1c&d). Co-injection with stromal cells was associated with significantly more tumour foci per animal (Hey.A8 versus Hey.A8+MSC, P=0.029; Hey.A8 versus Hey.A8+INOF P=0.021).
Figure 1. INOFs and MSCs promote EOC cell tumourigenicity in vivo.
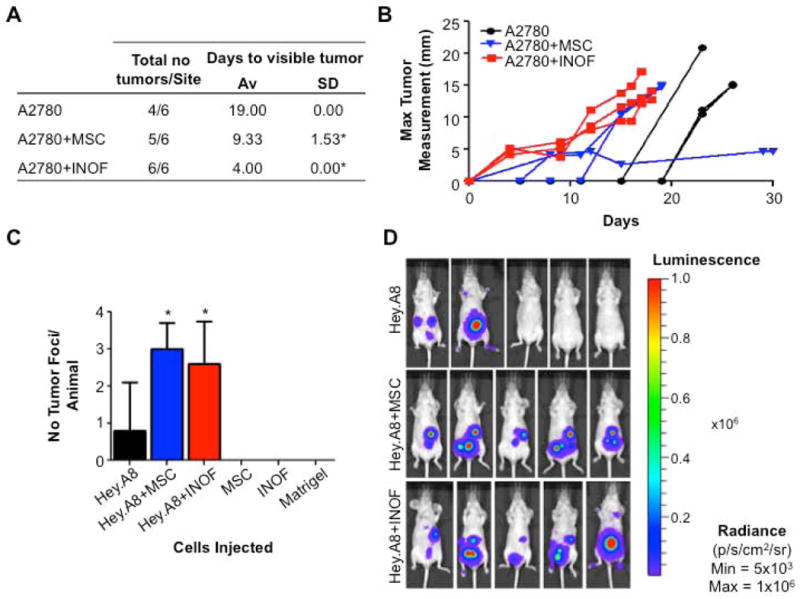
(a) A2780 cells were injected subcutaneously into 3 mice, with 2 injection sites per mouse, this table shows the number of tumours per injection site and the days taken for visible tumour to be detected. Co-injection of stromal cells was associated with a significant reduction in the time taken for palpable tumours to develop. (b) Measurement of maximum tumour diameter in each mouse. (c) In intraperitoneal xenografts, co-injection with stromal cells increases the number of tumour foci detectable at day 10. (d) Live animal Xenogen imaging. Tumours are detected in 2/5 animals that receive 3×106 Hey.A8luc cells alone. Co-injection with stromal cells results in tumour formation in 100% of animals, four animals shown per group. * P>0.05, two-tailed paired Student's T-test, α=0.05, compared to EOC cells injected alone. Error bars = s.d. Animals were examined post-mortem and tumor presence confirmed by macroscopic inspection and histological examination of the lesions. Tumor foci were absent in control mice injected with MSCs, INOFs or Matrigel alone.
We confirmed the ability of INOF and MSC cells to differentiate into CAF-like cells when exposed in vitro to conditioned medium from an epithelial ovarian cancer cell line (Hey.A8). Normal immortalized ovarian fibroblasts (INOFs) and two early passage primary mesenchymal stem cell lines (MSC1 and MSC2) were exposed to Hey.A8 conditioned medium. Vimentin and α-smooth muscle actin (αSMA), two markers commonly expressed by CAFs, showed increased expression in both MSC lines after long term (28 day) exposure to conditioned medium (Fig. 2a & b). INOFs showed no change in expression of vimentin after exposure to conditioned medium, but a striking upregulation of αSMA (Fig. 2b). There were negligible changes in vimentin and αSMA expression when INOFs and MSCs were exposed to conditioned medium from normal ovarian surface epithelial cells (OSECs). MSCs but not INOFs were significantly more proliferative in Hey.A8 conditioned medium compared to OSEC conditioned medium (Fig. 2c&d, P=0.033). In transwell migration assays MSCs were significantly more migratory when Hey.A8 conditioned medium was used as a chemoattractant compared to either OSEC conditioned or unconditioned media (Fig. 2e, P=0.017). INOFs migrated significantly more towards both Hey.A8 and OSEC conditioned medium (Fig. 2f, P=0.041 and P=0.003). Neither cell type showed significant changes in invasion in response to the different chemoattractants (data not shown). Thus both INOFs and MSCs have the ability to become CAF-like cells in vitro and in vivo.
Figure 2. In vitro genesis of cancer-associated fibroblasts.
