

INTRODUCTION
Mucopolysaccharidosis (MPS) IIIB is an autosomal recessive lysosomal storage disease (LSD) caused by defects of α-N-Acetylglucosaminidase (NaGlu), an enzyme, involved in degrading one of a group of biologically important glycosaminoglycans (GAG) in lysosomes.1 The primary pathology of MPS IIIB is lysosomal accumulation of heparan sulfate (HS), in somatic cells and the central nervous system (CNS), especially cells throughout the CNS, with complex secondary pathology manifestations in MPS IIIB mouse brain.2–8 MPS IIIB infants appear normal at birth but develop progressive severe neurological manifestations, causing high mortality and premature death. The somatic manifestation in MPS IIIB is mild relative to other MPS, such as MPS I, II and VII. No treatment is currently available for MPS IIIB. The disease is not amenable to either recombinant enzyme replacement therapy (ERT) or hematopoietic stem cell transplantation (HSCT), which have been used to treat mostly somatic disorders in patients with MPS I, II and IV.1,9,10 This is because the neurological pathology in MPS IIIB is global and the blood brain barrier (BBB) precludes effective central nervous system (CNS) access.
Effective treatments for the majority CNS diseases are rare, since the CNS is a unique system, located in a well protected environment, and isolated by a highly defined anatomical/functional barrier. The BBB is a cellular interface between the blood circulation and the CNS, formed mainly by capillary endothelial cells with tight junctions, and enhanced by surrounding cells.11,12 The BBB is completely formed at birth in humans. In general, the BBB protects the CNS by selectively regulating the transport of molecules/agents from the blood circulation into the CNS or vice versa. Likewise, it also prevents potential therapeutic materials from entering the CNS. The presence of the BBB is the most critical challenge to developing therapies for CNS diseases, especially global CNS disorders, since targeting the entire CNS can be most effectively achieved only by systemic delivery through vasculature.13
Over the years, many strategies have been developed to deliver therapeutic agents into the CNS, though detailed mechanisms of BBB function remain largely unknown.11,14 These strategies include osmotic disruption of the BBB, receptor-mediated delivery by drug manipulation, and direct delivery bypassing the BBB.13,15–17 Mannitol, a well-characterized osmotic agent, has long been administered by intravascular infusion in routine medical practice for various purposes, the most important of which has been the temporary opening of the BBB.13 Previous animal experiments have shown that disrupting the BBB with mannitol by an intra (carotid) arterial (IA) injection made the BBB permeable to a wide range of substances, including antibodies, enzymes and viral vectors.13,18–23 Clinical studies in patients with brain tumors showed improved survival through mannitol facilitated CNS delivery of chemotherapeutic drugs by IA injection via the carotid artery.15 Intravenous (IV) delivery offers potential benefits for treating global CNS diseases, since it would result in non-differential distribution throughout the CNS. Previous studies showed that the peak opening of BBB in rats was 5 minutes after an IA infusion of mannitol and the opening lasted 20–30 minutes.24 However, the optimal conditions for IV injection of mannitol to disrupt the BBB, in order to enhance CNS entry of IV-delivered substances, were unclear.
Gene therapy has great potential for treating LSDs, due to possible long-term transduction of affected cells, and the bystander effect of secreted lysosomal enzymes, including NaGlu.25–27 The adeno-associated viral (AAV) vector system has been widely studied as a gene delivery tool for treating various diseases, with demonstrated therapeutic effect. The recombinant AAV (rAAV) vectors have a broad spectrum of tissue tropism, which can be varied through the use of different serotypes. To date, no known pathogenesis has been linked to AAV in humans.28 Previous studies demonstrated that rAAV vectors target both neuronal and non-neuronal cells in the CNS of animals.21,29,30 A number of studies using rAAV vectors have shown therapeutic impacts for the CNS diseases of MPS IIIB, and other LSDs, in animal models.31–37 Recent AAV gene therapy clinical trials have shown benefits in patients with Parkinson’s disease and late infantile neuronal ceroid lipofuscinosis.38,39 However, in the majority of rAAV CNS gene therapy studies, vectors were delivered by direct intracranial injection, which has limited therapeutic potential for treating global CNS diseases.31 Intravenous rAAV injection into neonatal MPS I and MPS VII mice led to long-term correction of lysosomal accumulation in both somatic tissue and the CNS, which persisted into adulthood.40–43 However, the BBB may still be permeable in neonatal mice but closed at birth in humans. Previously, we demonstrated that pretreatment with an IV infusion of mannitol facilitated the CNS entry of IV-delivered rAAV serotype 2 (rAAV2) reporter vector, and resulted in a diffuse global distribution of transduced neuronal and non-neuronal cells throughout the CNS in adult mice, though the conditions for CNS delivery of rAAV by IV infusion were not fully optimized.21 In addition, IV vector delivery may be ideal for treating MPS IIIB, since the disease also affects virtually all somatic organs, though the neuropathology is the cause of fatality in patients.
In this study, we have optimized mannitol-facilitated CNS entry of IV-delivered rAAV vector to assess the therapeutic potential for treating the neurological disease in MPS IIIB mice. We performed IV injection of rAAV2 vector with a refined time course relative to the administration of mannitol, thus delineating the maximum potential for rAAV-CNS entry. Using these conditions, an IV infusion of a therapeutic rAAV2 vector significantly slowed disease progression and extended the survival of MPS IIIB mice.
RESULTS
Mannitol-facilitated rAAV2-CNS entry: optimized vector CNS entry and diffuse global distribution of transgene expression
Based on the hypothesis that, mannitol facilitated rAAV vector entry into the CNS via the BBB would depend on a combination of dynamic factors including the time of maximum opening of the BBB, the concentration of vector in the bloodstream, and the movement of fluid between the CNS and bloodstream, we directly assessed the optimum timing between mannitol infusion and vector injection. A self-complementary rAAV vector, scAAV2-CMV-GFP, was used in these biodistribution experiments, for its much higher transduction efficiency than conventional single-stranded vetor.44 An IV infusion of mannitol was administered into 6–8-week-old wildtype (wt) mice to temporarily disrupt the BBB and the vector (4×1011 VGP) was injected IV at various time points after mannitol pretreatment (n≥4/group). At 4 weeks post-injection (pi), the animals were sacrificed and tissues harvested for assessment of GFP expression by immunofluorescence (IF) staining.
In general, an IV administration of mannitol allowed the CNS entry of peripherally-delivered rAAV2 vector, with a diffuse global distribution of transgene expression throughout the CNS, including the brain and spinal cord (Fig.1A). The rAAV2 vector appeared to target both neuronal and non-neuronal cells, though transduced neurons predominated. Of the GFP-expressing brain cells counted, approximately 90% were neurons and the rest were glial cells. The transduction of neurons was non-preferential, and included both large and small neurons distributed ubiquitously throughout the CNS, without a discernable pattern.
Fig. 1. Mannitol-facilitated AAV-CNS entry, diffuse global CNS transduction and somatic transduction in mice.
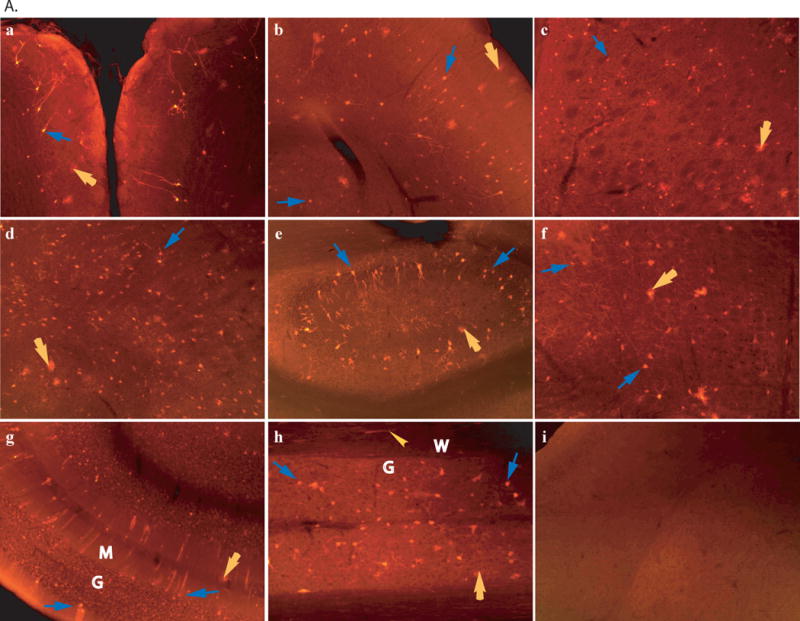
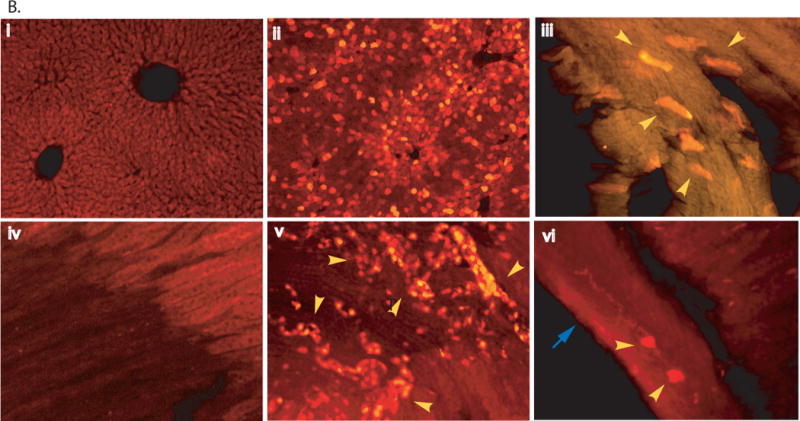
scAAV2-CMV-GFP viral vector (4×1011 viral particles) was injected into 6–8-week-old wt mice through tail vein, 8 minutes after an IV infusion of mannitol or without mannitol pretreatment. Vibratom tissue sections (50μm) were obtained at 4 weeks pi and probed for GFP expression by immunofluorescence using a polyclonal antibody against GFP and a secondary antibody conjugated with AlexaFluo568. A: Transgene expression in the CNS. a–h: samples of mice injected with AAV2 vector following mannitol pretreatment. a. olfactory; b. cerebral cortex; c. striatum; d. thalamus; e. hippocampus; f. brain stem; g. cerebellum. G: granule layer, M: molecular layer, blue arrows: Purkinje cells; h. spinal cord, G: grey matter, W: white matter; i. cerebral cortex of mice injected with AAV2 vector without mannitol pretreatment. GFP was stained in red. Blue arrows: neurons; Yellow arrows: glial cells; Yellow arrowhead: processes in white matter. Images: 10×. B: Somatic transduction. i. non-treated liver of non-treated mouse, ii. AAV2-treated live, red fluorescent cells are GFP-positive hepatocytes; iii. AAV2-treated heart (myocardium), yellow arrowhead: GFP-positive cardiac myocytes: iv. non-treated kidney (medulla), v. AAV2-treated kidney, yellow arrowhead: GFP-positive cuboidal epithelial cells; vi. AAV2-treated intestine, yellow arrowhead: GFP-positive enteric plexus neurons. Blue arrow: muscularis externa. i–ii: 10×; iii–vi: 20×.
