

Highlights
-
•
The anthraquinone atanyl blue PRL inhibits human cytomegalovirus replication.
-
•
The block to viral replication appears early after entry and substantially reduces viral immediate early gene expression.
-
•
In vitro, atanyl blue PRL inhibits the nuclease activity of purified viral alkaline nuclease, UL98.
-
•
The antiviral activity of atanyl blue PRL may be manifested through inhibition of UL98’s nuclease activity.
Keywords: Cytomegalovirus, Antivirals, Anthraquinone, Atanyl blue PRL, Alkaline nuclease
Abstract
Human cytomegalovirus (CMV) causes significant disease in immunocompromised patients and serious birth defects if acquired in utero. Available CMV antivirals target the viral DNA polymerase, have significant toxicities, and suffer from resistance. New drugs targeting different pathways would be beneficial. The anthraquinone emodin is proposed to inhibit herpes simplex virus by blocking the viral nuclease. Emodin and related anthraquinones are also reported to inhibit CMV. In the present study, emodin reduced CMV infectious yield with an EC50 of 4.9 μM but was cytotoxic at concentrations only twofold higher. Related anthraquinones acid blue 40 and alizarin violet R inhibited CMV at only high concentrations (238–265 μM) that were also cytotoxic. However, atanyl blue PRL inhibited infectious yield of CMV with an EC50 of 6.3 μM, significantly below its 50% cytotoxic concentration of 216 μM. Atanyl blue PRL reduced CMV infectivity and inhibited spread. When added up to 1 h after infection, it dramatically reduced CMV immediate early protein expression and blocked viral DNA synthesis. However, it had no antiviral activity when added 24 h after infection. Interestingly, atanyl blue PRL inhibited nuclease activities of purified CMV UL98 protein with IC50 of 4.5 and 9.3 μM. These results indicate that atanyl blue PRL targets very early post-entry events in CMV replication and suggest it may act through inhibition of UL98, making it a novel CMV inhibitor. This compound may provide valuable insights into molecular events that occur at the earliest times post-infection and serve as a lead structure for antiviral development.
1. Introduction
Human cytomegalovirus (CMV) causes a spectrum of diseases in immune compromised patients, including retinitis in HIV patients, pneumonitis in transplant patients, and serious birth defects characterized by sensorineural hearing loss and severe mental retardation when acquired during pregnancy. Drugs currently licensed for the treatment of systemic CMV infections have limited therapeutic effectiveness due to dose-limiting toxicities, and prolonged therapy often results in resistance (Biron, 2006). None are approved for treatment or prevention of congenital infections. Thus, there is a pressing need for more potent, less toxic therapeutics to combat CMV infections.
One possible target for development of such novel antivirals is the CMV UL98 alkaline nuclease encoded by the UL98 gene. Homologs of UL98 are encoded by all known herpesviruses. The alkaline nuclease encoded by herpes simplex virus type 1 (HSV-1), UL12, is necessary for efficient replication in cell culture (Martinez et al., 1996, Shao et al., 1993). Impaired replication has been linked to defects in DNA processing, capsid stability, and capsid nuclear egress (Martinez et al., 1996, Porter and Stow, 2004, Shao et al., 1993, Weller et al., 1990). Recently our group showed that a CMV UL98 null mutant is severely compromised for replication (Kuchta et al., 2012). At least one function of the CMV UL98 is likely to be similar to HSV-1 UL12 since the CMV UL98 gene can functionally complement the replication defect of HSV-1 UL12 null viruses (Gao et al., 1998). A recent study indicated that the nuclease activity of UL12 is important for HSV-1 neurovirulence in mice yet is largely dispensable for replication in cell culture (Fujii et al., 2013). In contrast, earlier studies indicated that the nuclease activity is important for in vitro replication since only alleles of HSV-1 UL12 that encoded a functional nuclease were able to complement replication of UL12 null mutant viruses (Goldstein and Weller, 1998, Henderson et al., 1998).
Consistent with UL12 nuclease activity playing important roles both in vitro and in vivo, recent studies found that the anthraquinone emodin (Fig. 1 ) inhibits DNase activity of the HSV-1 UL12 in vitro, blocks replication of HSV-1 and herpes simplex virus type 2 (HSV-2) in cell culture, and reduces viral pathogeneses in a mouse model (Hsiang and Ho, 2008, Xiong et al., 2011). Earlier reports indicated that emodin and other anthraquinone derivatives also have CMV inhibitory activities (Barnard et al., 1992, Barnard et al., 1995), although the mechanism of CMV inhibition has not been further studied. In the present study the anti-CMV activities of emodin and three related anthraquinones, atanyl blue PRL (also known as acid blue 129), acid blue 40, and alizarin violet R (Fig. 1) were evaluated. Atanyl blue PRL had anti-CMV activity and acted at an early post-entry stage of replication. Atanyl blue PRL also inhibited the nuclease activity of UL98, suggesting a potential mechanism of action in which UL98 activity is important early in the CMV replication cycle.
Fig. 1.
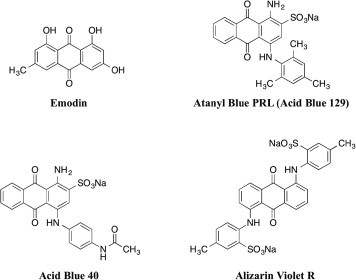
Structures of compounds used.
2. Materials and methods
2.1. Viruses and cell culture
Human MRC-5 fibroblasts (ATCC CCL-171) were propagated in modified Eagle medium (Gibco-BRL) supplemented with 10% fetal calf serum (HyClone Laboratories), 10,000 IU/L penicillin, and 10 mg/L streptomycin (Gibco-BRL) (MEM). CMV BADrUL131-Y4 (a gift from Thomas Shenk and Dai Wang) is a variant of CMV strain AD169 that contains a marker cassette encoding green fluorescent protein (GFP) under control of a simian virus 40 promoter inserted into UL21.5 (Wang and Shenk, 2005). CMV RC2626 is a variant of CMV strain Towne containing a luciferase expression cassette under control of a synthetic P1125 promoter (composed of seven tetracycline operator elements, a 23-bp TAATA-containing element from the adenovirus major late promoter, and a 17-bp initiator from the mouse TdT gene promoter) inserted into the US2-US6 region (McVoy and Mocarski, 1999). Expression of the relevant marker proteins (GFP or luciferase) encoded by these viruses can be detected as early as 24 h post infection (hpi). Viruses were propagated in MRC-5 cells and titered as described (Cui et al., 2012, Cui et al., 2008, Saccoccio et al., 2011b).
2.2. Compounds
Ganciclovir was purchased from InvivoGen. BAY 38-4766 was provided by Bayer® Pharmaceuticals (Tubingen, Germany). Emodin, atanyl blue PRL, and acid blue 40 were purchased from Sigma–Aldrich Co. Alizarin violet R was purchased from MP Biomedicals. Ganciclovir was dissolved in water at a concentration of 100 mM. The remaining compounds were solubilized in dimethyl sulfoxide (DMSO, Sigma–Aldrich) at a stock concentration of 100 mM. Compounds were stored at −20 °C.
2.3. Luciferase-based yield reduction assay
CMV yield was determined using a luciferase-based assay as described previously (Bhave et al., 2013). Briefly, 96-well plates containing confluent monolayers of MRC-5 fibroblasts were infected at low input multiplicity with luciferase-tagged CMV RC2626 (McVoy and Mocarski, 1999) and incubated for 1 hour. Various amounts of test compounds in MEM were then added. No-virus controls and no-drug controls consisting of medium containing 0.25% DMSO (equal to the maximum amount of DMSO present in test compound assays) were assayed on each plate and all conditions were assayed in triplicate. Cultures were maintained with no additional media changes until 5 days post infection (dpi), when 50 μl of supernatant from each well was transferred to black-walled, clear/flat-bottomed 96-well plates containing confluent MRC-5 monolayers and luciferase activity was measured 24 h later using the Steady-Glo luciferase assay reagent (Promega). Luciferase activity was measured in relative light units (RLU) using a Biotek Synergy HT Multimode Microplate Reader. Data were normalized by converting RLU to “percent maximum luminescence” for each experiment and fitted to four-parameter curves using Prism 5 (GraphPad Software, Inc.) to determine the half-maximal effective concentrations (EC50) of test compounds.