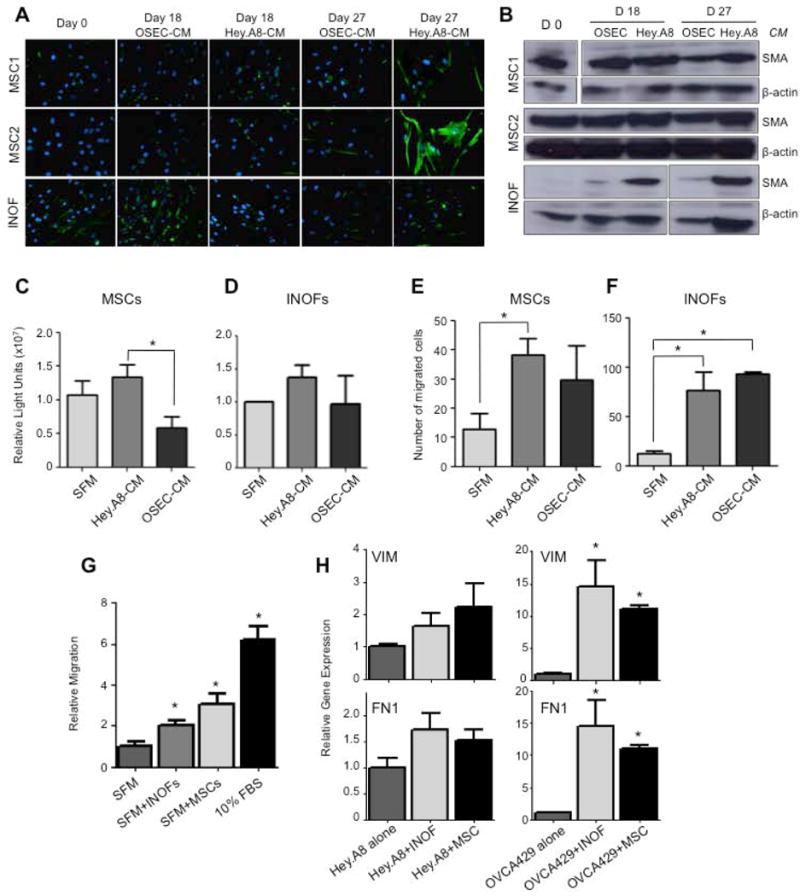
(a) Cancer associated fibroblast precursor cells, mesenchymal stem cells (MSCs, N=2) and immortalized normal ovarian fibroblasts (INOFs, N=1) were cultured in normal ovarian epithelial cell conditioned medium (OSEC-CM) or epithelial ovarian cancer cell conditioned medium (EOC-CM). After 0, 18 and 27 days cells were stained for vimentin (green), nuclei are stained blue with DAPI. MSCs upregulated vimentin when cultured in EOC-CM relative to OSEC-CM treated cells. INOFs already expressed high levels of this marker and no change in expression was observed. (b) After 28 days of culture in EOC-CM, MSCs and in particular INOFs, upregulated expression of α-smooth muscle actin (αSMA), as measured by Western blotting. When cultured in Hey.A8-CM we measured a 1.4-fold and 19.4-fold increase in αSMA expression in MSCs and INOFs respectively (compared to day 0 controls). Beta-actin was used as a loading control. Proliferation assays for (c) MSCs and (d) INOFs cultured in EOC-CM and OSEC-CM; MSCs are significantly more proliferative when cultured in EOC-CM. Migration assays measuring chemotaxis of stromal cells towards conditioned media. (e) MSCs migrate more towards EOC-CM than serum free media or OSEC-CM. (f) INOFs migrate significantly more towards EOC-CM and OSEC-CM than serum-free media. Data shown are mean ± standard deviation (s.d.) for three independent experiments. *P<0.05, two-tailed paired Student's T-test, α=0.05, compared to SFM control. (g) MSCs and INOFs produce secreted factors that promote migration of epithelial ovarian cancer cells. Hey.A8 EOC cells are 2-3 fold more migratory in the presence of INOFs and MSCs compared to control media. 10% fetal bovine serum (FBS) was used as a chemoattractant. (h) EOC cells upregulate mesechymal markers in the presence of co-cultured stromal cells. VIM, vimentin; FN1, fibronectin. *P>0.05, two-tailed paired Student's T-test, α=0.05, compared to EOC cells cultured alone. Error bars = s.d.
Secreted factors produced by INOFs and MSCs promote transformed phenotypes in EOC cells
We used co-culture assays to evaluate whether secreted factors produced by MSCs and INOFs had the ability to enhance the neoplastic phenotype of EOC cells independently of cell-cell contact. Co-culture of Hey.A8 cells with INOFs and MSCs had no affect on proliferation (data not shown), but the same cells were 2-3 fold more migratory in the presence of INOFs and MSCs compared to serum free media (Fig. 2g), and mesenchymal markers were upregulated when ovarian cancer cells were co-cultured with both types of stromal cells (Fig. 2h).
Molecular changes associated with differentiation of NOFs and MSCs into CAFs
To identify candidate stromal-derived biomarkers, gene expression microarray analysis was performed to evaluate the molecular changes induced during the development of MSC and INOF derived CAFs by co-culture with Hey.A8 EOC cells. We observed widespread changes in gene expression occurring within just 7 days of co-culture. Using a false discovery rate threshold of 0.01 we found 2,825 significant probes differentially expressed for INOFs/MSCs co-cultured with Hey.A8 cells compared with INOFs/MSCs cultured alone, and 1282 differentially expressed probes between Hey.A8-co-cultured INOFs/MSCs and OSEC-co-cultured INOFs/MSCs. Finally, 859 probes were differentially expressed between Hey.A8-co-cultured INOFs/MSCs and both sets of controls; the most significantly up- and down-regulated of these probes are listed in Table 1.
Table 1. Differentially expressed genes in co-cultured stromal cells.
This table shows the top 20 up- and downregulated genes in INOFs and MSCs co-cultured with Hey.A8 ovarian cancer cells compared to INOFs/MSCs co-cultured with normal ovarian surface epithelial cells. Genes are ranked by pooled fold change.