The GFP expressing glial cells were GFAP-negative (data not shown) and morphologically homogenous with a highly branched radial process network, the characteristic features of mature oligodendrocytes (Fig. 1A), though immuno-detection for oligodendrocyte-specific markers was not conducted. The GFP expression was also seen in other non-neuronal cells, such as meninges cells and cells of choroid plexus (data not shown). No detectable GFP expression was observed in the CNS of the control mice receiving an IV injection of AAV vector without mannitol pretreatment (Fig. 1A), or an IV infusion of PBS following mannitol injection.
Importantly, our time-course results also showed that the efficiency of rAAV2-CNS entry was exquisitely sensitive to the timing of the IV vector injection after mannitol pretreatment (Table 1). We observed that the maximal transduction in the CNS occurred when the IV vector injection was performed at 8 minutes after mannitol pretreatment, with detectable GFP expression in approximately 1.2×106 cells per mouse brain, which was approximately 10-fold more efficient than IV delivery of the vector at 5 or 10 minutes after the administration of mannitol. These data support the prediction that the timing of IV rAAV vector after mannitol pretreatment is indeed critical for the efficiency of mannitol-facilitated rAAV-CNS entry, and IV vector delivery at the optimal timing allows the maximal number of rAAV vector entering the CNS, indicating greater therapeutic potential for CNS gene therapy.
Table 1.
The efficiency of rAAV2-CNS entry and the timing of IV vector injection after mannitol pretreatment
| Timing of IV vector injection after mannitol infusion (min) | Number of GFP-positive cells/mouse brain |
|---|---|
| 5 | ~1.2×105 |
| 6 | ~5×105 |
| 7 | ~8×105 |
| 8 | 1.2−1.5×106 |
| 9 | ~6×105 |
| 10 | 1.2−1.6×105 |
| 15 | <1,000 |
| 20 | <500 |
scAAV2-CMV-GFP vector (4×1011 VGP) was IV injected into 6–8-wk-old wt mice (n≥4/group), following an IV mannitol infusion. Tissue samples were collected 4wk pi, and immunofluorescence for GFP was performed on serial vibratome brain sections (50μm) of entire brain. All GFP-positive cells on 1 of every 5 sections were counted. The total number of GFP-expressing cells per brain was calculated based on the cell counting results and the size of the brain. The data of each time point was generated with the results of 4 mouse brains (n=4).
Somatic transduction unaffected by mannitol
Immunofluorescence staining on multiple somatic tissue sections was conducted to assess the transduction efficiency and the tropism of rAAV2 in the peripheral system in the presence or absence of mannitol pre-treatment. We observed that an IV rAAV2 vector injection resulted in GFP expression in multiple peripheral tissues/organs, with or without mannitol pretreatment, and the timing of the vector injection did not yield significant observable differences in peripheral tissues in terms of the number of the cells transduced or the intensity of transgene expression (Fig. 1B). The GFP expression was detected in 40–50% of hepatocytes in liver, and the transduction appeared stronger in cells surrounding the portal vein than the central vein. Approximately 10–20% of cardiac myocytes were GFP-positive in heart (Fig. 1B). In kidney, GFP appeared to be expressed in cuboidal epithelial cells in the medulla, but not in cells of the cortex. In intestine, rAAV2 vector seemed to transduce mainly the neurons of the myenteric plexus and submucosal plexus (Fig. 1B), while no detectable GFP expression was observed in smooth muscle. It was difficult to determine whether cells of the mucosa of intestine were transduced, due to a strong autofluorescence background. The expression of GFP was also observed in lungs in both alveolar type I and II cells (<1%)(data not shown). Also, GFP expression in skeletal muscles was not clear due to the strong background of autofluorescence (data not shown). These data suggest that the mannitol pre-treatment does not alter vector distribution or tropism in somatic tissues.
An IV rAAV2 vector injection following mannitol pretreatment is therapeutically beneficial for treating the CNS disease in MPS IIIB mice
An infusion of rAAV2-CMV-hNaGlu vector (4×1011 VGP) was given to 4–6-week-old MPS IIIB mice via tail vein at either 8 (n=24) or 10 (n=20) minutes after mannitol pretreatment, to assess the therapeutic benefits of mannitol-facilitated CNS entry of IV-delivered rAAV vector, and the impact of the timing of IV vector injection on the therapeutic efficacy of the procedure. We had noted a 10-fold difference in vector delivery to the CNS at the 8 and 10 minute time points post-mannitol infusion in our time course experiment. We did not include vector-treated animals without mannitol pretreatment because we had observed that no vector entered the CNS under these conditions.
Behavioral correction
To evaluate the functional impact of the treatment, we performed behavioral testing on rAAV2-CMV-hNaGlu-treated (n=12–14/group) and control mice, which received vehicle only after mannitol pre-treatment (n=20/group), when they were 5.0–5.5 months old. We observed that an IV injection of rAAV vector at 8 minutes after mannitol pretreatment led to significantly improved behavioral performance of MPS IIIB mice in a Morris water maze and on an accelerating rotarod (Fig. 2a–c). These rAAV-treated MPS IIIB mice displayed a significant decrease in latency to find a hidden platform and an increase in swimming ability in the water maze, compared to non-treated MPS IIIB mice (Fig. 2a, b). These treated mice also showed significantly longer latency to fall from an accelerating rotarod than non-treated MPS IIIB mice. However, we did not see changes in behavioral performance in MPS IIIB mice receiving an IV infusion of rAAV vector at 10 minutes after mannitol pretreatment. These data indicate that the increase in rAAV-CNS entry using optimal delivery conditions offers greater therapeutic benefit for treating the neurological disease of MPS IIIB mice.
Fig. 2. Significant behavioral improvement and extended survival in MPS IIIB mice treated with an IV infusion of rAAV2 vector.
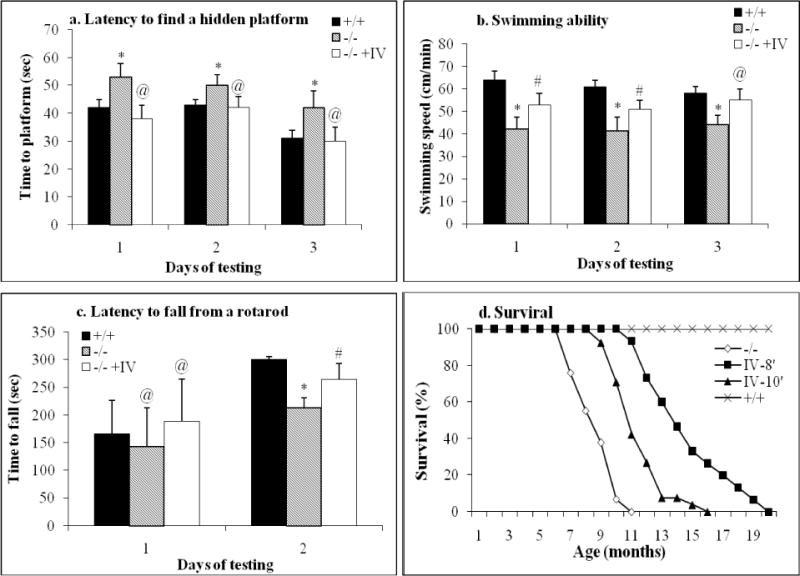
MPS IIIB mice (4–6-wk-old) were treated with an IV injection of rAAV2-CMV-hNaGlu, at 8 or 10 minutes after an IV infusion of mannitol. The mice were tested for behavioral performance at 5–5.5 months of age, and were observed for longevity. a. Latency to find a hidden platform in water maze. b. Swimming ability. c. Latency to fall from an accelerating rotarod. +/+: wt (n=20); −/−: MPS IIIB (n=24); −/− +IV: MPS IIIB mice given an IV vector injection at 8 minutes after mannitol pretreatment (n=14); d. Survival. The lifespan of MPS IIIB mice were significantly prolonged when treated with an IV injection of rAAV2 vector at 8 minutes (P<0.01) and 10 minutes (P<0.05) after mannitol pretreatment (n=16–20/group). IV-8’ and IV-10’: MPS IIIB mice given an IV vector injection at 8 or 10 minutes after mannitol pretreatment; *: P<0.05 (vs. −/− +IV); #: P<0.05 (vs. +/+); @: P>0.05 (vs. +/+).