For time of addition studies cultures were infected as described above and maintained in medium until compounds were added. At different times post infection compounds diluted in culture medium were added. Cultures were maintained with no additional media changes until 5 dpi. Two preincubation experiments were conducted in conjunction with time of addition studies. In the first, wells containing uninfected cells were incubated for 1 h with medium containing 28 μM atanyl blue PRL, then washed 3 times with medium to remove the atanyl blue PRL. The cultures were then infected as described above and maintained for 5 days in medium without atanyl blue PRL. In the second, a 10-fold higher concentration of virus inoculum was incubated for 1 h with medium containing 28 μM atanyl blue PRL, then diluted 10-fold with culture medium and added to wells containing cells.
2.4. Cytotoxicity assay
Black-walled, clear bottom 96-well plates containing confluent monolayers of MRC-5 cells were incubated in triplicate with test compounds as described above. After 5 days, 100 μl of each supernatant was removed and 100 μl CellTiter-Glo assay reagent (Promega) was added to each well. Luminescence was measured in RLUs as described above and the data were normalized and fitted to four-parameter curves to determine the 50% toxic doses (TD50) of test compounds.
2.5. GFP-based assays of gene expression, virus spread, and infectivity
Clear-walled, clear-bottom 96-well plates containing confluent monolayers of MRC-5 cells were infected at 37 °C with GFP-expressing CMV BADrUL131-Y4. At the time of virus infection, atanyl blue PRL in MEM was added to achieve various final concentrations. No-virus controls and no-drug controls consisting of medium containing 0.25% DMSO (equal to the maximum amount of DMSO present in test compound assays) were assayed on each plate and all conditions were assayed in triplicate. On specified dpi GFP fluorescence was quantified as relative fluorescent units (RFUs) using a Biotek Synergy HT Multimode Microplate Reader and used to calculate EC50 as described above. In some experiments individual GFP-positive cells were marked and photomicrographed every two days using a Nikon Eclipse TS100 UV inverted microscope.
To measure the effect of atanyl blue PRL on CMV infectivity, MRC-5 cultures in 96-well plates were infected with CMV BADrUL131-Y4 at a concentration empirically determined to result in 75% of wells becoming infected. Groups of 32 replicate wells were treated at the time of infection with MEM containing increasing concentrations of atanyl blue PRL. After 14 days wells were scored for the presence or absence of GFP fluorescence using a Nikon Eclipse TS100 inverted microscope. The percentage of positive wells for each group of 32 was plotted vs. atanyl blue PRL concentration and the EC50 was determined as described above.
2.6. Immunoblot analysis
Replicate 25 cm2 flasks containing confluent MRC-5 cells were mock infected or infected with BadrUL131-Y4 at a multiplicity of infection (MOI) of 0.8. At the time of infection replicate flasks were adjusted to a final concentration of 28 μM atanyl blue PRL or left untreated. Cells in replicate flasks were harvested daily on days 1–4 post infection and analyzed by immunoblotting as described previously (Ryckman et al., 2008, Saccoccio et al., 2011a). Primary antibodies included mouse monoclonal mAB810 (anti-IE1/2, Millipore) diluted 1:1000, mouse monoclonal I2 (anti-UL98 (Rice et al., 1984), a gift from Jay Nelson) diluted 1:1000, and rabbit polyclonal antisera to glycoprotein B (gB) (a gift from Larry Smith, Vical Inc.) diluted 1:2000. Secondary antibodies were horseradish peroxidase-conjugated polyclonal goat anti-mouse or goat anti-rabbit IgG (Thermo-Fisher) diluted 1:5000.
2.7. RT-PCR quantification of CMV DNA
Replicate 25 cm2 flasks containing confluent MRC-5 cells were mock infected or infected with BadrUL131-Y4 at an MOI of 0.5. One set of flasks contained MEM only throughout the experiment; a second contained MEM with 28 μM atanyl blue PRL from the time of infection; a third set contained MEM from the time of infection until 24 hpi, then MEM was replaced with MEM containing 28 μM atanyl blue PRL. Cells from replicate flasks were harvested at 3, 24, 48, 72, and 96 hpi by trypsinization and DNA was extracted as described previously (McVoy et al., 1997). DNA precipitates were dissolved in 20 μl water and DNA concentrations were measured using a nanodrop spectrophotometer.
qRT-PCR was performed by the Virginia Commonwealth University Nucleic Acid Research Facilities using a ViiA 7 Sequence Detection System (Life Technologies) and TaqMan® Universal PCR Master Mix Reagents (Life Technologies). The forward primer 5′-GAGCCCGACTTTACCATCCA, reverse primer 5′-CAGCCGGCGGTATCGA, and probe VIC-ACCGCAACAAGATT-MGBNFQ were derived from exon five of the UL122 gene as described previously (Boppana et al., 2010). 10 μl reactions contained 20 ng DNA, 0.9 μM forward and reverse primers, and 0.25 μM probe. Cycling conditions were: 50 °C for 2 min; 95 °C for 10 min; then 40 cycles of 95 °C for 15 s and 60 °C for 1 min. 18S ribosomal RNA was measured using primers provided in the TaqMan® Assay Reagents as an endogenous control to determine relative CMV DNA copies/cell. Each reaction was performed in triplicate.
2.8. Expression and purification of UL98
Recombinant UL98 was inducibly expressed in Escherichia coli BL21 (DE3) lys S and purified using methods modified from those previously described (Kuchta et al., 2012). The UL98 expression vector pD17–98(wt) (Kuchta et al., 2012) was modified to contain an N-terminal sequence encoding two tandem histidine (his6) tags. This was accomplished by annealing of synthetic linkers 5′p-TATGTCGTACTACCATCACCACCATCACCATTCTAGAGCTTGGCGTCACCCGCAGTTCGGAGGCCACCACCACCACCACCACAGCAGCGGCAGAGAAAACCTGTATTTTCAGGGCCATATGTGGGGCGTC-3′OH and 5′p-TCGAGACGCCCCACATATGGCCCTGAAAATACAGGTTTTCTCTGCCGCTGCTGTGGTGGTGGTGGTGGTGGCCTCCGAACTGCGGGTGACGCCAAGCTCTAGAATGGTGATGGTGGTGATGGTAGTACGACA-3′OH and ligation into NdeI/XhoI-restricted pD17-98(wt) to make plasmid p2XhisUL98(wt).
E. coli BL21 (DE3) lys S cells containing p2XhisUL98(wt) were cultured at 37 °C then induced with IPTG for 4 h at 25 °C as described previously (Kuchta et al., 2012). Bacteria from 3 L of culture were pelleted, snap frozen in liquid nitrogen, and stored at −80 °C. Frozen cells were thawed on ice in buffer A (20 mM Tris–Cl, pH 8.0, 10 mM imidazole, 10% glycerol, 300 mM NaCl, and 1% NP-40), extracted by ultrasonic disruption, and clarified by centrifugation at 70,000×g. The supernatant was diluted 1:3 with buffer A then applied to an AKTA fast protein liquid chromatography system equipped with a 6 ml Ni-agarose column (Ni–NTA Superflow, QIAGEN) equilibrated in buffer A. The column was washed with 40 ml of buffer A containing 20 mM imidazole and eluted with a 60 ml linear gradient of buffer A containing 20–500 mM imidazole. Fractions containing UL98 (material reactive to I2 monoclonal antibody by immunoblot analysis) were combined and dialyzed overnight in buffer B2 (20 mM Tris–Cl, pH 8.2, 50 mM NaCl, 1 mM EDTA, 2 mM 2-mercaptoethanol, 10% glycerol), then applied to a 1 ml Mono Q column equilibrated in buffer B2. The column was washed with 10 ml buffer B2 and proteins were eluted into 1 mL fractions with a 25 ml linear gradient of buffer B2 containing 0–500 mM KCl. Fractions were assayed for the presence of nuclease activity as described below and by immunoblotting with I2 monoclonal antibody. UL98 was present in fractions eluted with ∼340–350 mM KCl and stored for up to 1 month at 4 °C.