| Rank | Probe_Id | Symbol | Gene Name | Fold Change v IOSE | Fold Change v non-Co-cultured | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| P Value | MSC | INOF | Pooled | P Value | MSC | INOF | Pooled | ||||
| Upregulated Genes | |||||||||||
|
| |||||||||||
| 1 | 4290040 | NPPB | natriuretic peptide B | 5.5E-06 | 71.86 | 9.42 | 28.23 | 1.8E-07 | 258.78 | 38.01 | 109.94 |
| 2 | 2030309 | KISS1 | KiSS-1 metastasis-suppressor | 0.013 | 10.64 | 1.06 | 9.12 | 1.0E-05 | 16.46 | 4.00 | 10.23 |
| 3 | 1110373 | SERPING1 | serpin peptidase inhibitor, clade G (C1 inhibitor), member 1 | 5.2E-06 | 11.15 | 3.60 | 7.90 | 0.003 | 25.53 | 1.70 | 9.89 |
| 4 | 1470148 | PTGIS | prostaglandin I2 (prostacyclin) synthase | 0.007 | 9.68 | 1.20 | 7.16 | 5.0E-05 | 9.45 | 2.57 | 6.14 |
| 5 | 7150762 | C15orf48 | chromosome 15 open reading frame 48 | 0.005 | 15.22 | 1.37 | 7.11 | 0.012 | 14.81 | 1.07 | 9.05 |
| 6 | 1430487 | MGP | matrix Gla protein | 0.008 | 8.36 | 1.16 | 5.83 | n.s. | 10.59 | 0.39 | 3.73 |
| 7 | 6020468 | MGP | matrix Gla protein | 0.006 | 7.59 | 1.19 | 5.46 | 2.6E-04 | 5.97 | 1.83 | 4.17 |
| 8 | 3460743 | PPP1R14A | protein phosphatase 1, regulatory (inhibitor) subunit 14A | 3.4E-04 | 7.73 | 1.90 | 4.87 | 1.5E-05 | 3.77 | 2.04 | 3.17 |
| 9 | 2940291 | MEGF6 | multiple EGF-like-domains 6 | 0.013 | 5.16 | 1.06 | 4.59 | 8.5E-05 | 4.06 | 1.76 | 2.68 |
| 10 | 940735 | CCDC81 | coiled-coil domain containing 81 | 7.3E-05 | 6.31 | 2.10 | 4.49 | 0.083 | 10.74 | 0.60 | 4.82 |
| 11 | 130044 | ACTG2 | actin, gamma 2, smooth muscle | 0.001 | 6.19 | 1.61 | 4.42 | 8.9E-07 | 7.55 | 3.65 | 4.82 |
| 12 | 6620292 | TINAGL1 | tubulointerstitial nephritis antigen-like 1 | 0.018 | 5.24 | 0.99 | 4.34 | 2.2E-07 | 4.00 | 2.59 | 3.30 |
| 13 | 3710154 | CYP26B1 | cytochrome P450, family 26, subfamily B, polypeptide 1 | 0.015 | 5.32 | 1.03 | 3.81 | 5.8E-04 | 12.57 | 2.00 | 3.72 |
| 14 | 60470 | TIMP3 | TIMP metallopeptidase inhibitor 3 | 0.008 | 3.67 | 1.11 | 3.63 | 4.6E-06 | 3.45 | 2.12 | 2.69 |
| 15 | 6100717 | COL11A1 | collagen, type XI, alpha 1 | 2.8E-05 | 4.34 | 2.07 | 3.49 | 4.4E-06 | 5.26 | 2.46 | 4.27 |
| 16 | 10543 | DHRS3 | dehydrogenase/reductase (SDR family) member 3 | 4.6E-09 | 3.17 | 3.40 | 3.28 | 4.9E-08 | 2.20 | 2.14 | 2.17 |
| 17 | 2450110 | CRYAB | crystallin, alpha B | 2.2E-04 | 3.38 | 1.51 | 3.27 | 1.1E-08 | 2.35 | 2.46 | 2.41 |
| 18 | 4610431 | EPDR1 | ependymin related protein 1 | 2.4E-08 | 3.35 | 2.73 | 3.25 | 0.001 | 8.58 | 1.70 | 5.46 |
| 19 | 4590008 | CRISPLD2 | cysteine-rich secretory protein LCCL domain containing 2 | 1.4E-08 | 3.00 | 3.77 | 3.22 | 1.0E-04 | 3.58 | 1.72 | 2.59 |
| 20 | 4180079 | CNN1 | calponin 1, basic, smooth muscle | 2.6E-05 | 4.00 | 1.95 | 3.20 | 2.9E-07 | 3.51 | 6.68 | 4.16 |
|
| |||||||||||
| Downregulated Genes | |||||||||||
|
| |||||||||||
| 1 | 3360224 | MMP1 | matrix metallopeptidase 1 | 0.024 | 0.18 | 1.08 | 0.22 | 0.012 | 0.33 | 0.94 | 0.39 |
| 2 | 1240440 | TXNIP | thioredoxin interacting protein | 4.9E-10 | 0.22 | 0.25 | 0.22 | 6.2E-06 | 0.42 | 0.21 | 0.37 |
| 3 | 6370221 | DIRAS3 | DIRAS family, GTP-binding RAS-like 3 | 0.011 | 0.18 | 0.91 | 0.23 | 0.001 | 0.70 | 0.65 | 0.68 |
| 4 | 630315 | DHRS9 | dehydrogenase/reductase (SDR family) member 9 | 0.029 | 0.19 | 1.07 | 0.29 | 0.030 | 0.43 | 1.01 | 0.56 |
| 5 | 840204 | HAS2 | hyaluronan synthase 2 | 0.016 | 0.20 | 0.98 | 0.33 | n.s. | 0.23 | 2.16 | 0.41 |
| 6 | 7050575 | PPAP2B | phosphatidic acid phosphatase type 2B | 0.003 | 0.33 | 0.83 | 0.35 | 0.001 | 0.16 | 0.66 | 0.17 |
| 7 | 5810047 | F2RL1 | coagulation factor II (thrombin) receptor-like 1 | 0.025 | 0.11 | 1.10 | 0.35 | 0.069 | 0.21 | 1.28 | 0.60 |
| 8 | 6220019 | PENK | proenkephalin | 0.014 | 0.27 | 0.98 | 0.38 | 0.016 | 0.17 | 0.93 | 0.26 |
| 9 | 6290142 | LOC100008589 | 28S ribosomal RNA | 0.015 | 0.27 | 0.72 | 0.39 | n.s. | 0.84 | 0.58 | 0.69 |
| 10 | 7210681 | LIPG | lipase, endothelial | 2.1E-04 | 0.16 | 0.50 | 0.40 | 2.3E-05 | 0.45 | 0.34 | 0.35 |
| 11 | 2100767 | COLEC12 | collectin sub-family member 12 | 0.019 | 0.38 | 0.97 | 0.41 | 0.008 | 0.26 | 0.91 | 0.28 |
| 12 | 1050068 | F2RL1 | coagulation factor II (thrombin) receptor-like 1 | 0.029 | 0.11 | 1.13 | 0.41 | 0.040 | 0.17 | 1.19 | 0.58 |
| 13 | 1300470 | MAMDC2 | MAM domain containing 2 | 0.002 | 0.81 | 0.39 | 0.42 | 4.2E-05 | 0.59 | 0.49 | 0.50 |
| 14 | 2120452 | MLPH | melanophilin | 0.004 | 0.39 | 0.87 | 0.42 | 1.5E-04 | 0.44 | 0.72 | 0.46 |
| 15 | 770730 | ANXA2 | annexin A2 | 1.8E-04 | 0.28 | 0.55 | 0.43 | 5.9E-04 | 0.19 | 0.58 | 0.36 |
| 16 | 4260017 | CDK5RAP2 | CDK5 regulatory subunit associated protein 2 | 0.006 | 0.29 | 0.88 | 0.43 | 8.7E-04 | 0.31 | 0.72 | 0.43 |