Significantly extended survival
Longevity observations were carried out to assess whether an IV rAAV delivery following mannitol pretreatment had an impact on the survival of MPS IIIB mice. As shown in Fig. 2d, an IV infusion of rAAV vector at either 8 or 10 minutes after mannitol administration significantly increased the lifespan of MPS IIIB mice. The rAAV-treated MPS IIIB mice lived 12.6–20.9 months (16.5±2.2 months, P<0.01, n=20) and 9.5–15.9 months (11.5±2.1, P<0.05, n=14) for the 8 and 10 minute treated groups, respectively, compared to the vehicle-treated MPS IIIB mice, which lived 7.9–11.9 months (10.1±1.2 months, n=20). The difference in lifespan between the 8-minute-treated group and the 10-minute-treated group was also statistically significant (P<0.05). These results suggest that, while both vector delivery conditions were therapeutically beneficial, the optimization of the time interval between IV infusion of mannitol and IV injection of rAAV vector significantly improved the survival of MPS IIIB mice. It is worth noting that late stage symptoms, such as urine retention, rectal prolase and protruding penis, emerged 1–2 months before the endpoint in all MPS IIIB mice, treated and non-treated, indicating that the treatment significantly slowed, but did not stop the disease progression.
Global distribution of rNaGlu and decrease in lysosomal storage in the brain
At 3 months pi, and at the end of the longevity observation when animals had developed irreversible symptoms of neurological dysfunction, tissue samples were assayed to assess the expression and distribution of recombinant hNaGlu and the correction of lysosomal storage in the rAAV treated MPS IIIB mouse brains.
By IF staining using a polyclonal antibody against hNaGlu, the transgene expression was detected in cells throughout the brain of MPS IIIB mice given an IV vector injection at 8 minutes after mannitol pretreatment (Fig. 3A), although the number of cells with detectable rNaGlu in the brain parenchyma is low (approximately 1%). The distribution of rNaGlu-positive brain cells appeared mostly to be non-preferential, except for cells of the choroid plexus, meninges, and brain parenchyma adjacent to the ventricles, where a group of rNaGlu-positive cells could be seen (Fig. 3A). Previously, we observed the presence of autofluorescent materials in cells of the MPS IIIB mouse brain (unpublished data), which may be due to the secondary storage pathology of the disease.3,45 In this study, the rNaGlu was observed in cells both with and without autofluorescent materials, and appeared not to be co-localized with the autofluorescent materials. In addition, the intracellular distribution of rNaGlu appeared to be to follow two different patterns, either diffuse in the cytoplasm or as granules. The rNaGlu was also found in the processes of brain cells. We also observed that the intensity of rNaGlu signals varied in rNaGlu-positive cells. We did not see detectable rNaGlu by IF staining in the brains of MPS IIIB mice treated with an IV rAAV injection at 10 minutes after mannitol administration, or in non-treated mice.
Fig. 3. Expression of recombinant NaGlu and decrease in lysosomal storage pathology in MPS IIIB mouse brain. A.
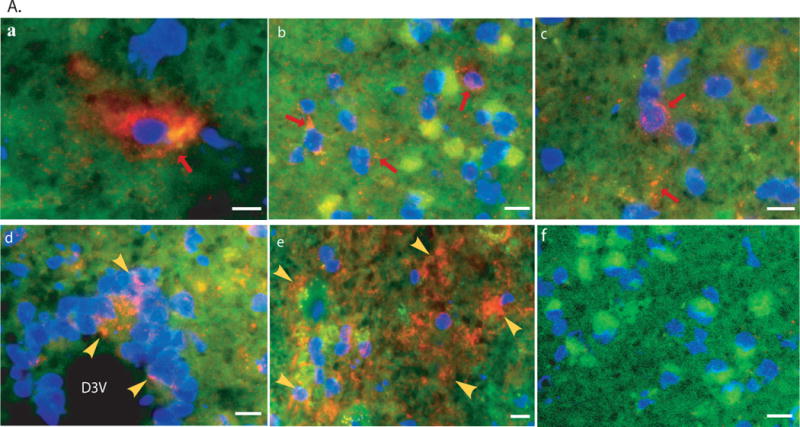
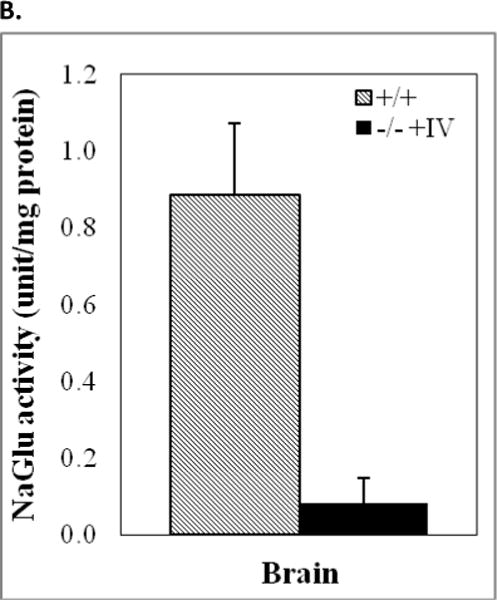
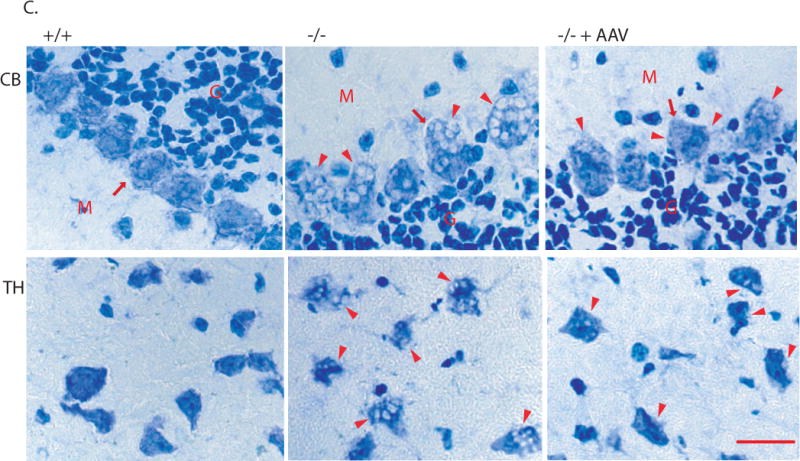
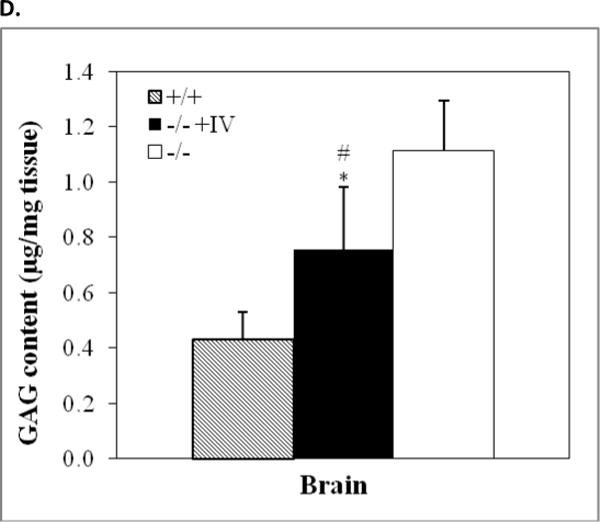
Immunofluorescence staining for rNaGlu expression in the brain of MPS IIIB mouse (17-month-old) treated with an IV injection of rAAV-hNaGlu vector at 8 minutes after mannitol pretreatment, using a polyclonal antibody against hNaGlu and a secondary antibody labeled with AlexaFluo568 (a–e). a. brain stem, b. hypothalamus, c. thalamus, d. Dorsal 3rd ventricle and periventricular thalamic area, e. choroid plexus in lateral ventricle, f. thalamus of non-treated MPS IIIB mouse. Red arrows: NaGlu-positive neurons/glia and process. Yellow arrowheads: NaGlu-positive ependymal cells (d) and choroid plexus cells (e). D3V: dorsal 3rd ventricle. Nuclei are labeled blue. Green fluorescence was used to separate autofluorecence from specific fluorescence signals (red). Scale bars: 10μm. B. NaGlu activity in brain (endpoint, n>8/group). C. Histopathology staining with toluidine blue. +/+: wt; −/−: non-treated; −/−+AAV: AAV-treated. CB: cerebella; TH: Thalamus. M: molecular layer; G: granule layer, red arrows: Purkinje cells; red arrowhead: enlarged lysosomes. Scale bar: 20μm. D. GAG contents in the brain of rAAV-treated MPS IIIB mice (3 months pi, n>8/group): expressed as μg/mg tissue (wet). *: P<0.05 (vs. −/−), #: P<0.05 (vs. +/+).
The NaGlu enzymatic activity in MPS IIIB brain tissues was measured to assess the efficiency and persistence of rNaGlu expression in the CNS after rAAV gene delivery. As expected, relatively low levels of enzyme activity were detected in the brains of MPS IIIB mice given an IV vector injection at 8 minutes after mannitol pretreatment (Fig. 3B). We did not observe detectable NaGlu activity in the brains of MPS IIIB mice receiving an IV infusion of rAAV at 10 minutes after the administration of mannitol. In addition, we did not observe detectable difference in expression and distribution of rNaGlu in the brain at 3 months pi and the endpoint, suggesting the long-term and stable CNS transduction.
Examining for histopathology in CNS sections, we observed decreases in the size and number of vacuolated lysosomes throughout the brains of both groups of MPS IIIB mice IV-treated with rAAV2 following mannitol pretreatment, compared to the non-treated MPS IIIB mice, although a complete clearance of vacuoles was not seen (Fig. 3C). Also, we detected significantly less GAG content in the brains of MPS IIIB mice given an IV vector infusion at 8 minutes after mannitol pretreatment, at 3 months pi (Fig. 3D) but not at the endpoint of the longevity observations, when irreversible symptoms of neurological dysfunction had developed. There was no significant reduction in GAG contents in the brain of 10-minute-treated group.