2.9. Nuclease inhibition
Two assays were used to measure inhibition of UL98 nuclease activity. The first measured combined endonuclease and exonuclease activity as release of trichloroacetic acid (TCA) soluble-radioactivity from uniformly 14C-labeled E. coli duplex DNA as described previously (Hoffmann and Cheng, 1978). A unit of enzyme activity was defined as the amount of enzyme required to digest 1 ng of labeled DNA to TCA-solubility in 10 min at 37 °C. Various compound concentrations were added to reaction mixes and assays were initiated by the addition of purified UL98 (1100 units). Control reactions contained 5% DMSO without compound. After incubation for 15 min at 37 °C, reactions were terminated by addition of TCA to a 5% final concentration and 50 μg unlabeled, sonicated, and denatured salmon sperm DNA as carrier. The second assay used a doubly-quenched fluorescent hairpin probe to measure predominantly exonuclease activity in a fluorescent resonance emission transfer (FRET) assay. A synthetic 29-mer oligonucleotide was labeled at the 5′ end with FAM fluorophore, between nucleotides 9 and 10 with ZEN quencher, and at the 3′ end with Iowa Black dark quencher (IDT). The probe was heated to 95 °C for 2 min and quenched on ice to form a hairpin containing a 4-bp 5′ overhang. Exonucleolytic release of the 5′ FAM-labeled nucleotide resulted in fluorescence. 70 μl assays containing 400 nM hairpin DNA substrate were incubated with 10 μl purified enzyme (1100 units as described above) for 15 min at 37 °C and terminated by the addition of EDTA to 16 mM final concentration. 20 μl aliquots were transferred in triplicate to round-bottom black-walled 384-well pates and RFUs were measured using a SpectraMax M5 fluorometer (Molecular Devices). Controls containing substrates incubated without enzyme were used to determine background values, which were subtracted from experimental readings. Data for each inhibitor concentration were expressed as the percentage of maximal activities obtained from untreated controls (DNA plus enzyme in 5% DMSO). Means (±1 standard deviation) from three independent assays were plotted and fitted to four-parameter curves to determine 50% inhibitory concentrations (IC50).
2.10. Molecular interaction analysis of atanyl blue PRL and acid blue 40 with UL98
Structures of the compounds were sketched using SYBYL-X 2.1 (Tripos Inc.); Gasteiger–Hückel charges were assigned and subjected to energy minimization under the Tripos force-field (10,000 iterations, termination gradient of 0.01 kcal/mol-Å). A previously reported model of UL98 in complex with substrate DNA (Kuchta et al., 2012) was used to perform docking studies using GOLD v5.2. The binding site was defined to encompass all atoms within 10 Å radius of Oδ1 of D254, which is located at the center of the active site. Default genetic algorithm parameters were used and a total of 50 solutions per compound were generated (no early termination, no constraints). The most plausible binding mode was identified using the HINT force-field (Kellogg and Abraham, 2000). The ligand in its most favorable binding conformation was complexed with the protein and subjected to minimization (Powell minimization, 2500 iterations, termination gradient of 0.01 kcal/mol-Å) to remove steric clashes and optimize the protein–ligand interactions within the active site.
3. Results
3.1. Antiviral activities of emodin and other anthraquinone derivatives
To further explore the anti-CMV activity of emodin and related anthraquinones (Fig. 1), a luciferase-based yield reduction assay (Bhave et al., 2013) was used. MRC-5 cells were infected with a luciferase-tagged CMV at an MOI of 0.03 and increasing concentrations of emodin were added 1 hpi. Five dpi virus yields were measured by transfer of culture supernatants to uninfected MCR-5 cells and measurement of luciferase activity in these cultures 24 h later. Cytotoxicity was evaluated by adding the same concentrations of emodin to uninfected MRC-5 cells for 5 days and then measuring intracellular ATP levels as a function of luciferase activity using the CellTiter-Glo assay. Luciferase data were used to determine emodin’s 50% effective concentration (EC50) for anti-CMV activity and 50% cytotoxic dose (TD50) (Table 1 ). While the EC50 of 4.9 μM confirmed that emodin inhibits CMV replication, emodin concentrations less than twofold higher were cytotoxic (Fig. 2 A and Table 1). Thus, emodin’s anti-CMV activity is likely to be non-specific and arise simply due to toxic effects on host cells.
Table 1.
Anti-CMV activities and cytotoxicities of anthraquinone derivatives (μM ± SD).
| Compound | This study |
Previously reporteda |
||||
|---|---|---|---|---|---|---|
| Anti-CMVb | Cytotoxicityc | SIf | Anti-CMVd | Cytotoxicitye | SIf | |
| Emodin | 4.9 ± 0.38 | 9.2 ± 1.3 | 1.9 | 4.1 | 9.6 | 2.3 |
| Acid blue 40 | 205 ± 27 | 231 ± 27 | 1.1 | 10 | 380 | 38 |
| Alizarin violet R | 183 ± 19 | 342 ± 40 | 1.9 | 10 | 300 | 30 |
| Atanyl blue PRL | 6.3 ± 1.5 | 216 ± 12 | 34 | 7 | 275 | 40 |
EC50 from luciferase-based CMV yield assays.
TD50 from CellTiter-Glo cytotoxicity assays.
EC50 from plaque reduction assays.
IC50 from morphological cytotoxicity assays.
Selectivity index (SI, determined as TD50/EC50).
Fig. 2.
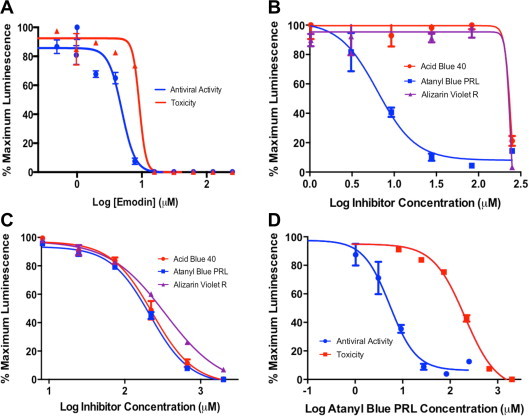
Luciferase-based viral yield and cytotoxicity assays. (A) Antiviral activity of emodin (blue line) was determined by infecting confluent MRC-5 monolayers in 96-well plates with luciferase-tagged CMV RC2626 at an MOI of 0.03 in the presence of increasing concentrations emodin. After 5 days supernatants were transferred to 96-well plates containing fresh MRC-5 monolayers and 24 h later the cultures were lysed and luciferase activities determined as a measure of virus in the transferred culture supernatants. Cytotoxicity (red line) was determined by incubating replicate uninfected cultures with emodin for 5 days, then determining luciferase activities as a measure of cell viability using the CellTiter-Glo assay. (B) Antiviral activities of the indicated compounds were determined as described for panel A. (C) Cytotoxicities of the indicated compounds were determined as described for panel A. (D) Results from panels B and C are graphed together to illustrate the difference between antiviral activity (blue) and cytotoxicity (red) of atanyl blue PRL. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Several other anthraquinone derivatives were previously reported to possess anti-CMV activity (Barnard et al., 1995). Atanyl blue PRL, acid blue 40, and alizarin violet R (Fig. 1) were evaluated using the assays described above. EC50 for acid blue 40 and alizarin violet R were high (205 μM and 183 μM, respectively) and closely corresponded to their cytotoxicities (Fig. 2 B and C, Table 1). Thus, while less toxic than emodin, these two compounds also appeared to lack virus-specific inhibitory activity. In contrast, atanyl blue PRL exhibited anti-CMV activity similar to emodin (EC50 = 6.3 μM) but with a cytotoxicity similar to that of acid blue 40 and alizarin violet R (TD50 = 216 μM) (Fig. 2D, Table 1). The resulting selectivity index (SI) of 34 (Table 1) suggests that atanyl blue PRL has CMV inhibitory activity.