| 17 | 6220097 | PPAP2B | phosphatidic acid phosphatase type 2B | 0.022 | 0.39 | 1.01 | 0.44 | 0.009 | 0.20 | 0.89 | 0.24 |
| 18 | 6980411 | LOC100134259 | uncharacterised miscRNA | 0.004 | 0.37 | 0.87 | 0.44 | 3.3E-04 | 0.44 | 0.71 | 0.49 |
| 19 | 870630 | LYPD1 | LY6/PLAUR domain containing 1 | 2.6E-05 | 0.32 | 0.54 | 0.45 | 1.3E-05 | 0.37 | 0.60 | 0.49 |
| 20 | 6290341 | EFEMP1 | EGF containing fibulin-like extracellular matrix protein 1 | 0.002 | 0.54 | 0.43 | 0.46 | n.s. | 1.65 | 0.67 | 1.02 |
Genes that showed the greatest increase in expression in Hey.A8 co-cultured INOFs/MSCs compared to OSEC co-cultured INOFs/MSCs included natriuretic peptide B (NPPB, P=5.5×10-6), a cardiac hormone; the KiSS-1 metastasis suppressor gene (KISS1, P=0.013); serpin peptidase inhibitor, clade G (C1 inhibitor), member 1 (SERPING1, P=5.2×10-6), a regulator of complement activation; and prostaglandin I2 synthase (PTGIS, P=6.9×10-3), a potent vasodilator and inhibitor of platelet aggregation. Downregulated genes included matrix metallopeptidase 1 (MMP1, P=0.024), a known tumour suppressor gene (TXNIP, P=4.9×10-10), and genes involved in differentiation: ANXA2, (P=1.8×10-4), and CDK5RAP2, (P= 5.6×10-3) which have a role in the development of osteoclasts and neuronal cells, respectively.
Gene Ontology enrichment analyses indicated that genes encoding proteins that are secreted or expressed on the cell membrane are the most commonly enriched pathways in our CAF models. Extracellular matrix genes showed the greatest changes in expression between Hey.A8 co-cultured INOFs/MSCs and controls (adjusted P=2.11×10-8) (Fig. 3, Supplementary Table 3). Terms associated with adhesion, cell locomotion, migration and female pregnancy were also among the enriched biological processes terms (adjusted P=0.005). In the molecular function category, the most enriched term was ‘integrin binding’ (P=2.81×10-4). KEGG pathway analysis identified genes enriched in cell-matrix interactions (adjusted P=0.009), and molecules associated with the mitogen-activated protein kinase and p53 signaling pathways, although these latter terms were not significant after adjustment (Supplementary Table 4).
Figure 3. Gene Ontology (GO) Analyses.
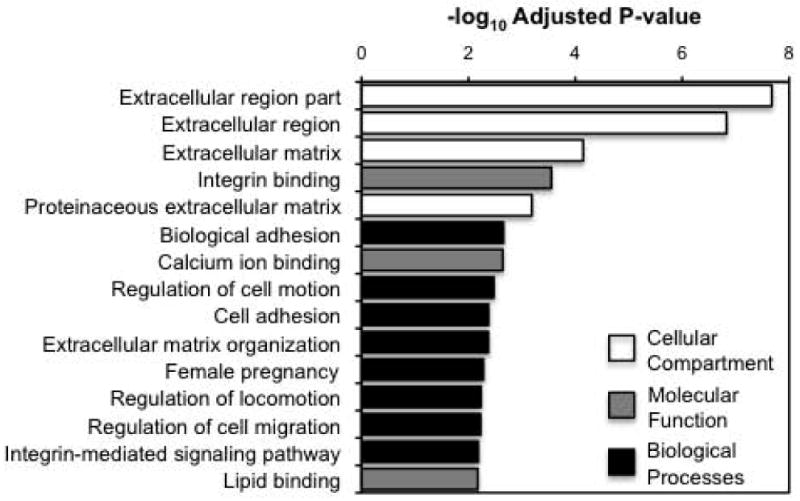
Combined analysis of enriched GO terms, all GO terms within the biological processes, cellular compartments and molecular function categories were ranked by adjusted p-values. The 20 most significant terms are shown. The 20 most significantly enriched terms for each individual category are listed in Supplementary Table 1.
NPPB is a novel candidate stromal-derived biomarker that can be detected in the blood of EOC patients
Using in vitro transwell co-culture models, we validated the expression of the gene that showed the largest fold-changes in both INOF and MSCs: NPPB. We analysed NPPB expression in four stromal cell lines (INOFs, MSC1, MSC2 and a skin fibroblast line) co-cultured with 7 different with EOC and two normal OSEC lines. NPPB expression was typically higher in stromal cells co-cultured with EOC cell lines compared to the same cells co-cultured with OSECs (Fig. 4A). Changes in expression of NPPB were absent or negligible in skin fibroblasts, suggesting that this marker is specifically upregulated in ovarian CAFs. Immunohistochemical staining in primary tissues verified that NPPB is also expressed in the stroma of primary human ovarian cancers. In a tissue microarray of 145 EOC specimens; NPPB was expressed by stromal fibroblasts in 87/145 (60%) ovarian cancers (Figure 4) but expression was absent or weak normal ovarian stromal tissues (n=4). NPPB expression in tumours was positively associated with stage (P=0.032, Fisher's Exact Test) and serous histology (P=0.010, Fisher's Exact Test) (Table 2). We did not see a survival association when we performed Kaplan-Meier estimates with a log rank test, which may be due to the small number of cases that could be included in this analysis (n=65; NPPB positive, n=42; NPPB negative, n=23). NPPB is secreted with an N-terminal fragment (NT-proBNP) that is biological inactive but has a longer half-life than the active NPPB protein. NT-proBNP levels were measured by electrochemiluminescence assays in eight patients with ovarian cancer who did not have congestive or acute heart failure (BNP/NT-proBNP levels are used to diagnose these conditions in the clinic). In patients not suffering from congestive heart failure, levels of NT-proBNP 100pg/ml or greater are considered to be elevated 26. In 4/8 patients (without cardiac failure), levels of NT-proBNP were elevated in the blood, suggesting NPPB is a novel candidate stromal-derived biomarker for EOC.