These data suggest that mannitol-facilitated CNS entry of IV-delivered rAAV vector, led to the long-term restoration of functional NaGlu in the CNS, and subsequent correction of lysosomal storage, improvements in behavioral performance and survival of MPS IIIB mice. Further, the low levels of detectable NaGlu activity were sufficient to provide a significant, dose-dependent therapeutic effect.
The expression of rNaGlu and correction of lysosomal storage in the somatic system
Multiple somatic tissues were tested for rNaGlu expression by NaGlu activity assay and immunofluorescence staining, to evaluate the effects of IV rAAV delivery on somatic tissues/organs in MPS IIIB mice treated with rAAV-CMV-NaGlu infusion. At the endpoint of longevity observation, NaGlu enzyme activity was detected in all tissues assayed, including liver (10–100% of wt level), kidney (1–3%), spleen (1–13%), heart (2–30%), lung (2–15%), intestine (1–5%) and skeletal muscle (10–30%) (Fig. 4a). Consistent with our GFP studies, neither the presence or absence of mannitol pretreatment, nor the timing of the IV rAAV injection after mannitol infusion appeared to have a significant impact on rNaGlu expression in these somatic tissues. We did not observe a significant difference in NaGlu activity in somatic tissues at 3 months pi and the endpoint. No detectable NaGlu activity was seen in the somatic tissues of non-treated MPS IIIB control mice.
Fig.4. Expression of hNaGlu and correction of lysosomal storage in somatic tissues.
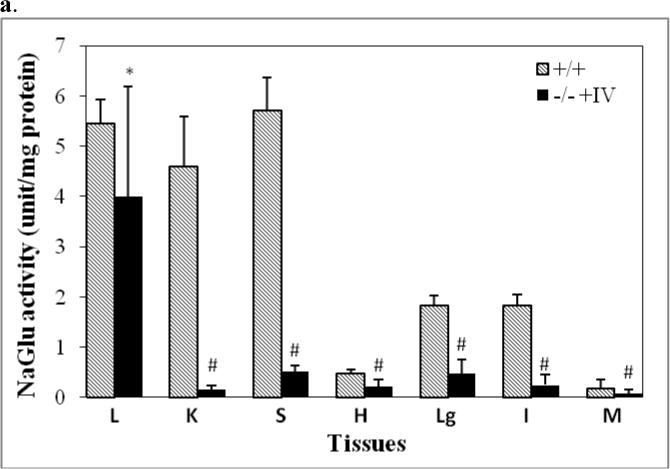
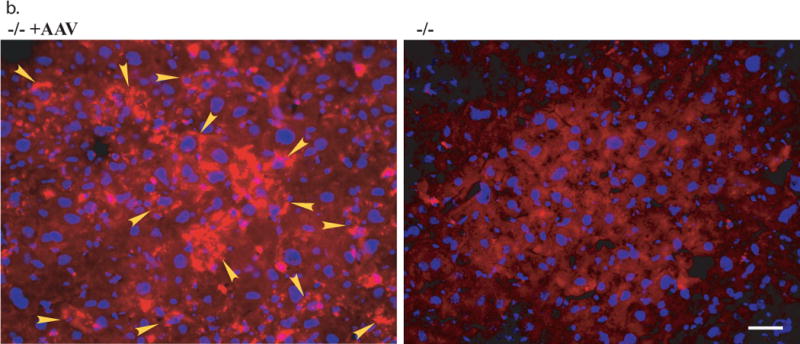
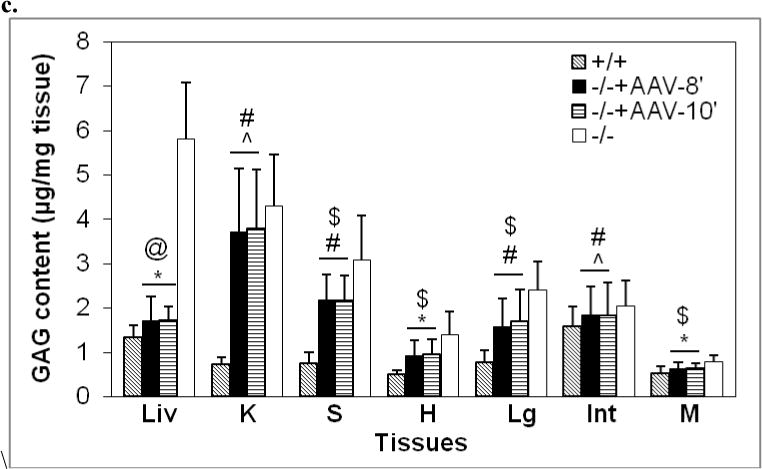
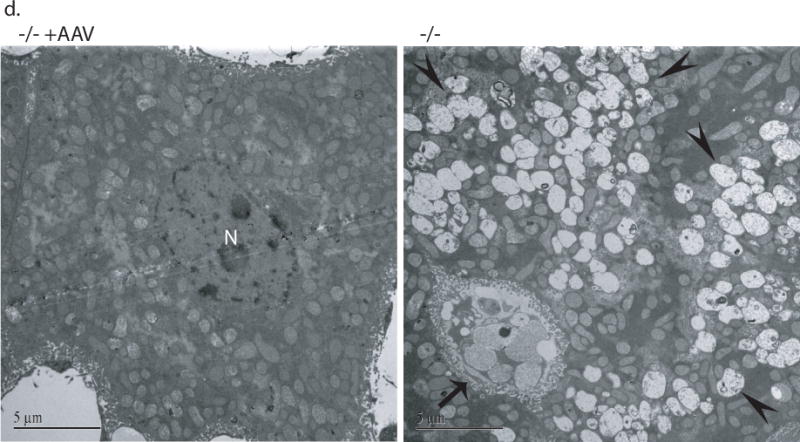
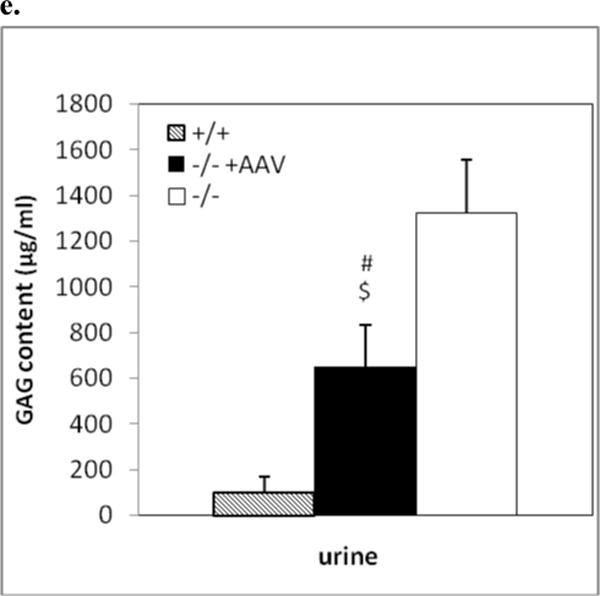
Somatic tissues (endpoint, n>8/group) were assayed for NaGlu expression (a, b) and correction of lysosomal storage (c, d). a. NaGlu activity is expressed as unite (U)/mg protein. 1U = 1nmol 4MU released/h at 37°C. +/+: wt mice; −/−:/ non-treated MPS IIIB mice; −/− +AAV: AAV-treated MPS IIIB mice; −/− +AAV-8’ and −/− +AAV-10’: MPS IIIB mice IV injected with AAV vector at 8 or 10 min after mannitol infusion. L: liver, K: Kidney; S: spleen; H: heart; Lg: lung; I: intestine; M: skeletal muscle. b. Immunofluorescence staining for hNaGlu on cryostat liver sections of AAV-treated (−/− +AAV, 17-month-old) and non-treated (−/−) MPS IIIB mice. Scale bar: 50μm. c. GAG content in somatic tissues. d. Ultrastructural correction of lysosomal storage in liver of AAV-treated MPS IIIB mice. Arrow: a Kupffer cell. Arrow heads: enlarged lysosomes. e. Urine GAG content (3 months pi, n>4/group). GAG content was expressed as μg/mg tissue (wet) or μg/ml (urine). $: P<0.05 (vs. −/−). @: P<0.01 (vs. −/−); *: P<0.05 (vs. +/+); #: P<0.01 (vs. +/+); ˆ: P>0.05 (vs. −/−).
Immunoflurescence was performed to determine the distribution of rNaGlu expression using a polyclonal antibody for hNaGlu. The rNaGlu was detected in 15–20% of cells in the livers of rAAV-treated MPS IIIB mice, and the rNaglu-positive cells appeared to be predominantly hepatocytes (Fig. 4b). The intensity of fluorescent staining varied among different cells and in interstitial spaces, possibly as the result of variable levels of rNaGlu expression and secretion from transduced cells, and uptake of rNaGlu by non-transduced cells, though we could not distinguish the transduced cells from the cells that may have taken up the secreted enzyme. Again, mannitol pretreatment and the timing of the IV rAAV injection after mannitol infusion did not lead to visible effects on the distribution and the intensity of rNaGlu staining in liver. The rNaGlu was not detected in the livers of wt or non-treated MPS IIIB control mice (Fig. 4b). In somatic tissues other than liver, we did not observe convincing rNaGlu expression by IF staining in rAAV-treated mice, even though NaGlu enzyme activity was detected. This may be due to a relatively low sensitivity with this antibody compared to the enzyme activity assays. These data are consistent with the property of the liver to take up the vast majority of IV injected rAAV2 vector, with or without mannitol pre-treatment.21
Multiple somatic tissues were analyzed using the GAG content assay, histopathology, and/or transmission electron microscopy (TEM), to assess the rAAV2-mediated correction of lysosomal storage in MPS IIIB mice by an IV vector injection. We observed a complete (100%) correction of GAG storage in liver (P<0.01), and reduction of GAG accumulation in spleen, heart, lung, and skeletal muscle (P<0.05) in rAAV-treated MPS IIIB mice (Fig. 4c). We did not see significant reductions in GAG content in intestine or kidney (P>0.05). Further, we observed the clearance of abnormal lysosomal storage in the livers of treated MPS IIIB mice by TEM (Fig. 4d) and histopathology (data not shown), confirming the complete correction of lysosomal accumulation in this organ. No obvious histopathological correction of lysosomal storage was observed in other somatic tissues. Again, mannitol pretreatment and the timing of the IV rAAV injection after mannitol administration had no discernable impact on the correction of lysosomal storage in somatic tissues (Fig. 4c). The sham-treatment with mannitol and PBS did not lead to a decrease in GAG content in any somatic tissue of MPS IIIB mice. In addition, the urine GAG content of IV-treated MPS IIIB mice was significantly decreased (Fig. 4e). These results were consistent with the efficiency of rNaGlu expression in somatic system.