3.2. Atanyl blue PRL blocks viral-encoded GFP expression, virus spread, and infectivity
To determine the impact of atanyl blue PRL on viral gene expression, cell cultures were infected with a CMV genetically engineered to express GFP. An MOI of 1 was used in order to initially infect approximately two-thirds of cells in the culture. Increasing concentrations of atanyl blue PRL were added immediately after infection and GFP fluorescence was quantified at 4 dpi. Atanyl blue PRL reduced GFP expression from infected cells in a dose–responsive fashion (Fig. 3 A). The EC50 calculated from these GFP data (6.2 μM) was comparable to the luciferase-based anti-CMV activity (6.3 μM) (Table 2 ).
Fig. 3.
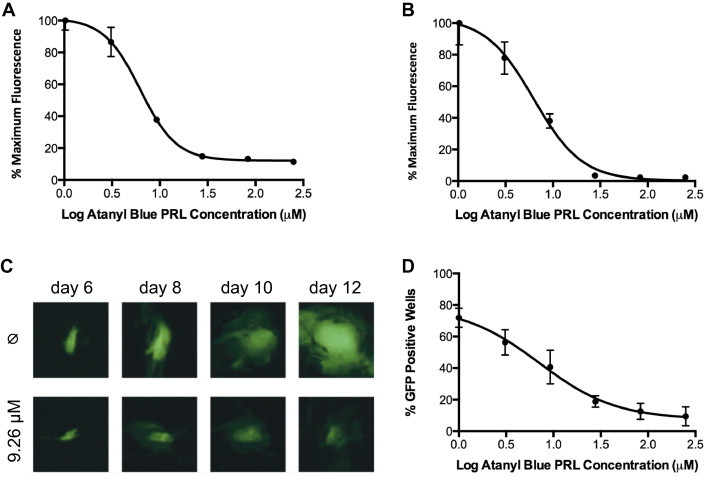
Effects of atanyl blue PRL on CMV-encoded GFP expression, spread, and infectivity. (A) 96-well plates containing confluent monolayers of MRC-5 cells were infected at an MOI of 1 with GFP-tagged CMV BADrUL131-Y4 in the presence of increasing concentrations of atanyl blue PRL. Four dpi GFP fluorescence was determined as a measure of viral gene expression. (B) The same experiment was conducted using an MOI of 0.1 such that only once cell in ten was initially infected. GFP fluorescence was determined every 2 days as a measure of viral spread to cells that were initially uninfected; data shown are from 20 dpi. (C) 96-well plates containing confluent monolayers of MRC-5 cells were infected with BADrUL131-Y4 at an MOI of 0.01 in the absence (Ø) or presence of 9 μM atanyl blue PRL. GFP-positive cells were marked and photographed 6 dpi and at 2-day intervals thereafter; representative images are shown. (D) Thirty-two replicate wells containing MRC-5 cells were infected with an inoculum of BADrUL131-Y4 sufficient to infect 75% of the wells in the absence of inhibitor. Replicate sets of 32 wells were infected and treated with increasing concentrations of atanyl blue PRL. After 14 days wells containing GFP-positive cells were scored as having undergone successful viral infection and spread. The number of positive wells out of 32 was converted to % GFP positive wells.
Table 2.
CMV-inhibitory activities of atanyl blue PRL (μM ± SD).
| Assay | Activity (EC50 or IC50) |
|---|---|
| Infectious yield (luciferase-based, 5 dpi, MOI = 0.03) | 6.3 ± 1.5 |
| Viral gene expression (GFP-based, 4 dpi, MOI = 1) | 6.2 ± 0.9 |
| Spread (GFP-based, 20 dpi, MOI = 0.1) | 6.3 ± 0.8 |
| Infectivity (% GFP-positive wells) | 7.1 ± 6.0 |
| Inhibition of UL98 total nuclease activity (14C release) | 4.5 ± 0.7 |
| Inhibition of UL98 5′ exonuclease activity (fluorescence) | 9.3 ± 2.3 |
A similar experiment was conducted to measure the effect of atanyl blue PRL on CMV spread. In this case cultures were infected at an MOI of 0.1 such that ∼10% of cells were initially infected. GFP was measured 20 dpi, allowing progeny time to spread to the remaining cells in the culture. Atanyl blue PRL reduced GFP expression in a dose–responsive fashion in these experiments as well (Fig. 3B), with an EC50 of 6.3 μM that was identical to that obtained from the higher MOI experiment, above.
To visualize the effect of atanyl blue PRL on CMV spread, cultures were infected at an MOI of 0.01 and were either mock treated with 5% DMSO or were treated with 9 μM atanyl blue PRL. At 6 dpi individual GFP-positive cells were identified and marked so that they could be relocated and photographed every 2 days thereafter. In the absence of inhibitor, GFP-positive foci expanded to form large syncytia, presumably by infecting and subsequently fusing with adjacent cells. In contrast, in the presence of atanyl blue PRL, GFP-positive foci expanded to very few cells (Fig. 3C).
It was noted that atanyl blue PRL did not eliminate GFP expression observed at 6 dpi, but greatly reduced the number of GFP-positive cells per well compared to the vehicle control (data not shown). This suggested the possibility that atanyl blue PRL reduced CMV infectivity by promoting abortive infections that failed to reach the stage at which GFP is expressed. To quantify the impact of atanyl blue PRL on CMV infectivity, MRC-5 cells in 96-well plates were infected with GFP-tagged CMV at a dilution empirically determined to result in infection of only 75% of wells. Under these conditions most of the infected wells received only one or two infectious virions. Increasing concentrations of atanyl blue PRL were added to replicate sets of 32 wells. After 14 days wells were scored for the absence or presence of GFP (indicating successful viral entry and gene expression). Although no drug controls confirmed that 75% of wells received at least one infectious virus that led to GFP expression by 14 dpi, the percentage of GFP-positive wells when treated with atanyl blue PRL decreased in a dose-dependent manner. The EC50 calculated from these data (7.1 μM) was similar to those obtained from virus yield or GFP expression assays (Fig. 3D, Table 2), suggesting that atanyl blue PRL blocks CMV at an early stage in the viral replication cycle.
3.3. Time of addition studies indicate that atanyl blue PRL acts early in viral replication
To better determine the stage of viral replication affected by atanyl blue PRL, the compound was added to virus or cells prior to infection, to cells and virus at the time of infection, or to infected cells at different times post infection. Based on the results shown in Fig. 2D a concentration of 28 μM atanyl blue PRL was selected as the lowest concentration that results in effective inhibition of viral yield. Ganciclovir, which inhibits CMV DNA synthesis, and BAY 38-4766, which inhibits CMV DNA packaging (Buerger et al., 2001), were also tested as known late-acting controls. As expected, ganciclovir and BAY 38-4766 could be added as late as 72 hpi and still substantially reduce virus yield detected on day 5. In contrast, antiviral activity of atanyl blue PRL was lost when added 24 hpi or later (Fig. 4 A). To refine the window of activity the experiment was repeated with atanyl blue PRL added at 0, 6, 12, 24, and 48 hpi. As before, atanyl blue PRL was ineffective when added at 24 hpi but retained partial effectiveness when added at 6 or 12 hpi (Fig. 4B).
Fig. 4.
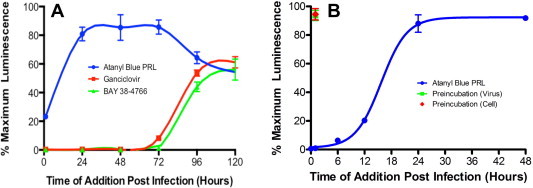
Time of addition studies. (A) MRC-5 monolayers in 96-well plates were infected with luciferase-tagged CMV RC2626 at an MOI of 0.03. Atanyl blue PRL, ganciclovir, or BAY 38–4766 were added without removal of existing medium to final concentrations of 28 μM, 10 μM, or 8 μM, respectively, at 0, 24, 48, 72, 96, or 120 hpi and the cultures were maintained without further medium changes until 5 dpi. (B) The experiment in A was repeated but atanyl blue PRL was added to a final concentration of 28 μM at 0, 6, 12, 24, or 48 hpi. In the same experiment a preincubation (cells) group consisted of wells that were preincubated for 1 h with 28 μM atanyl blue PRL, washed three times with medium, then infected with virus as above and incubated without atanyl blue PRL for 5 days. In a preincubation (virus) group a 10-fold higher concentration of virus inoculum was preincubated with 28 μM atanyl blue PRL for 1 h, diluted 10-fold with medium, added to cells as above, and incubated without atanyl blue PRL for 5 days. Virus yields were determined 5 dpi by transfer of culture supernatants to fresh MRC-5 monolayers and quantitation of luciferase activity after incubation for 24 h.