Figure 4. Validation of NPPB expression in EOC models and primary tumours.
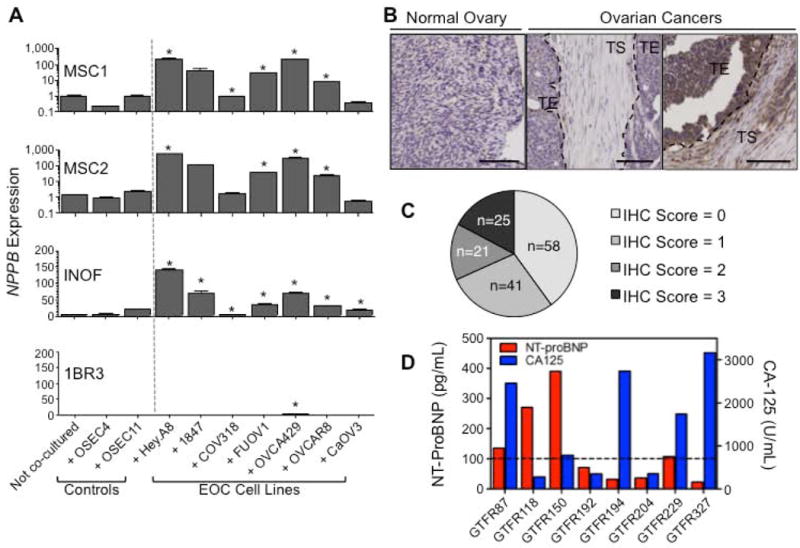
NPPB is the most significantly upregulated gene (ranked by fold change). Expression of (a) NPPB in stromal cells co-cultured for 7 days with 2 normal OSEC lines and 7 EOC cell lines. NPPB expression in stromal cells is significantly upregulated in response to co-culture with at least 50% of EOC cell lines; with expression levels of NPPB increasing up to 350-fold when INOFs and MSCs were co-cultured with EOC cells compared to controls. Expression of NPPB was significantly lower in stromal cells co-cultured with OSECs than the same cells co-cultured with the majority of EOC cell lines. Expression of NPPB is absent/negligible in 1BR3 skin fibroblasts, regardless of culture conditions. Note that for NPPB expression MSC values are shown on a log scale, whereas INOF and 1BR3 data are on a linear scale. In the microarrays and by qPCR, MSCs tended to show larger changes in gene expression than INOFs. (*P<0.05, two-tailed paired Student's T-test, α=0.05, compared to at least 2/3 controls: non-co-cultured stromal cells and OSEC-co-cultured stromal cells). Error bars = s.d. (b) NPPB protein is expressed in ovarian tumours. Right panel, example of a tumour staining positive for the active, secreted NPPB peptide. Expression is seen in the tumour stromal (TS) cells as well as, in some cases, diffuse staining in the tumour epithelium (TE), which may be due to diffusion of the secreted protein. Middle panel shows an example of a tumour that does not express NPPB. Left panel, NPPB expression was not detected in normal ovarian stromal cells. (c) Analysis of NPPB protein expression in tumour stromal cells of 145 primary EOCs stained by immunohistochemistry. (d) Measurement of NT-proBNP levels in patients with ovarian cancer. NT-proBNP is elevated in 4/8 EOC patients independently of cardiac failure. Dashed line = cutoff for elevated NT-proBNP (100pg/ml).
Table 2. Immunohistochemical staining of NPPB in EOC specimens.
145 tumours were stained for NPPB. Patient characteristics can be found in Supplementary Table 2. P-values indicate the results of Fisher's Exact tests. Statistically significant associations are shown in bold.
|
|
||||
|---|---|---|---|---|
| NPPB Negative | NPPB Positive | P-value | ||
| All tumours | 99 | 46 | NA | |
| Grade | G1 | 9 | 5 | |
| G2/G3 | 50 | 81 | 0.085 | |
| Stage | Stage I/II | 20 | 16 | |
| Stage III/IV | 38 | 71 | 0.032 | |
| Histology | Serous† | 31 | 60 | |
| Non-serous | 26 | 19 | 0.060 | |
Low grade serous tumours were excluded from this analysis,
Discussion
The tumour microenvironment offers potential as a therapeutic target and source of biomarkers for many tumour types. For epithelial ovarian cancer (EOC) there is a particularly urgent need for new minimally invasive approaches to tumour detection and monitoring, ideally for biomarkers that can be measured in the blood. CA-125 is a biomarker currently used clinically to detect EOC. However CA-125 has limited specificity as it can be expressed by many other normal and transformed gynecological epithelial cells, including common conditions such as endometrial cancer and endometriosis. CA-125 also has limited sensitivity as not all EOCs express CA-125, particularly at the early stages and so we explored the potential for ovarian cancer stromal fibroblasts to be a novel source of biomarkers for this disease. Since the origins of cancer-associated fibroblasts (CAFs) in epithelial ovarian cancers is poorly understood, we began by comparing the potential of normal ovarian fibroblasts and mesenchymal stem cells to become CAFs. In this study we both co-cultured epithelial and stromal cells directly, and indirectly, by producing conditioned media. In contrast to previous reports, which only use serum free media as a control we also prepared conditioned media from normal ovarian surface epithelial cells. We did find that OSEC conditioned media had a modest effect on the expression of CAF markers by stromal cells, highlighting the importance of including normal epithelial cells as a control. Normal OSEC conditioned medium however, did not induce high levels of CAF marker expression in MSCs nor CAFs, and CAF-like phenotypes were strongest when INOFs and MSCs were co-cultured with EOC cells. These data agree with previous reports that cancer cells induce CAF phenotypes in CAF precursor cells 11, 15. An alternative interpretation of these data is that signaling from normal OSECs inhibits CAF development, but when the tumor suppressive signaling from a normal microenvironment is lost, during tumor development, inhibition of CAF development is lost.