Differential distribution of vector genome in tissues/organs
Quantitative real-time PCR was performed to assess the tissue targeting efficiency of rAAV2-CMV-hNaglu vector. Table 2 shows a differential distribution of the vector genome in different tissues/organs of MPS IIIB mice treated with an IV vector injection at 8 minutes after mannitol pretreatment. The vector genome was mostly seen in liver (2.76±1.42 copies/cell), followed by brain (0.013±0.004 copies/cell), and very low copies of vector genome were detected in other tissues/organs (Table 2). This differential vector distribution in rAAV2-treated MPS IIIB mice correlated with the distribution of rNaGlu, and with our time course experiment using GFP-expressing vector. Further, our data also showed consistent vector genome distribution in treated mice at 3 months pi and the endpoint, supporting a stable long-term transduction. Vector genome analysis was not performed with tissues from mice in biodistribution studies using rAAV-GFP vector and MPS IIIB mice IV injected with rAAV2-hNaglu at 10 minutes after mannitol infusion.
Table 2.
rAAV2 vector genome in tissues
| Tissues | rAAV Vector genome (copies/cell) |
|---|---|
| Liver | 2.76±1.42 |
| Kidney | 0.002±0.0003 |
| Spleen | 0.006±0.003 |
| Heart | 0.004±0.002 |
| Lung | 0.001±0.0004 |
| Intestine | 0.0009±0.0006 |
| Skeletal muscle | 0.0004±0.0002 |
| Brain | 0.013±0.004 |
Genomic DNA isolated from tissues of rAAV-treated MPS IIIB was assayed by quantitative real- to determine the number of vector genome per cell in each tissue. The data here are means±SD (n=4).
DISCUSSION
In this study, we were able to show association between the optimal timing of an IV rAAV vector injection for maximal mannitol-facilitated rAAV-CNS entry, and increased therapeutic efficacy in mice. The optimal timing for IV injection of AAV vector was 8 minutes after an IV infusion of mannitol, which was 10-fold more efficient in CNS transduction than IV vector injection at 5 or 10 minutes after mannitol pretreatment. Mannitol is a known osmotic BBB disruption agent,13 and IV infusion of mannitol has been used safely for decades in routine medical practice.46 Many previous studies have shown the increase in access of peripherally delivered substances to the CNS in experimental animals and human patients, following an IA infusion of mannitol.13,15,47 However, using mannitol pretreatment does not necessarily enable efficient CNS entry of peripherally delivered materials, since the mannitol-facilitated BBB disruption is transient with a very narrow open window.13,24,48 The timing of therapeutic delivery after mannitol administration is considered critical for an efficient CNS entry of peripherally delivered therapeutic substances, by allowing a high concentration of therapeutic materials in the blood circulation at the time of peak BBB opening. Previous studies using IA infusion of mannitol through the carotid artery in rat showed that the peak opening of BBB was approximately 5 minutes after mannitol administration, and the opening lasted for 20–30 min.24,48 However, no detailed study had been reported on the timing for optimal BBB opening after a systemic IV infusion of mannitol, though IV therapeutic administration is considered the only route to reach the entire CNS. We therefore believe that our data is an important addition to CNS therapeutic delivery, which may also be applied to not only the delivery of rAAV viral vectors, but also other therapeutic reagents for a broad range of neurological diseases.
We also demonstrated here a ubiquitously diffuse global AAV transduction throughout the CNS, including the brain and spinal cord. The resulting non-preferential distribution of AAV transgene expression throughout the CNS reflected the typical distribution pattern of vascular delivery, which has been considered the only route to achieve a global CNS therapeutic delivery. With approximately 106 CNS cells transduced, it may be sufficient for treating many global neurological disorders, such as lysosomal storage diseases, in which the deficient lysosomal enzymes are secreted and can be taken up by neighboring cells (by-stander effect). In addition, our data demonstrated that AAV2 vector targeted predominantly neurons, as well as some non-neuronal cells. The transduction of neurons appears to be non-preferential. In contrast, the transduction of glial cells seems to be cell-specific, targeting only oligodendrocyte-like cells, though it is unclear whether this is a receptor- or promoter-specific phenomenon. We anticipate that using the optimal condition for peripheral rAAV2 delivery to the CNS may offer great potential in developing therapies for global/broad CNS diseases involving brain and spinal cord with manifestation in neurons and/or glial cells, such as lysosomal storage diseases and amyotrophic lateral sclerosis (ALS).
The peripherally delivered AAV2 vector also exhibited broad tropism in peripheral tissues. As expected, the transduction in liver was much more efficient than in other tissues, presumably due to the nature of the abundant blood supply, and high accessibility and susceptibility of this organ to AAV. This may negatively affect the CNS transduction efficiency of peripherally delivered AAV by depleting the vector from the blood. However, we believe that this peripheral targeting may play an important role in gene therapy for many diseases, such as the majority of lysosomal storage diseases, which manifest in both the CNS and somatic tissues. Moreover, these data also suggest the importance of mastering the timing of conducting IV vector injection after mannitol pretreatment, to ensure a peak vector concentration in the blood flow by the time when the BBB opening reaches its peak.
We also demonstrated that the optimal IV-mannitol rAAV2 delivery (8-min) regimen is therapeutically beneficial with clinical significance for treating the CNS disease of MPS IIIB in mice. This is the first study achieving a successful long-term therapeutic neurologic correction of MPS IIIB by a single IV vector delivery in adult mice. Using the optimized procedure, we were able to significantly extend the survival and improve the behavioral performance of MPS IIIB mice. We also demonstrated a global distribution of rNaGlu throughout the brain, though at very low level. It is apparent that this low-level of rNaGlu, attributed to the transduction of a relatively small number of CNS cells with low copy numbers of the vector, was sufficient to correct the neuropathology to a significant extent and delay the CNS disease progression, although the treatment did not yield a cure for the disease. While lysosomal storage lesions and GAG content were significantly reduced in the CNS, especially at early time points, all of the affected mice had high levels of GAG in the brain at the endpoint, though this was significantly delayed. We have not been able to determine the factors involved in this. It is likely that these CNS therapeutic impacts were achieved largely through the by-stander effect of the enzyme, since the diffuse global distribution of rNaGlu-expressing cells is likely to offer the secreted enzyme broad access to non-transduced cells. Previous successful IV gene delivery studies for CNS correction in LSDs have mostly been performed in neonatal animal models, before the closure of their BBB,40,42,49,50 which is not applicable in humans, in whom the BBB is physically closed at birth. We believe that our newly optimized IV rAAV vector delivery regimen may contribute greatly to therapeutic options for MPS IIIB and many other CNS diseases by offering a safe procedure with significant therapeutic potential.
An IV rAAV2 vector injection at 10 minutes after mannitol pretreatment also lead to an increase in survival (though smaller), but failed to show an impact on the behavior of MPS IIIB mice at the age of testing. The neurological manifestation is considered the primary cause of high mortality and premature death in MPS patients and animals. We believe that the significantly extended survival here was the result of limited neurological correction by an undetectably low level of rNaGlu, since the CNS transduction efficiency of the 10-min regimen is 10-fold lower than the optimized procedure.
As expected, we also demonstrated here a long-term complete correction of lysosomal storage in liver, and partial correction in multiple other somatic tissues, after IV infusion of the rAAV2-NaGlu vector, as observed in our previous studies combining an IV (10-min regimen) and an intracisternal injection of rAAV2 vector. The timing of IV vector injection after mannitol infusion had no significant impact on the therapeutic efficiency in peripheral tissues. The rNaGlu expression in only 15–20% of liver cells was sufficient to clear the lysosomal storage of GAGs in the organ, indicating the significant contribution of the by-stander effect of the secreted rNaGlu. Although the neuropathology is the cause of high mortality of the disease, the somatic correction may also be beneficial for treating MPS IIIB, since broad somatic manifestations inevitably occur in all patients and animals with MPS IIIB. Many factors may contribute to the differential lysosomal storage correction pattern in somatic tissues, with liver being the only organ showing efficient correction in ERT clinical studies on different MPS patients and LSD animal models given IV infusion of recombinant enzymes.51–55 However, the strong tissue tropism of rAAV2 may be a more important factor in the differential therapeutic efficacy of our IV-rAAV2 gene delivery.
Importantly, even though the timing of vector delivery and mannitol injection did not affect the distribution of vector or lysosomal storage correction in peripheral tissues, it did have a significant impact on longevity in MPS IIIB mice. This supports our conclusion that the greatest contributor to therapeutic efficacy is vector transduction in the CNS, which is the only outcome affected by mannitol. The time point for maximal therapeutic benefit also coincided with the maximal CNS delivery in our GFP vector time course experiment.