The above results suggested that atanyl blue PRL inhibits an important step in viral replication that is sufficiently completed by 24 hpi. However, because viral attachment and entry are largely completed by that time, it was possible that viral particles were inactivated by exposure to atanyl blue PRL before or during attachment and entry. To test this possibility the viral inoculum was preincubated with 28 μM atanyl blue PRL for 1 h, then diluted 10-fold and added to cells. Pretreatment had no impact on viral yield 5 days later (Fig. 4B), indicating that transient exposure to an effective concentration of atanyl blue PRL does not irreversibly inactivate infectivity of virions. In a similar experiment, cells were pretreated with 28 μM atanyl blue PRL for 1 h, then washed just prior to infection to remove atanyl blue PRL. Pretreatment of cells had no impact on viral yield 5 days later (Fig. 4B), indicating that atanyl blue PRL does not induce changes in the cellular environment that limit subsequent viral entry or replication.
3.4. Atanyl blue PRL reduces levels of CMV immediate early proteins
Following entry, the CMV genome is deposited within the nucleus, where abundant transcription initiates at the major immediate early promoter (MIEP), resulting in rapid accumulation of several immediate early (IE) proteins. These in turn are required for expression of early proteins, which include factors required for viral DNA synthesis. This in turn triggers expression of late proteins that are required to assemble progeny virions. The time of addition studies above suggested that atanyl blue PRL may block replication prior to IE protein expression. To address this question, cells infected in the presence or absence of 28 μM atanyl blue PRL were assayed by immuoblotting to determine levels of IE proteins. As shown in Fig. 5 A, atanyl blue PRL profoundly reduced the levels of CMV IE proteins throughout the course of infection. Levels of the early protein UL98 and late protein gB were also reduced (data not shown).
Fig. 5.
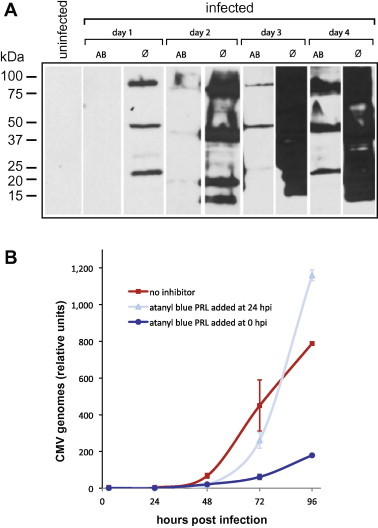
Impact of atanyl blue PRL on CMV IE protein and DNA synthesis. (A) MRC-5 cells were infected at an MOI of 0.8 in the absence (Ø) or presence of 28 μM atanyl blue PRL (AB). Cell lysates were prepared on the indicated dpi and analyzed by immunoblotting using antibodies specific to IE1/2. Images are composites from separate gels. Treated and untreated samples from a given day were analyzed on the same gel and are quantitatively comparable; however, samples from different days were analyzed on separate gels and therefore do not accurately reflect quantitative changes over time. (B) MRC-5 cells were infected with an MOI of 0.5. Replicate cultures were treated with no inhibitor, with 28 μM atanyl blue PRL from the time of infection, or with 28 μM atanyl blue PRL starting at 24 hpi. DNA was extracted from replicate cultures at 0, 24, 48, 72, and 96 hpi and relative CMV DNA levels were determined by RT-qPCR.
3.5. Atanyl blue PRL does not directly inhibit CMV DNA synthesis
By 24 hpi the CMV genome has circularized and the initial stages of DNA replication have begun; however, significant amplification of CMV DNA does not occur until 36–48 hpi (McVoy and Adler, 1994). That addition of atanyl blue PRL at 24 hpi had no impact on yield strongly suggested that atanyl blue PRL does not affect CMV DNA synthesis or inhibit the viral DNA polymerase or other replication factors. To directly determine the impact of atanyl blue PRL on CMV DNA synthesis, CMV-infected cultures were either left untreated or treated with 28 μM atanyl blue PRL from the time of infection or starting at 24 hpi. Replicate cultures were harvested at 3, 24, 48, 72, and 96 hpi and relative copy numbers of intracellular CMV DNA were determined by qPCR. In untreated cultures, CMV DNA replication began between 24 and 48 hpi and copy number increased steadily thereafter (Fig. 5B). However, when atanyl blue PRL was present from the time of infection, CMV DNA copy number remained low throughout the time course. Addition of atanyl blue PRL at 24 hpi delayed but did not prevent the onset of CMV DNA synthesis. Copy number began to increase at 72 hpi and exceeded the mock-treated control at 96 hpi (Fig. 5B). These results confirm that atanyl blue PRL does not directly inhibit viral DNA syntheses.
3.6. Atanyl blue PRL inhibits the CMV alkaline nuclease UL98
Inhibition of the viral alkaline nuclease by emodin has been proposed as the mechanism of emodin’s antiviral activity against HSV-1 and HSV-2 (Hsiang and Ho, 2008). In prior studies we expressed and purified the CMV alkaline nuclease, UL98, and characterized its nuclease activities in vitro (Kuchta et al., 2012). To determine if atanyl blue PRL inhibits the nuclease activity of recombinant UL98, we performed two different assays. In the first, we measured the effect of increasing concentrations of the inhibitor on the ability of purified UL98 to degrade a uniformly labeled DNA substrate to a TCA-soluble form (∼6-mer or less). This assay measures a combination of endonuclease and 5′-to-3′ exo activities (Hoffmann and Cheng, 1978). Atanyl blue PRL inhibited the release of soluble radioactivity with an IC50 of 4.5 μM (Fig. 6 A). In contrast, the related compound acid blue 40 had little or no effect on the nuclease activity of UL98 at concentrations 10-fold higher (IC50 = 99 μM). The second assay used a FRET assay to measure removal of a 5′ labeled fluorophore from a 3′ and internally-quenched hairpin DNA substrate by 5′ to 3′ exonuclease activity. In this assay atanyl blue PRL inhibited UL98’s nuclease activity with an IC50 of 9.3 μM, while again acid blue 40 had little inhibitory activity (IC50 = 149 μM, Fig. 6B). These results confirm that atanyl blue PRL inhibits the nuclease activity of purified UL98.
Fig. 6.
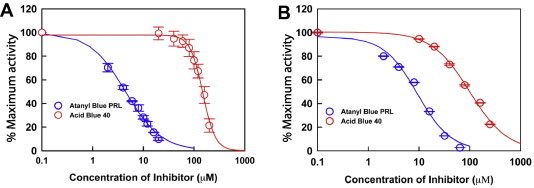
Atanyl blue PRL inhibits UL98 nuclease activity. (A) Purified recombinant UL98 was incubated with 14C-labeled E. coli DNA in the presence of increasing concentrations of atanyl blue PRL or acid blue 40. Total nuclease activity was measured as TCA-soluble counts. (B) Purified recombinant UL98 was incubated with a synthetic hairpin DNA containing a 5′ FAM fluorophore and two quenchers (internal and 3′) in the presence of increasing concentrations of atanyl blue PRL or acid blue 40. 5′-to-3′ exonuclease activity was measured as increases in fluorescence resulting from release of FAM. Data for each inhibitor concentration were expressed as the percentage of maximal activities obtained from untreated controls (DNA plus enzyme in 5% DMSO).