In our in vitro models both resident normal ovarian fibroblasts and bone marrow derived mesenchymal stem cells have the ability to acquire the expression of CAF markers and promote neoplastic behavior, moreover in vivo, both INOFs and CAFs could promote engraftment of EOC cells. Our findings may therefore suggest that ovarian tumour fibroblasts may come from multiple origins, although further analyses of human tumors would be required to validate this hypothesis. It may be that at different stages of tumourigenesis, stromal cells are recruited from different sources depending on the gene expression programs activated in the tumour epithelium at that time. Given that EOCs are also highly heterogenous tumours, in terms of histology, clinical outcomes, molecular profiles and origins, it is also highly plausible that the molecular and biological characteristics of EOC CAFs also display a high degree of inter-patient variability. Our model most closely mimics Type 2 ovarian cancers, which consists of high-grade serous and endometrioid tumours, and, consistent with this, NPPB expression was associated with advanced stage and high-grade serous histology.
Deregulated genes from ovarian CAFs were highly enriched for genes encoding proteins expressed within the extracellular milieu or on the plasma membrane. This supports our hypothesis that CAFs represent a new source of tumour biomarkers as this group of genes represents the most promising candidates for blood-borne biomarkers – for example CA-125 is a mucin expressed on the surface of ovarian cancer cells and shed into the bloodstream where it is measured to monitor ovarian cancer tumour burden. When cultured with EOC cells, CAF precursors produced an abundance of secreted molecules including NPPB, which increased in expression over three hundred-fold in CAFs compared to controls. NPPB is a secreted peptide that acts as a cardiac hormone produced by the heart and is used clinically in the diagnosis of acute and congestive heart failure. NPPB has not previously been implicated in ovarian cancer pathogenesis, although interestingly, one study did report elevated NPPB levels in lung cancer patients 27. We did not detect expression of the known NPPB receptor (NPR1) on the EOC cell lines used in this study (data not shown), leading us to hypothesize that NPPB may bind a novel receptor on EOC cells or alternatively may target other cell types within the tumour stroma. We were able to detect elevated levels of NPPB in the blood of 50% of ovarian cancer patients, by measuring levels of the 76 amino-acid N-terminal fragment that is secreted together with NPPB. This fragment is more stable and therefore represents a potentially more sensitive screening biomarker than active NPPB. Elevation of NPPB in patients with heart failure could limit the use of this biomarker for detection of EOC, but other indirect screening biomarkers related to NPPB may also exist since NPPB is a target for the prolyl oligopeptidase FAP, a CAF marker not expressed by normal stromal cells and so the products of FAP mediated proteolysis of NPPB may represent alternative, highly specific tumour biomarkers 28. Large clinical screening trials would be needed to comprehensively evaluate the potential of NPPB and related proteins to serve as novel EOC biomarkers that may increase the sensitivity and/or specificity of CA-125.
Other differentially expressed markers we identified included serpin peptidase inhibitor, clade G (C1 inhibitor), member 1 (SERPING1) a potent vasodilator and inhibitor of platelet aggregation. Plausibly, SERPING1 could promote vascular permeability in the tumour milieu, thus enhancing tumour cell nourishment. Expression of SERPING1 has previously been found to be upregulated in myoepithelial cells in the stroma of breast cancers. A group of pregnancy-associated biomarkers was also upregulated in the ovarian CAF models, including pregnancy-associated plasma protein A and pregnancy-specific beta-1 glycoprotein-1, -2, -4, -5 and -6 (PSG1,2,4-6). Previous studies have shown that other pregnancy-associated placental proteins are elevated in the blood of ovarian cancer patients 29, 30. The PSG genes are a subgroup of glycoproteins within the carcinoembryonic antigen (CEA) family of genes. The PSG genes identified in this present study have not previously been implicated in EOC, and so may represent a novel class of clinical biomarkers for this disease. In support of this, studies have shown that CEA is overexpressed in many solid tumours and may have clinical utility as part of a multi-biomarker assay for the detection of epithelial ovarian carcinoma 31.
Cancer biomarker and therapeutic discovery efforts are usually based on in vitro models of tumour epithelium; but our data show that extracellular and secreted protein cancer biomarkers are produced in abundance by the tumour-associated mesenchyme. Analyses of a skin fibroblast cell line suggested that these proteins were specific to ovarian CAF precursor cells, although additional stromal models would need to be evaluated to confirm this finding. Several biomarkers showed substantial differential expression (>10-fold differences compared to controls) between CAFs and controls. If this replicated in vivo, it is likely that many of these secreted molecules enter the blood stream, which will enable their clinical detection. Our data also suggest that production of extracellular antigens by CAFs may be an early event during ovarian CAF development. Widespread changes in stromal cell gene expression occurred shortly after co-culture with EOC cells but before significant upregulation of classical CAF markers, suggesting that stromal biomarkers may perhaps be able to detect ovarian cancers at the earliest, most treatable, stages of disease. Currently the vast majority of invasive ovarian cancers are diagnosed at the late-stages, primarily because there are no early stage biomarkers with sufficient sensitivity and specificity. A panel of blood-based biomarkers that could reliably detect early-stage ovarian cancers of all histologies could potentially be used for population-based ovarian cancer screening of at risk women and could have a significant impact on reducing mortality from the disease. The biomarker currently used in the clinic, CA-125, is excellent at detecting tumour recurrence but cannot reliably detect early-stage tumours and currently so the majority of cases are diagnosed at the advanced stages (III and IV) when 5-year survival rates are only ∼30%. While NPPB was associated with higher tumour stage, and was expressed by only 28% of early-stage EOCs, some of the other biomarkers we identified may be more commonly expressed by early-stage EOCs and so could potentially be used as early-stage biomarkers.
CAF-like phenotypes were rapidly acquired and since it is unlikely that multiple genetic aberrations could have accumulated in this short timeframe, this suggests that epigenetic mechanisms may drive the development of CAFs in EOC. This is consistent with previous observations that mutations in the stroma of ovarian tumours are rare 12, 13. Although the mechanisms underlying CAF development have yet to be fully elucidated, by establishing ovarian cancer associated fibroblasts we have identified NPPB as a novel candidate CAF-specific marker for ovarian cancer. Future studies will focus on evaluating the utility of NPPB and other candidate stromal genes as blood serum biomarkers for the detection of ovarian cancers.