The neurologic benefit of the optimized IV rAAV delivery strategy in treating MPS IIIB was surprisingly similar to what we achieved in our previous studies with high levels of rNaGlu expression in broad brain areas, after a combination of an IV (10 min post mannitol) and an intracisternal vector delivery (same rAAV2 vector), though neither treatment yielded a complete cure. It is therefore important to consider additional factors that may be critical for therapeutic efficacy. First, it has been observed that rNaGlu expressed from the hNaGlu cDNA construct is poorly manose-6-phosphorylated,56,57 possibly due to lack of regulatory signals required for post-transcriptional modification. Although the rNaGlu can correct the enzymatic and lysosomal storage phenotype in human MPS IIIB skin fibroblasts,32,56,57 improper processing of the protein may impede its efficiency through other aspects of its enzymatic function, such as secretion, uptake, intracellular transport, and catalytic capacity. The detailed mechanisms involved in NaGlu catalytic processing in brain may be different from that in liver, a metabolic organ, which may explain the more efficient hepatic correction of lysosomal storage.
Additionally, the neuropathology of MPS IIIB involves complex components secondary to the initial pathology of lysosomal accumulation of HS, most of which are still being characterized. Many secondary neuropathological sequelae emerge at an early stage of the disease and may contribute significantly to the CNS disease progression.1,3–8,45 This includes a strong inflammatory and autoimmune component, which will probably need to be treated in conjunction with gene therapy. Although our gene delivery demonstrated a significant neurological effect, further improvements to the therapeutic efficacy will require a better understanding of NaGlu enzymatic function and MPS IIIB neuropathology.
We are confident that we have developed a safe and clinically significant rAAV CNS gene therapy procedure for MPS IIIB. It is worth noting that IV infusion of mannitol has been routine medical practice for decades with a long history of safe use in treating various neurological and non-neurological conditions,46 and the dose of mannitol used in this study was scaled directly from that for human application. Side effects occur in humans only when multiple doses of mannitol are repeatedly administered within a relatively short time.46 Potentially more significant challenge facing this procedure is the high rate of pre-existing immunity to AAV2 in the human population. We are currently testing alternative non-human AAV serotypes for delivery across the BBB to deal with this issue.
In summary, we have optimized our IV/mannitol facilitated rAAV delivery procedure to achieve 10-fold higher vector transduction in the CNS than our previous studies. Using this optimized procedure, a single IV rAAV2 injection following an IV infusion of mannitol was therapeutically beneficial, with significant long-term impacts in treating the neurological disorders of MPS IIIB. This approach is safe and readily adaptable to any clinical setting, and may benefit the development of CNS therapeutic delivery not only for MPS IIIB but also other global CNS diseases in general.
MATERIALS AND METHODS
Recombinant AAV (rAAV) viral vector
A self-complementary AAV (scAAV) serotype 2 viral vector, scAAV2-CMV-GFP,21 was used in biodistribution experiments. A conventional single-stranded rAAV2 vector,32,34 rAAV2-CMV-hNaGlu, was used in therapeutic experiments. The AAV vectors contained minimal elements for transgene expression, including AAV2 terminal repeats, a human cytomegalovirus (CMV) immediate early promoter, SV40 splice donor/acceptor signal, a green fluorescent protein (GFP) gene or a human NaGlu coding sequence cDNA, and SV40 polyadenylation (Poly A) signal. The rAAV viral vectors were produced in 293 cells using three-plasmid co-transfection, and purified following previously published procedures.58
Animals
A MPS IIIB knock-out mouse model2 was maintained on an inbred background (C57BL/6) of backcrosses of heterozygotes and housed in the Laboratory Animal Facility of Department of Laboratory Animal Medicine (DLAM) at the University of North Carolina at Chapel Hill and at Nationwide Children’s Hospital. All care and procedures were in accordance with the Guide for the Care and Use of Laboratory Animals [DHHS Publication No. (NIH) 85–23]. The genotypes of progeny mice were identified by PCR. MPS IIIB mice and their wt littermates were used for the experiments in this study.
Biodistribution assessment
In biodistribution experiments, 6–8-week-old wt mice were given an IV infusion of scAAV2-CMV-GFP vector. Intravenous (IV) rAAV vector injections in mice were performed, based on the procedure previously developed in our laboratory.21 The mice were anesthetized by an intraperitoneal (IP) injection of Avertin (0.3–0.4μg/g body weight), followed by an IV infusion of mannitol (1–2mg/g bw) (25%, for IV use only, Abbott Laboratories, NDC0074-4031-01), to disrupt the BBB. A dose of scAAV2-CMV-GFP viral vector [4×1011 viral genome-containing particles (VGP) in 150–200 μl PBS] was then delivered into each mouse by tail vein injection over a period of 1 min, at 5, 6, 7, 8, 9, 10, 15 or 20 min after the administration of mannitol. Control mice were treated with an IV injection of the vector only without mannitol pretreatment, or with an IV infusion of PBS after mannitol pretreatment.
Tissue samples were collected for analysis 4–5 weeks after the IV vector injection. The mice were anesthetized with 2.5% Avertin and then perfused transcardially with cold PBS (0.1M, pH7.4), followed by 4% paraformaldehyde in phosphate buffer (0.1M, pH7.4). The entire brain and spinal cord, as well as multiple somatic tissues (including liver, kidney, spleen, heart, lung, intestine and skeletal muscles), were collected and fixed in 4% paraformaldehyde overnight at 4°C before being further processed.
Immunofluorescence for GFP
Tissue sections (50μm) of mouse brains (180–200 transverse sections/brain), spinal cord (longitudinal sections), and somatic tissues were obtained using a vibratome. The IF staining was performed to detect the AAV mediated GFP expression, using a polyclonal antibody against GFP (Invitrogen, A-6455) and a secondary antibody conjugated with Alaxa Fluo 568 (Molecular Probes, A11011), based on manufacture’s instruction. Double immunofluorescence staining was conducted following the same procedures using the polyclonal antibody against GFP and a monoclonal antibody against glial fibrillary acidic protein (GFAP, Chemicon, MAB360), to identify the transduced glial cells. The sections were then visualized under a fluorescence microscope. The number of GFP-positive cells in mouse brain was determined by counting all GFP-positive neurons and glial cells on 1 of every 5 sections. Cell counting did not include cells of meninges, choroids plexus and ependymal cells, since these structures were poorly preserved during tissue processing. The total number of GFP-expressing cells per brain was calculated based on the cell counting results and the size of the brain.
Therapeutic rAAV vector delivery
In therapeutic experiments, 4–6-week-old MPS IIIB mice were anesthetized, as described above, and IV injected with rAAV2-CMV-hNaGlu vector (4×1011 VGP in 100–150 μl PBS) at 8 or 10 min after mannitol pretreatment. Controls were wt and MPS IIIB sham-treated with an IV infusion of PBS after mannitol pretreatment.
Longevity assessment
Following the rAAV vector injection(s), mice were continuously observed for the development of endpoint symptoms, or occasionally until death from natural causes occurred. All MPS IIIB mice, rAAV-treated or non-treated (sham-treated), were euthanized when the symptoms of late stage clinical manifestation, such as urine retention, rectal prolapse, protruding penis became irreversible. The wt control mice were observed until they were 24 months or older. Behavioral tests (see below) were conducted to study the therapeutic benefits of the treatments on the neurological disorder in MPS IIIB mice when they were approximately 5.0–5.5 months old. At the end of the experiments, multiple somatic tissues and the entire brains of the mice were collected on dry ice or embedded in OCT compound and stored at −70°C, before being processed for analyses.
Behavioral studies
Behavioral tests were conducted in the animal behavioral testing laboratory of the Vivarium at TRINCH. Testing groups included rAAV treated MPS IIIB mice (n=12–14/group), wt (n=20) and sham-treated MPS IIIB mice (n=20). Testing began when mice were approximately 5.0–5.5 months in age.
Hidden task in the Morris water maze
The water maze consisted of a large circular pool (diameter = 122 cm) filled with water (45 cm deep, 24–26° C) containing 1% white TEMPERA paint, located in a room with numerous visual cues.59 Mice were tested for their ability to find a hidden escape platform (20×20cm) 0.5cm under the water surface. Each animal was given four trials per day, across three days. For each trial, the mouse was placed in the pool at one of four randomly ordered locations, and then given 60 seconds to swim to the hidden escape platform. If the mouse found the platform, the trial ended, and the animal was allowed to remain 10 seconds on the platform before the next trial began. If the platform was not found, the mouse was placed on the platform for 10 seconds, and then given the next trial. Measures were taken of latency to find the platform (sec), swimming distance (cm) and swimming velocity (cm/min) via an automated tracking system (San Diego Instruments).
Rotarod
Mice were tested on an accelerating rotarod (Med Associate, Inc.) to assess motor coordination.60 Revolutions per minute (rpm) was set at an initial value of 3, with a progressive increase to a maximum of 30 rpm across five minutes (the maximum trial length). For the first test session, animals were given three trials, with 45 seconds between each trial. Two additional trials were given 48 hours later. Measures were taken for latency to fall from the top of the rotating barrel.
Tissue analysis
NaGlu activity assay
The NaGlu enzyme activity assay was carried out following a previously published procedure with modification.2,61 The assay measures 4-methylumbelliferone (4MU), a fluorescent product formed by hydrolysis of the substrate 4-methylumbellireyl-N-acetyl-α-D-glucosaminide. The NaGlu activity is expressed as unit/mg protein. 1 unit is equal to 1nmol 4MU released/h at 37°C.
GAG content measurement
Extraction of GAG from tissues was conducted following previously published procedures62 with modification.34,62 Dimethylmethylene blue (DMB) assay was used to measure GAG content.63 The GAG samples (from 0.5–1.0 mg tissue) were mixed with H2O to 40μl before adding 35nM DMB (Polysciences Cat# 03610-1) in 0.2mM sodium formate buffer (SFB, pH 3.5). The product was measured using a spectrophotometer (OD535). The GAG content was expressed as μg/mg tissue. Urine GAG content was also measured. Heparan sulfate (Sigma, H9637) was used as standard control.