4. Discussion
Anthraquinones and anthraquinone derivatives are bioactive compounds found in extracts derived from bacteria, fungi, lichens, and plants. A number of compounds in this class have been shown to inactivate or inhibit entry and/or replication of a wide variety of viruses, including herpesviruses (HSV-1, HSV-2, pseudorabies virus, varicella zoster virus), retroviruses (including HIV), vesicular stomatitis virus, influenza virus, parainfluenza virus, vaccinia virus, SARS coronavirus, hepatitis B virus, poliovirus, and Coxsackievirus (Andersen et al., 1991, Cohen et al., 1996, Esposito et al., 2011, Esposito et al., 2012, Higuchi et al., 1991, Ho et al., 2007, Liu et al., 2013, Schinazi et al., 1990, Schwarz et al., 2011, Semple et al., 2001, Shuangsuo et al., 2006, Sydiskis et al., 1991). A variety of mechanisms have been described, including inactivation through disruption of viral envelopes (Alves et al., 2004, Andersen et al., 1991, Sydiskis et al., 1991), disruption of virion/receptor interactions (Ho et al., 2007), disruption of ion channels (Schwarz et al., 2011), inhibition of host protein kinase 2 (Battistutta et al., 2000, Yim et al., 1999), and inhibition of reverse transcriptase and RNase H (Esposito et al., 2011, Esposito et al., 2012, Higuchi et al., 1991, Schinazi et al., 1990).
In the 1990s emodin and certain other anthraquinone derivatives were reported to have anti-CMV activities (Barnard et al., 1992, Barnard et al., 1995). More recent studies found that emodin has activity against replication of HSV-1 and HSV-2 in cell culture and has therapeutic benefit in a mouse model of HSV pathogenesis (Hsiang and Ho, 2008, Xiong et al., 2011). These studies prompted our investigations into the anti-CMV activities of emodin and related anthraquinone derivatives. The EC50 of emodin for inhibiting CMV in our studies (4.9 μM) were remarkably similar to that reported by Barnard et al. (4.1 μM) (Barnard et al., 1992) (Table 1). However, the high toxicities resulting in low SIs from both studies (Table 1) suggest that emodin’s CMV inhibition likely arises through non-specific disruption of cellular metabolic processes rather than inhibition of processes uniquely required for virus replication. In a subsequent study, Barnard et al. examined the anti-CMV activities of several anthraquinone derivatives (Barnard et al., 1995). For acid blue 40 and alizarin violet R they reported EC50 of 10 μM. We were unable to confirm these results. In our assays acid blue 40 and alizarin violet R had EC50 of 205 and 183 μM and SIs of 0.9 and 1.4 (Table 1), respectively, again suggesting a lack of specific antiviral activity. However, our EC50, TD50, and SI for atanyl blue PRL (6.3 μM, 216 μM, and 34 μM, respectively) were very similar to those reported by Barnard et al. (7 μM, 275 μM, and 40 μM) (Barnard et al., 1995) (Table 1).
In the present study we have extended these previous findings by demonstrating that atanyl blue PRL impairs CMV infectivity by severely reducing IE gene expression. Rapid and abundant accumulation of IE proteins through active transcription from the MIEP is crucial for driving CMV-infected cells forward in the lytic replication cycle. Conversely, repression of the MEIP leads to abortive infection and, in certain cell types, may promote establishment of latency. Repressive chromatinization of the MIEP is thought to occur by default upon delivery of the viral genome to the nucleus (Ioudinkova et al., 2006, Reeves et al., 2005a, Reeves et al., 2005b, Sinclair, 2010); in order to enter lytic replication, the viral tegument protein pp71 must traffic to the nucleus where it acts to de-repress IE transcription by inducing degradation of Daxx (Saffert and Kalejta, 2006). Atanyl blue PRL could interfere with IE protein expression at any point between attachment and IE transcription/translation. That exposure of the inoculum to an inhibitory dose of atanyl blue PRL prior to infection had no impact on viral yield suggests that atanyl blue PRL does not block viral entry by directly interacting with and irreversibly inactivating virions. Moreover, that atanyl blue PRL retains significant potency when added as late as 12 hpi further suggests that it does not interfere with viral attachment and entry, as these steps are largely completed within 1–3 hpi. Further studies will be needed to determine the exact stage at which atanyl blue PRL acts. It could interfere with capsid transport to nuclear pores, release of viral DNA, circularization of DNA, de-repression of the MIEP, or IE transcription/translation.
Two lines of evidence suggest that non-specific effects of atanyl blue PRL on viral DNA synthesis, RNA transcription, or protein synthesis can be ruled out. First, Barnard et al. measured the effects of atanyl blue PRL on cellular DNA, RNA, and protein synthesis and observed IC50 > 1000 μM (Barnard et al., 1995). Second, atanyl blue PRL added at 24 hpi did not reduce viral DNA levels or infectious virus yield at 5 dpi (Figs. 4 and 5B). At 24 hpi, viral DNA synthesis and transcription/translation of late proteins, which are necessary for assembly of viral progeny, are just beginning. Inhibition of either activity starting at 24 hpi should substantially reduce viral yield, as evidenced by the potent inhibitory effects of ganciclovir when added at 24, 48, and even 72 hpi (Fig. 4A).
Hsiang and Ho showed that emodin inhibits the nuclease activity of recombinant HSV-1 alkaline nuclease in vitro (Hsiang and Ho, 2008). While this infers that emodin may inhibit HSV-1 replication by targeting UL12’s nuclease activity, this hypothesis has not been confirmed by mapping emodin resistance to amino acid changes in UL12. Moreover, a recent report indicates that genetic inactivation of UL12’s nuclease activity has little effect on HSV-1 replication in some cells in culture (Fujii et al., 2013). As for HSV-1, the CMV alkaline nuclease UL98 is important for efficient replication in cell culture – a UL98 null mutant is severely replication deficient (Kuchta et al., 2012). However, the importance of UL98’s nuclease activity has not yet been evaluated by targeted mutation of the UL98 active site. Thus, it is possible that atanyl blue PRL inhibits CMV replication through inhibition of UL98.
Molecular modeling studies were conducted to gain insight into the structural features of the ligand and protein active-site that allow inhibition of UL98 activity by atanyl blue PRL but not acid blue 40. Both compounds were docked into the active site of a UL98 model that was built using KSHV-SOX (PDB 3fhd) as a template (Kuchta et al., 2012). The most favorable binding modes for each compound (Fig. 7 ) were in concordance with that of the 5′-end of DNA substrate, with both ligands showing significant interactions with the active site residues previously verified using mutagenesis (Kuchta et al., 2012). The sulfonate group at 2-position interacts with R164, T171, S252, Q464 (analogous to the interactions made by 5′ phosphate group). The oxygen at 9-position coordinates to the putative active site metal (occupying a position analogous to the P2 group). The 4-position mesitylene moiety of atanyl blue PRL forms favorable hydrophobic interactions with W160, H161 and M165. In contrast, the 4-position N-phenylacetamide moiety of acid blue 40 forms weaker hydrophobic interactions owing to the polar amide substitution on its phenyl ring. The lipophilic potential surface maps of the ligands within the protein active site clearly show the favorable hydrophobic–hydrophobic complementarity between atanyl blue PRL and UL98, compared to the unfavorable polar–hydrophobic complementarity between acid blue 40 and UL98. Another key non-covalent interaction observed is the π–π stacking interaction between the guanidinium ion of R164 and the 4-position aromatic substituent. For atanyl blue PRL the stacking interaction is further stabilized by the electron-donating substituents on its phenyl ring. These additional favorable interactions observed for atanyl blue PRL may explain its better inhibition profile compared to acid blue 40.
Fig. 7.