Supplementary Material
Supplementary Table 1. Cell culture media. Details of the cell culture media used for growth of ovarian cancer cell lines and normal ovarian cells. FBS, foetal bovine serum.
Supplementary Table 2. Patient characteristics in the tissue microarray.
Supplementary Table 3. Gene ontology (GO) analyses of gene differentially expressed in co-cultured stromal cells. DAVID was used to test for enrichment of Biological Processes, Cellular Compartments and Molecular Function GO terms. Genes differentially expressed by EOC-co-cultured stromal cells were enriched for pathways involved in female pregnancy, and tended to be associated with the extracellular milieu. Statistically significant terms are shown (P<0.05, adjusted).
Supplementary Table 4. Gene ontology (GO) analyses of gene differentially expressed in co-cultured stromal cells. DAVID was used to test for enrichment of KEGG pathway analysis terms. Statistically significant terms are shown (P<0.05, unadjusted).
Acknowledgments
K.L. is funded by National Institute of Health grant 5 U19 CA148112-02 and an Ann Schreiber Research Training Program of Excellence award from the Ovarian Cancer Research Fund. B.G. was supported by a studentship from the MRC. The majority of this research was performed at the Keck School of Medicine, University of Southern California, Los Angeles, California, USA, within the Norris Comprehensive Cancer Centre (NCI CCSG grant P30CA014089). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. Some of this work undertaken at UCLH/UCL and was partly funded by the Department of Health's NIHR Biomedical Research Centre funding scheme. This work as also partly funded by the Wright Foundation through the USC CTSI. We also thank Tassja J Spindler for a critical review of this manuscript.
Footnotes
Author Contributions: K.L and B.G designed and performed experiments and analysed data. N.L and S.S assisted with experiments. J.M. analysed gene expression microarray data. P.M.F, T.P and J.K performed analyses of protein expression in tumours. Y.G.L collected blood sera from EOC patients. K.L and S.A.G wrote the manuscript, all authors contributed to the final draft of the paper.
Contributor Information
Kate Lawrenson, Email: kate.lawrenson@med.usc.edu.
Barbara Grun, Email: b.grun@ucl.ac.uk.
Nathan Lee, Email: nathanl@usc.edu.
Paulette Mhawech-Fauceglia, Email: mhawechf@usc.edu.
Steve Swenson, Email: sswenson@usc.edu.
Yvonne G. Lin, Email: ylinliu@usc.edu.
Tanja Pejovic, Email: pejovict@ohsu.edu.
Joshua Millstein, Email: joshua.millstein@usc.edu.
Simon A Gayther, Email: gayther@usc.edu.
References
- 1.http://seer.cancer.gov
- 2.Network CGAR. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Labiche A, Heutte N, Herlin P, Chasle J, Gauduchon P, Elie N. Stromal compartment as a survival prognostic factor in advanced ovarian carcinoma. Int J Gynecol Cancer. 2010;20:28–33. doi: 10.1111/IGC.0b013e3181bda1cb. [DOI] [PubMed] [Google Scholar]
- 4.Hao M, Zhang L, An G, Meng H, Han Y, Xie Z, Xu Y, Li C, Yu Z, Chang H, Qiu L. Bone marrow stromal cells protect myeloma cells from bortezomib induced apoptosis by suppressing microRNA-15a expression. Leuk Lymphoma. 2011;52:1787–94. doi: 10.3109/10428194.2011.576791. [DOI] [PubMed] [Google Scholar]
- 5.Nefedova Y, Landowski TH, Dalton WS. Bone marrow stromal-derived soluble factors and direct cell contact contribute to de novo drug resistance of myeloma cells by distinct mechanisms. Leukemia. 2003;17:1175–82. doi: 10.1038/sj.leu.2402924. [DOI] [PubMed] [Google Scholar]
- 6.Müerköster S, Wegehenkel K, Arlt A, Witt M, Sipos B, Kruse ML, Sebens T, Klöppel G, Kalthoff H, Fölsch UR, Schäfer H. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1beta. Cancer Res. 2004;64:1331–7. doi: 10.1158/0008-5472.can-03-1860. [DOI] [PubMed] [Google Scholar]
- 7.Singh RR, Kunkalla K, Qu C, Schlette E, Neelapu SS, Samaniego F, Vega F. ABCG2 is a direct transcriptional target of hedgehog signaling and involved in stroma-induced drug tolerance in diffuse large B-cell lymphoma. Oncogene. 2011;30:4874–86. doi: 10.1038/onc.2011.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang G, Rosen DG, Zhang Z, Bast RC, Mills GB, Colacino JA, Mercado-Uribe I, Liu J. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci U S A. 2006;103:16472–7. doi: 10.1073/pnas.0605752103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai J, Tang H, Xu L, Wang X, Yang C, Ruan S, Guo J, Hu S, Wang Z. Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness. Carcinogenesis. 2012;33:20–9. doi: 10.1093/carcin/bgr230. [DOI] [PubMed] [Google Scholar]
- 10.R S, Morikawa T, Shee K, Barzily-Rokni M, Rong Qian Z, Du J, Davis A, Mongare MM, Gould J, Frederick DT, Cooper ZA, Chapman PB, et al. Nature. AOP; 2012. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, Andreeff M, Marini FC. Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS One. 2012;7:e30563. doi: 10.1371/journal.pone.0030563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M, Trivett M, Thompson ER, Ramakrishna M, Gorringe KL, Polyak K, Haviv I, et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet. 2008;40:650–5. doi: 10.1038/ng.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akahane T, Hirasawa A, Tsuda H, Kataoka F, Nishimura S, Tanaka H, Tominaga E, Nomura H, Chiyoda T, Iguchi Y, Yamagami W, Susumu N, et al. The origin of stroma surrounding epithelial ovarian cancer cells. Int J Gynecol Pathol. 2013;32:26–30. doi: 10.1097/PGP.0b013e3182518533. [DOI] [PubMed] [Google Scholar]