Histopathology
Histopathology was performed following standard methods. Tissue samples were embedded in OCT compound, and fast frozen on dry ice for cryostat sectioning. Thin sections (8 μm) were fixed with 4% paraformaldehyde in phosphate buffer (0.1 M, pH 7.2) at 4°C for 15 min and stained with 1% toluidine blue at 37°C for 30 min. The sections were mounted, and visualized under light microscope.
Immunofluorescence for hNaGlu
A polyclonal antibody against hNaGlu (a kind gift from Dr. EF Neufeld, UCLA) was used to probe the rAAV-mediated hNaGlu expression in tissues using IF staining. The IF staining was performed on thin cryostat sections (8μm) from liver and brain samples of experimental subjects, using the primary antibody against hNaGlu and a secondary antibody conjugated with AlexaFluo568 (Molecular Probes, A11011), following the procedures recommended by the manufacturers. The sections were then visualized under a fluorescence microscope.
Quantitative real time PCR
Genomic DNA was isolated from tissue samples of treated and non-treated MPS IIIB mice. Brain genomic DNA was isolated from midbrain. A pair of primers for CMV promoter was used as marker to detect rAAV vector genome. The genomic DNA samples were analyzed by quatitative real time PCR, using SYBR® Green PCR Master Mix (Applied Biosystems) and Applied Biosystems 7000 Real-Time PCR System, following the procedures recommended by the manufacturer. Genomic DNA from non-treated MPS IIIB mouse tissues was used as controls for background check on lack of contamination.
Acknowledgments
We would like to thank Dr. Elizabeth F. Neufeld for generously providing us the hNaGlu antibody. This study was sponsored by Ben’s Dream – The Sanfilippo Research Foundation, NIDDK (R01 DK63972) and TRINCH.
References
- 1.Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic & molecular basis of inherited disease. 8. McGraw-Hill; New York, St Louis; San Francisco: 2001. pp. 3421–3452. [Google Scholar]
- 2.Li HH, Yu WH, Rozengurt N, Zhao HZ, Lyons KM, Anagnostaras S, et al. Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding alpha-N-acetylglucosaminidase. Proc Natl Acad Sci U S A. 1999;96:14505–14510. doi: 10.1073/pnas.96.25.14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGlynn R, Dobrenis K, Walkley SU. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J Comp Neurol. 2004;480:415–426. doi: 10.1002/cne.20355. [DOI] [PubMed] [Google Scholar]
- 4.Villani GR, Gargiulo N, Faraonio R, Castaldo S, Gonzalez YRE, Di Natale P. Cytokines, neurotrophins, and oxidative stress in brain disease from mucopolysaccharidosis IIIB. J Neurosci Res. 2007;85:612–622. doi: 10.1002/jnr.21134. [DOI] [PubMed] [Google Scholar]
- 5.Dirosario J, Divers E, Wang C, Etter J, Charrier A, Jukkola P, et al. Innate and adaptive immune activation in the brain of MPS IIIB mouse model. J Neurosci Res. 2008 doi: 10.1002/jnr.21912. [DOI] [PubMed] [Google Scholar]
- 6.Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci U S A. 2003;100:1902–1907. doi: 10.1073/pnas.252784899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walkley SU. Pathogenic mechanisms in lysosomal disease: a reappraisal of the role of the lysosome. Acta Paediatr Suppl. 2007;96:26–32. doi: 10.1111/j.1651-2227.2007.00202.x. [DOI] [PubMed] [Google Scholar]
- 8.Li HH, Zhao HZ, Neufeld EF, Cai Y, Gomez-Pinilla F. Attenuated plasticity in neurons and astrocytes in the mouse model of Sanfilippo syndrome type B. J Neurosci Res. 2002;69:30–38. doi: 10.1002/jnr.10278. [DOI] [PubMed] [Google Scholar]
- 9.Muenzer J, Fisher A. Advances in the treatment of mucopolysaccharidosis type I. N Engl J Med. 2004;350:1932–1934. doi: 10.1056/NEJMp048084. [DOI] [PubMed] [Google Scholar]
- 10.Rohrbach M, Clarke JT. Treatment of lysosomal storage disorders : progress with enzyme replacement therapy. Drugs. 2007;67:2697–2716. doi: 10.2165/00003495-200767180-00005. [DOI] [PubMed] [Google Scholar]
- 11.Misra A, Ganesh S, Shahiwala A, Shah SP. Drug delivery to the central nervous system: a review. J Pharm Pharm Sci. 2003;6:252–273. [PubMed] [Google Scholar]
- 12.Pardridge WM. Molecular biology of the blood-brain barrier. Mol Biotechnol. 2005;30:57–70. doi: 10.1385/MB:30:1:057. [DOI] [PubMed] [Google Scholar]
- 13.Rapoport SI. Osmotic opening of the blood-brain barrier: principles, mechanism, and therapeutic applications. Cell Mol Neurobiol. 2000;20:217–230. doi: 10.1023/A:1007049806660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Dalwadi G, Benson HA. Drug delivery across the blood-brain barrier. Curr Drug Deliv. 2004;1:361–376. doi: 10.2174/1567201043334542. [DOI] [PubMed] [Google Scholar]
- 15.Doolittle ND, Miner ME, Hall WA, Siegal T, Jerome E, Osztie E, et al. Safety and efficacy of a multicenter study using intraarterial chemotherapy in conjunction with osmotic opening of the blood-brain barrier for the treatment of patients with malignant brain tumors. Cancer. 2000;88:637–647. doi: 10.1002/(sici)1097-0142(20000201)88:3<637::aid-cncr22>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 16.Gaillard PJ, Visser CC, de Boer AG. Targeted delivery across the blood-brain barrier. Expert Opin Drug Deliv. 2005;2:299–309. doi: 10.1517/17425247.2.2.299. [DOI] [PubMed] [Google Scholar]
- 17.Pardridge WM. Molecular Trojan horses for blood-brain barrier drug delivery. Curr Opin Pharmacol. 2006;6:494–500. doi: 10.1016/j.coph.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Barranger JA, Rapoport SI, Fredericks WR, Pentchev PG, MacDermot KD, Steusing JK, et al. Modification of the blood-brain barrier: increased concentration and fate of enzymes entering the brain. Proc Natl Acad Sci U S A. 1979;76:481–485. doi: 10.1073/pnas.76.1.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burger C, Nguyen FN, Deng J, Mandel RJ. Systemic mannitol-induced hyperosmolality amplifies rAAV2-mediated striatal transduction to a greater extent than local co-infusion. Mol Ther. 2005;11:327–331. doi: 10.1016/j.ymthe.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 20.Doran SE, Ren XD, Betz AL, Pagel MA, Neuwelt EA, Roessler BJ, et al. Gene expression from recombinant viral vectors in the central nervous system after blood-brain barrier disruption. Neurosurgery. 1995;36:965–970. doi: 10.1227/00006123-199505000-00012. [DOI] [PubMed] [Google Scholar]
- 21.Fu H, Muenzer J, Samulski RJ, Breese G, Sifford J, Zeng X, et al. Self-complementary adeno-associated virus serotype 2 vector: global distribution and broad dispersion of AAV-mediated transgene expression in mouse brain. Mol Ther. 2003;8:911–917. doi: 10.1016/j.ymthe.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 22.Neuwelt EA, Barnett PA, Hellstrom KE, Hellstrom I, McCormick CI, Ramsey FL. Effect of blood-brain barrier disruption on intact and fragmented monoclonal antibody localization in intracerebral lung carcinoma xenografts. J Nucl Med. 1994;35:1831–1841. [PubMed] [Google Scholar]
- 23.Zlokovic BV, Apuzzo ML. Cellular and molecular neurosurgery: pathways from concept to reality–part II: vector systems and delivery methodologies for gene therapy of the central nervous system. Neurosurgery. 1997;40:805–812. doi: 10.1097/00006123-199704000-00028. discussion 812–803. [DOI] [PubMed] [Google Scholar]
- 24.Ikeda MF, Bhattacharjee AKF, Kondoh TF, Nagashima TF, Tamaki N. Synergistic effect of cold mannitol and Na(+)/Ca(2+) exchange blocker on blood-brain barrier opening. Biochem Biophys Res Commun. 2002;291:669–674. doi: 10.1006/bbrc.2002.6495. [DOI] [PubMed] [Google Scholar]
- 25.Fratantoni JC, Hall CW, Neufeld EF. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science (New York, NY. 1968;162:570–572. doi: 10.1126/science.162.3853.570. [DOI] [PubMed] [Google Scholar]
- 26.Leinekugel P, Michel S, Conzelmann E, Sandhoff K. Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet. 1992;88:513–523. doi: 10.1007/BF00219337. [DOI] [PubMed] [Google Scholar]
- 27.Abkowitz JL, Persik MT, Catlin SN, Guttorp P. Simulation of hematopoiesis: implications for the gene therapy of lysosomal enzyme disorders. Acta Haematol. 1996;95:213–217. doi: 10.1159/000203880. [DOI] [PubMed] [Google Scholar]
- 28.Berns KI, Linden RM. The cryptic life style of adeno-associated virus. Bioessays. 1995;17:237–245. doi: 10.1002/bies.950170310. [DOI] [PubMed] [Google Scholar]
- 29.Kaplitt MG, Leone P, Samulski RJ, Xiao X, Pfaff DW, O’Malley KL, et al. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat Genet. 1994;8:148–154. doi: 10.1038/ng1094-148. [DOI] [PubMed] [Google Scholar]