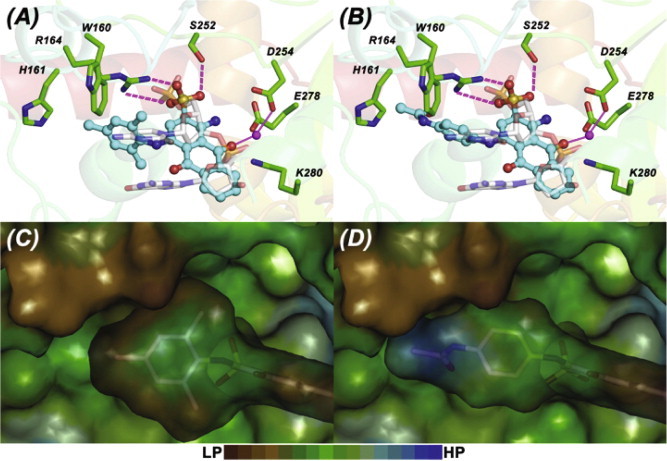
Docked poses of atanyl blue PRL and acid blue 40 in the active site of UL98. Modeled molecular interactions of atanyl blue PRL (A) and acid blue 40 (B) (ligand – ball and stick representation; carbon – cyan) with the active site residues of UL98 (stick representation; carbon – green). The double-stranded DNA substrate is shown as transparent sticks (carbon – white) and the active-site Mg2+ ion shown as magenta sphere. H-bond interactions of the sulfonate with R164 and S252 of UL98 are shown as magenta dashed-lines and coordination interactions of metal ion with carboxylates of D254 and E278 of UL98 and the 9-position oxo group of ligand are shown as solid magenta lines. Lipophilic surface potential maps of the 4-position substitutions of atanyl blue PRL (C) and acid blue 40 (D) within the active site of UL98. The scale at the bottom indicates the color gradient – brown (lipophilic LP/hydrophobic) to blue (hydrophilic HP/polar). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
As for HSV-1, a mechanism mediated by nuclease inhibition requires confirmation through mapping of atanyl blue PRL resistance to mutations in UL98. If atanyl blue PRL inhibits CMV replication through inhibition of UL98, then our results would indicate that UL98 is present and functionally important very early after infection. Consistent with this, low levels of UL98 have been observed in infected cells as early as 5 hpi (Adam et al., 1995), coinciding with the time window that atanyl blue PRL is active. However, a role for UL98 early but not late in CMV replication would contrast with proposed roles for the HSV-1 UL12 alkaline nuclease late in the HSV-1 life cycle in supporting recombination-dependent DNA replication (Reuven et al., 2003), DNA packaging, capsid formation, and nuclear egress (Martinez et al., 1996, Porter and Stow, 2004, Shao et al., 1993, Weller et al., 1990). While it is possible that UL98 and UL12 serve very different functions in replication of their respective viruses, it is also possible that atanyl blue PRL’s inhibition of UL98 nuclease activity is coincidental and that its true mechanism of CMV inhibition involves a different viral or cellular target that is important early in CMV replication.
5. Conclusions
The anthraquinone atanyl blue PRL inhibits CMV replication in cell culture. It acts very early in the CMV replication cycle to block IE protein synthesis, but time of addition studies suggest that it acts after viral entry. Consequently, atanyl blue PRL may be useful as a pharmacological probe to better understand the post-entry molecular events that culminate in robust IE transcription/translation. Atanyl blue PRL may also be useful as a lead structure for further antiviral drug discovery/development.
Atanyl blue PRL also inhibits the nuclease activity of CMV UL98 in vitro. It remains to be established whether its anti-CMV activity is mediated through inhibition of UL98. However, such a finding would implicate the nuclease as an important factor in very early events that determine the fate of infected cells. This would signal a profound revision of our understanding of how herpesvirus alkaline nucleases function during viral replication.
Acknowledgments
This work was supported by grants R01AI088750 and R21AI073615 (to M.A.M) from the National Institutes of Health. We thank Dai Wang and Thomas Shenk for BAC clone BADrUL131-Y4 and Jay Nelson for monoclonal antibody I2. We also thank Ruth Carvalho and the Virginia Commonwealth University Nucleic Acid Research Facilities for performing qPCR reactions.
References
- Adam B.L., Jervey T.Y., Kohler C.P., Wright G.L., Jr., Nelson J.A., Stenberg R.M. The human cytomegalovirus UL98 gene transcription unit overlaps with the pp28 true late gene (UL99) and encodes a 58-kilodalton early protein. J. Virol. 1995;69:5304–5310. doi: 10.1128/jvi.69.9.5304-5310.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves D.S., Perez-Fons L., Estepa A., Micol V. Membrane-related effects underlying the biological activity of the anthraquinones emodin and barbaloin. Biochem. Pharmacol. 2004;68:549–561. doi: 10.1016/j.bcp.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Andersen D.O., Weber N.D., Wood S.G., Hughes B.G., Murray B.K., North J.A. In vitro virucidal activity of selected anthraquinones and anthraquinone derivatives. Antiviral Res. 1991;16:185–196. doi: 10.1016/0166-3542(91)90024-l. [DOI] [PubMed] [Google Scholar]
- Barnard D.L., Fairbairn D.W., O’Neill K.L., Gage T.L., Sidwell R.W. Anti-human cytomegalovirus activity and toxicity of sulfonated anthraquinones and anthraquinone derivatives. Antiviral Res. 1995;28:317–329. doi: 10.1016/0166-3542(95)00057-7. [DOI] [PubMed] [Google Scholar]
- Barnard D.L., Huffman J.H., Morris J.L., Wood S.G., Hughes B.G., Sidwell R.W. Evaluation of the antiviral activity of anthraquinones, anthrones and anthraquinone derivatives against human cytomegalovirus. Antiviral Res. 1992;17:63–77. doi: 10.1016/0166-3542(92)90091-i. [DOI] [PubMed] [Google Scholar]
- Battistutta R., Sarno S., De Moliner E., Papinutto E., Zanotti G., Pinna L.A. The replacement of ATP by the competitive inhibitor emodin induces conformational modifications in the catalytic site of protein kinase CK2. J. Biol. Chem. 2000;275:29618–29622. doi: 10.1074/jbc.M004257200. [DOI] [PubMed] [Google Scholar]
- Bhave S., Elford H., McVoy M.A. Ribonucleotide reductase inhibitors hydroxyurea, didox, and trimidox inhibit human cytomegalovirus replication in vitro and synergize with ganciclovir. Antiviral Res. 2013;100:151–158. doi: 10.1016/j.antiviral.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron K.K. Antiviral drugs for cytomegalovirus diseases. Antiviral Res. 2006;71:154–163. doi: 10.1016/j.antiviral.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Boppana S.B., Ross S.A., Novak Z., Shimamura M., Tolan R.W., Jr., Palmer A.L., Ahmed A., Michaels M.G., Sanchez P.J., Bernstein D.I., Britt W.J., Fowler K.B., National Institute on Deafness, Other Communication Disorders, C.M.V., Hearing Multicenter Screening Study Dried blood spot real-time polymerase chain reaction assays to screen newborns for congenital cytomegalovirus infection. JAMA. 2010;303:1375–1382. doi: 10.1001/jama.2010.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerger I., Reefschlaeger J., Bender W., Eckenberg P., Popp A., Weber O., Graeper S., Klenk H.D., Ruebsamen-Waigmann H., Hallenberger S. A novel nonnucleoside inhibitor specifically targets cytomegalovirus DNA maturation via the UL89 and UL56 gene products. J. Virol. 2001;75:9077–9086. doi: 10.1128/JVI.75.19.9077-9086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P.A., Hudson J.B., Towers G.H. Antiviral activities of anthraquinones, bianthrones and hypericin derivatives from lichens. Experientia. 1996;52:180–183. doi: 10.1007/BF01923366. [DOI] [PubMed] [Google Scholar]
- Cui X., Adler S.P., Davison A.J., Smith L., Habib el S.E., McVoy M.A. Bacterial artificial chromosome clones of viruses comprising the Towne cytomegalovirus vaccine. J. Biomed. Biotechnol. 2012;2012:428498. doi: 10.1155/2012/428498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X., Meza B.P., Adler S.P., McVoy M.A. Cytomegalovirus vaccines fail to induce epithelial entry neutralizing antibodies comparable to natural infection. Vaccine. 2008;26:5760–5766. doi: 10.1016/j.vaccine.2008.07.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito F., Corona A., Zinzula L., Kharlamova T., Tramontano E. New anthraquinone derivatives as inhibitors of the HIV-1 reverse transcriptase-associated ribonuclease H function. Chemotherapy. 2012;58:299–307. doi: 10.1159/000343101. [DOI] [PubMed] [Google Scholar]