- 14.Jazedje T, Perin PM, Czeresnia CE, Maluf M, Halpern S, Secco M, Bueno DF, Vieira NM, Zucconi E, Zatz M. Human fallopian tube: a new source of multipotent adult mesenchymal stem cells discarded in surgical procedures. J Transl Med. 2009;7:46. doi: 10.1186/1479-5876-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, Andreeff M, Marini F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One. 2009;4:e4992. doi: 10.1371/journal.pone.0004992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shangguan L, Ti X, Krause U, Hai B, Zhao Y, Yang Z, Liu F. Inhibition of TGF-β/Smad Signaling by BAMBI Blocks Differentiation of Human Mesenchymal Stem Cells to Carcinoma-Associated Fibroblasts and Abolishes Their Pro-Tumor Effects. Stem Cells. 2012 doi: 10.1002/stem.1251. [DOI] [PubMed] [Google Scholar]
- 17.McLean K, Gong Y, Choi Y, Deng N, Yang K, Bai S, Cabrera L, Keller E, McCauley L, Cho KR, Buckanovich RJ. Human ovarian carcinoma–associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J Clin Invest. 2011;121:3206–19. doi: 10.1172/JCI45273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li NF, Wilbanks G, Balkwill F, Jacobs IJ, Dafou D, Gayther SA. A modified medium that significantly improves the growth of human normal ovarian surface epithelial (OSE) cells in vitro. Lab Invest. 2004;84:923–31. doi: 10.1038/labinvest.3700093. [DOI] [PubMed] [Google Scholar]
- 19.Lawrenson K, Grun B, Benjamin E, Jacobs IJ, Dafou D, Gayther SA. Senescent fibroblasts promote neoplastic transformation of partially transformed ovarian epithelial cells in a three-dimensional model of early stage ovarian cancer. Neoplasia. 2010;12:317–25. doi: 10.1593/neo.91948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korch C, Spillman MA, Jackson TA, Jacobsen BM, Murphy SK, Lessey BA, Jordan VC, Bradford AP. DNA profiling analysis of endometrial and ovarian cell lines reveals misidentification, redundancy and contamination. Gynecol Oncol. 2012;127:241–8. doi: 10.1016/j.ygyno.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rasband W, Rasband WS. ImageJ. U. S. National Institutes of Health, Bethesda; Maryland, USA: 1997-2012. http://imagej.nih.gov/ij/ [Google Scholar]
- 22.Dunning MJ, Smith ML, Ritchie ME, Tavaré S. beadarray: R classes and methods for Illumina bead-based data. Bioinformatics. 2007;23:2183–4. doi: 10.1093/bioinformatics/btm311. [DOI] [PubMed] [Google Scholar]
- 23.J M, D V, JR L, H D, SH F, EE S, J B. Permutation-based yet computationally parsimonious FDR point and confidence interval estimators. Joint Statistical Meetings. 2010:4184–97. [Google Scholar]
- 24.Huang dW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 25.Wysham WZ, Mhawech-Fauceglia P, Li H, Hays L, Syriac S, Skrepnik T, Wright J, Pande N, Hoatlin M, Pejovic T. BRCAness profile of sporadic ovarian cancer predicts disease recurrence. PLoS One. 2012;7:e30042. doi: 10.1371/journal.pone.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCullough PA. B-type natriuretic peptides. A diagnostic breakthrough in heart failure. Minerva Cardioangiol. 2003;51:121–9. [PubMed] [Google Scholar]
- 27.Aujollet N, Meyer M, Cailliod R, Combier F, Coignet Y, Campard S, Facy O, Bernard A, Girard C. High N-terminal pro-B-type natriuretic peptide: a biomarker of lung cancer? Clin Lung Cancer. 2010;11:341–5. doi: 10.3816/CLC.2010.n.043. [DOI] [PubMed] [Google Scholar]
- 28.Keane FM, Nadvi NA, Yao TW, Gorrell MD. Neuropeptide Y, B-type natriuretic peptide, substance P and peptide YY are novel substrates of fibroblast activation protein-α. FEBS J. 2011;278:1316–32. doi: 10.1111/j.1742-4658.2011.08051.x. [DOI] [PubMed] [Google Scholar]
- 29.Göcze PM, Szabò DG, Than GN, Krommer KF, Csaba IF, Bohn H. Placental protein 4 as a possible tumor marker in ovarian tumors. Gynecol Obstet Invest. 1991;32:107–11. doi: 10.1159/000293006. [DOI] [PubMed] [Google Scholar]
- 30.Lenhard M, Tsvilina A, Schumacher L, Kupka M, Ditsch N, Mayr D, Friese K, Jeschke U. Human chorionic gonadotropin and its relation to grade, stage and patient survival in ovarian cancer. BMC Cancer. 2012;12:2. doi: 10.1186/1471-2407-12-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yurkovetsky Z, Skates S, Lomakin A, Nolen B, Pulsipher T, Modugno F, Marks J, Godwin A, Gorelik E, Jacobs I, Menon U, Lu K, et al. Development of a multimarker assay for early detection of ovarian cancer. J Clin Oncol. 2010;28:2159–66. doi: 10.1200/JCO.2008.19.2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Cell culture media. Details of the cell culture media used for growth of ovarian cancer cell lines and normal ovarian cells. FBS, foetal bovine serum.
Supplementary Table 2. Patient characteristics in the tissue microarray.
Supplementary Table 3. Gene ontology (GO) analyses of gene differentially expressed in co-cultured stromal cells. DAVID was used to test for enrichment of Biological Processes, Cellular Compartments and Molecular Function GO terms. Genes differentially expressed by EOC-co-cultured stromal cells were enriched for pathways involved in female pregnancy, and tended to be associated with the extracellular milieu. Statistically significant terms are shown (P<0.05, adjusted).
Supplementary Table 4. Gene ontology (GO) analyses of gene differentially expressed in co-cultured stromal cells. DAVID was used to test for enrichment of KEGG pathway analysis terms. Statistically significant terms are shown (P<0.05, unadjusted).