- 30.McCown TJ, Xiao X, Li J, Breese GR, Samulski RJ. Differential and persistent expression patterns of CNS gene transfer by an adeno-associated virus (AAV) vector. Brain Res. 1996;713:99–107. doi: 10.1016/0006-8993(95)01488-8. [DOI] [PubMed] [Google Scholar]
- 31.Sly WS, Vogler C. Brain-directed gene therapy for lysosomal storage disease: going well beyond the blood- brain barrier. Proc Natl Acad Sci U S A. 2002;99:5760–5762. doi: 10.1073/pnas.102175599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu H, Samulski RJ, McCown TJ, Picornell YJ, Fletcher D, Muenzer J. Neurological correction of lysosomal storage in a mucopolysaccharidosis IIIB mouse model by adeno-associated virus-mediated gene delivery. Mol Ther. 2002;5:42–49. doi: 10.1006/mthe.2001.0514. [DOI] [PubMed] [Google Scholar]
- 33.Desmaris N, Verot L, Puech JP, Caillaud C, Vanier MT, Heard JM. Prevention of neuropathology in the mouse model of Hurler syndrome. Ann Neurol. 2004;56:68–76. doi: 10.1002/ana.20150. [DOI] [PubMed] [Google Scholar]
- 34.Fu H, Kang L, Jennings JS, Moy SS, Perez A, Dirosario J, et al. Significantly increased lifespan and improved behavioral performances by rAAV gene delivery in adult mucopolysaccharidosis IIIB mice. Gene Ther. 2007;14:1065–1077. doi: 10.1038/sj.gt.3302961. [DOI] [PubMed] [Google Scholar]
- 35.Liu G, Martins I, Wemmie JA, Chiorini JA, Davidson BL. Functional correction of CNS phenotypes in a lysosomal storage disease model using adeno-associated virus type 4 vectors. J Neurosci. 2005;25:9321–9327. doi: 10.1523/JNEUROSCI.2936-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heuer GG, Passini MA, Jiang K, Parente MK, Lee VM, Trojanowski JQ, et al. Selective neurodegeneration in murine mucopolysaccharidosis VII is progressive and reversible. Ann Neurol. 2002;52:762–770. doi: 10.1002/ana.10373. [DOI] [PubMed] [Google Scholar]
- 37.Cressant A, Desmaris N, Verot L, Brejot T, Froissart R, Vanier MT, et al. Improved behavior and neuropathology in the mouse model of Sanfilippo type IIIB disease after adeno-associated virus-mediated gene transfer in the striatum. J Neurosci. 2004;24:10229–10239. doi: 10.1523/JNEUROSCI.3558-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet. 2007;369:2097–2105. doi: 10.1016/S0140-6736(07)60982-9. [DOI] [PubMed] [Google Scholar]
- 39.Worgall S, Sondhi D, Hackett NR, Kosofsky B, Kekatpure MV, Neyzi N, et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Human gene therapy. 2008;19:463–474. doi: 10.1089/hum.2008.022. [DOI] [PubMed] [Google Scholar]
- 40.Sands MS, Barker JE. Percutaneous intravenous injection in neonatal mice. Lab Anim Sci. 1999;49:328–330. [PubMed] [Google Scholar]
- 41.Hartung SD, Frandsen JL, Pan D, Koniar BL, Graupman P, Gunther R, et al. Correction of metabolic, craniofacial, and neurologic abnormalities in MPS I mice treated at birth with adeno-associated virus vector transducing the human alpha-L-iduronidase gene. Mol Ther. 2004;9:866–875. doi: 10.1016/j.ymthe.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi H, Carbonaro D, Pepper K, Petersen D, Ge S, Jackson H, et al. Neonatal gene therapy of MPS I mice by intravenous injection of a lentiviral vector. Mol Ther. 2005;11:776–789. doi: 10.1016/j.ymthe.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 43.Mango RL, Xu L, Sands MS, Vogler C, Seiler G, Schwarz T, et al. Neonatal retroviral vector-mediated hepatic gene therapy reduces bone, joint, and cartilage disease in mucopolysaccharidosis VII mice and dogs. Mol Genet Metab. 2004;82:4–19. doi: 10.1016/j.ymgme.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 44.McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8:1248–1254. doi: 10.1038/sj.gt.3301514. [DOI] [PubMed] [Google Scholar]
- 45.Ryazantsev S, Yu WH, Zhao HZ, Neufeld EF, Ohmi K. Lysosomal accumulation of SCMAS (subunit c of mitochondrial ATP synthase) in neurons of the mouse model of mucopolysaccharidosis III B. Molecular genetics and metabolism. 2007;90:393–401. doi: 10.1016/j.ymgme.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diringer MN, Zazulia AR. Osmotic therapy: fact and fiction. Neurocrit Care. 2004;1:219–233. doi: 10.1385/NCC:1:2:219. [DOI] [PubMed] [Google Scholar]
- 47.Shapiro WR, Green SB, Burger PC, Selker RG, VanGilder JC, Robertson JT, et al. A randomized comparison of intra-arterial versus intravenous BCNU, with or without intravenous 5-fluorouracil, for newly diagnosed patients with malignant glioma. J Neurosurg. 1992;76:772–781. doi: 10.3171/jns.1992.76.5.0772. [DOI] [PubMed] [Google Scholar]
- 48.Cosolo WC, Martinello P, Louis WJ, Christophidis N. Blood-brain barrier disruption using mannitol: time course and electron microscopy studies. The American journal of physiology. 1989;256:R443–447. doi: 10.1152/ajpregu.1989.256.2.R443. [DOI] [PubMed] [Google Scholar]
- 49.Daly TM, Ohlemiller KK, Roberts MS, Vogler CA, Sands MS. Prevention of systemic clinical disease in MPS VII mice following AAV-mediated neonatal gene transfer. Gene Ther. 2001;8:1291–1298. doi: 10.1038/sj.gt.3301420. [DOI] [PubMed] [Google Scholar]
- 50.Daly TM, Vogler C, Levy B, Haskins ME, Sands MS. Neonatal gene transfer leads to widespread correction of pathology in a murine model of lysosomal storage disease. Proc Natl Acad Sci U S A. 1999;96:2296–2300. doi: 10.1073/pnas.96.5.2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gliddon BL, Hopwood JJ. Enzyme-replacement therapy from birth delays the development of behavior and learning problems in mucopolysaccharidosis type IIIA mice. Pediatr Res. 2004;56:65–72. doi: 10.1203/01.PDR.0000129661.40499.12. [DOI] [PubMed] [Google Scholar]
- 52.Kakkis ED. Enzyme replacement therapy for the mucopolysaccharide storage disorders. Expert Opin Investig Drugs. 2002;11:675–685. doi: 10.1517/13543784.11.5.675. [DOI] [PubMed] [Google Scholar]
- 53.Muenzer J, Lamsa JC, Garcia A, Dacosta J, Garcia J, Treco DA. Enzyme replacement therapy in mucopolysaccharidosis type II (Hunter syndrome): a preliminary report. Acta Paediatr Suppl. 2002;91:98–99. doi: 10.1111/j.1651-2227.2002.tb03115.x. [DOI] [PubMed] [Google Scholar]
- 54.Harmatz P, Whitley CB, Waber L, Pais R, Steiner R, Plecko B, et al. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) J Pediatr. 2004;144:574–580. doi: 10.1016/j.jpeds.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 55.Vogler C, Sands MS, Levy B, Galvin N, Birkenmeier EH, Sly WS. Enzyme replacement with recombinant beta-glucuronidase in murine mucopolysaccharidosis type VII: impact of therapy during the first six weeks of life on subsequent lysosomal storage, growth, and survival. Pediatr Res. 1996;39:1050–1054. doi: 10.1203/00006450-199606000-00019. [DOI] [PubMed] [Google Scholar]
- 56.Zhao KW, Neufeld EF. Purification and characterization of recombinant human alpha-N-acetylglucosaminidase secreted by Chinese hamster ovary cells. Protein Expr Purif. 2000;19:202–211. doi: 10.1006/prep.2000.1230. [DOI] [PubMed] [Google Scholar]
- 57.Weber B, Hopwood JJ, Yogalingam G. Expression and characterization of human recombinant and alpha-N-acetylglucosaminidase. Protein Expr Purif. 2001;21:251–259. doi: 10.1006/prep.2000.1361. [DOI] [PubMed] [Google Scholar]
- 58.Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, et al. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 59.Warburton EC, Baird A, Morgan A, Muir JL, Aggleton JP. The conjoint importance of the hippocampus and anterior thalamic nuclei for allocentric spatial learning: evidence from a disconnection study in the rat. J Neurosci. 2001;21:7323–7330. doi: 10.1523/JNEUROSCI.21-18-07323.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lijam N, Paylor R, McDonald MP, Crawley JN, Deng CX, Herrup K, et al. Social interaction and sensorimotor gating abnormalities in mice lacking Dvl1. Cell. 1997;90:895–905. doi: 10.1016/s0092-8674(00)80354-2. [DOI] [PubMed] [Google Scholar]
- 61.Thompson JN, Nowakoski RW. Enzymatic diagnosis of selected mucopolysaccharidoses: Hunter, Morquio type A, and Sanfilippo types A, B, C, and D, and procedures for measurement of 35SO4-glycosaminoglycans. In: Hommes FA, editor. Techniques in diagnostic human biochemical genetics – a laboratory manual. Wiley-Liss; New York: 1991. pp. 567–586. [Google Scholar]
- 62.van de Lest CH, Versteeg EM, Veerkamp JH, van Kuppevelt TH. Quantification and characterization of glycosaminoglycans at the nanogram level by a combined azure A-silver staining in agarose gels. Anal Biochem. 1994;221:356–361. doi: 10.1006/abio.1994.1425. [DOI] [PubMed] [Google Scholar]
- 63.de Jong JG, Wevers RA, Laarakkers C, Poorthuis BJ. Dimethylmethylene blue-based spectrophotometry of glycosaminoglycans in untreated urine: a rapid screening procedure for mucopolysaccharidoses. Clin Chem. 1989;35:1472–1477. [PubMed] [Google Scholar]