- Esposito F., Kharlamova T., Distinto S., Zinzula L., Cheng Y.C., Dutschman G., Floris G., Markt P., Corona A., Tramontano E. Alizarine derivatives as new dual inhibitors of the HIV-1 reverse transcriptase-associated DNA polymerase and RNase H activities effective also on the RNase H activity of non-nucleoside resistant reverse transcriptases. FEBS J. 2011;278:1444–1457. doi: 10.1111/j.1742-4658.2011.08057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H., Mugitani M., Koyanagi N., Liu Z., Tsuda S., Arii J., Kato A., Kawaguchi Y. Role of the nuclease activities encoded by herpes simplex virus 1 UL12 in viral replication and neurovirulence. J. Virol. 2013 doi: 10.1128/JVI.03621-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M., Robertson B.J., McCann P.J., O’Boyle D.R., Weller S.K., Newcomb W.W., Brown J.C., Weinheimer S.P. Functional conservations of the alkaline nuclease of herpes simplex type 1 and human cytomegalovirus. Virology. 1998;249:460–470. doi: 10.1006/viro.1998.9344. [DOI] [PubMed] [Google Scholar]
- Goldstein J.N., Weller S.K. The exonuclease activity of HSV-1 UL12 is required for in vivo function. Virology. 1998;244:442–457. doi: 10.1006/viro.1998.9129. [DOI] [PubMed] [Google Scholar]
- Henderson J.O., Ball-Goodrich L.J., Parris D.S. Structure-function analysis of the herpes simplex virus type 1 UL12 gene: correlation of deoxyribonuclease activity in vitro with replication function. Virology. 1998;243:247–259. doi: 10.1006/viro.1998.9054. [DOI] [PubMed] [Google Scholar]
- Higuchi H., Mori K., Kato A., Ohkuma T., Endo T., Kaji H., Kaji A. Antiretroviral activities of anthraquinones and their inhibitory effects on reverse transcriptase. Antiviral Res. 1991;15:205–216. doi: 10.1016/0166-3542(91)90067-2. [DOI] [PubMed] [Google Scholar]
- Ho T.Y., Wu S.L., Chen J.C., Li C.C., Hsiang C.Y. Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antiviral Res. 2007;74:92–101. doi: 10.1016/j.antiviral.2006.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann P.J., Cheng Y.C. The deoxyribonuclease induced after infection of KB cells by herpes simplex virus type 1 or type 2. I. Purification and characterization of the enzyme. J. Biol. Chem. 1978;253:3557–3562. [PubMed] [Google Scholar]
- Hsiang C.Y., Ho T.Y. Emodin is a novel alkaline nuclease inhibitor that suppresses herpes simplex virus type 1 yields in cell cultures. Br. J. Pharmacol. 2008;155:227–235. doi: 10.1038/bjp.2008.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioudinkova E., Arcangeletti M.C., Rynditch A., De Conto F., Motta F., Covan S., Pinardi F., Razin S.V., Chezzi C. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene. 2006;384:120–128. doi: 10.1016/j.gene.2006.07.021. [DOI] [PubMed] [Google Scholar]
- Kellogg G.E., Abraham D.J. Hydrophobicity: is LogP(o/w) more than the sum of its parts? Eur. J. Med. Chem. 2000;35:651–661. doi: 10.1016/s0223-5234(00)00167-7. [DOI] [PubMed] [Google Scholar]
- Kuchta A.L., Parikh H., Zhu Y., Kellogg G.E., Parris D.S., McVoy M.A. Structural modelling and mutagenesis of human cytomegalovirus alkaline nuclease UL98. J. Gen. Virol. 2012;93:130–138. doi: 10.1099/vir.0.034876-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Wei F., Chen L.J., Xiong H.R., Liu Y.Y., Luo F., Hou W., Xiao H., Yang Z.Q. In vitro and in vivo studies of the inhibitory effects of emodin isolated from Polygonum cuspidatum on Coxsackievirus B(4) Molecules. 2013;18:11842–11858. doi: 10.3390/molecules181011842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez R., Sarisky R.T., Weber P.C., Weller S.K. Herpes simplex virus type 1 alkaline nuclease is required for efficient processing of viral DNA replication intermediates. J. Virol. 1996;70:2075–2085. doi: 10.1128/jvi.70.4.2075-2085.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVoy M.A., Adler S.P. Human cytomegalovirus DNA replicates after early circularization by concatemer formation, and inversion occurs within the concatemer. J. Virol. 1994;68:1040–1051. doi: 10.1128/jvi.68.2.1040-1051.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVoy M.A., Mocarski E.S. Tetracycline-mediated regulation of gene expression within the human cytomegalovirus genome. Virology. 1999;258:295–303. doi: 10.1006/viro.1999.9724. [DOI] [PubMed] [Google Scholar]
- McVoy M.A., Nixon D.E., Adler S.P. Circularization and cleavage of guinea pig cytomegalovirus genomes. J. Virol. 1997;71:4209–4217. doi: 10.1128/jvi.71.6.4209-4217.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter I.M., Stow N.D. Replication, recombination and packaging of amplicon DNA in cells infected with the herpes simplex virus type 1 alkaline nuclease null mutant ambUL12. J. Gen. Virol. 2004;85:3501–3510. doi: 10.1099/vir.0.80403-0. [DOI] [PubMed] [Google Scholar]
- Reeves M.B., Lehner P.J., Sissons J.G., Sinclair J.H. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J. Gen. Virol. 2005;86:2949–2954. doi: 10.1099/vir.0.81161-0. [DOI] [PubMed] [Google Scholar]
- Reeves M.B., MacAry P.A., Lehner P.J., Sissons J.G., Sinclair J.H. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. U.S.A. 2005;102:4140–4145. doi: 10.1073/pnas.0408994102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuven N.B., Staire A.E., Myers R.S., Weller S.K. The herpes simplex virus type 1 alkaline nuclease and single-stranded DNA binding protein mediate strand exchange in vitro. J. Virol. 2003;77:7425–7433. doi: 10.1128/JVI.77.13.7425-7433.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice G.P., Schrier R.D., Oldstone M.B. Cytomegalovirus infects human lymphocytes and monocytes: virus expression is restricted to immediate-early gene products. Proc. Natl. Acad. Sci. U.S.A. 1984;81:6134–6138. doi: 10.1073/pnas.81.19.6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckman B.J., Rainish B.L., Chase M.C., Borton J.A., Nelson J.A., Jarvis M.A., Johnson D.C. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J. Virol. 2008;82:60–70. doi: 10.1128/JVI.01910-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccoccio F.M., Gallagher M.K., Adler S.P., McVoy M.A. Neutralizing activity of saliva against cytomegalovirus. Clin. Vaccine Immunol.: CVI. 2011;18:1536–1542. doi: 10.1128/CVI.05128-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccoccio F.M., Sauer A.L., Cui X., Armstrong A.E., Habib E.S., Johnson D.C., Ryckman B.J., Klingelhutz A.J., Adler S.P., McVoy M.A. Peptides from cytomegalovirus UL130 and UL131 proteins induce high titer antibodies that block viral entry into mucosal epithelial cells. Vaccine. 2011 doi: 10.1016/j.vaccine.2011.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffert R.T., Kalejta R.F. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 2006;80:3863–3871. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinazi R.F., Chu C.K., Babu J.R., Oswald B.J., Saalmann V., Cannon D.L., Eriksson B.F., Nasr M. Anthraquinones as a new class of antiviral agents against human immunodeficiency virus. Antiviral Res. 1990;13:265–272. doi: 10.1016/0166-3542(90)90071-e. [DOI] [PubMed] [Google Scholar]
- Schwarz S., Wang K., Yu W., Sun B., Schwarz W. Emodin inhibits current through SARS-associated coronavirus 3a protein. Antiviral Res. 2011;90:64–69. doi: 10.1016/j.antiviral.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple S.J., Pyke S.M., Reynolds G.D., Flower R.L. In vitro antiviral activity of the anthraquinone chrysophanic acid against poliovirus. Antiviral Res. 2001;49:169–178. doi: 10.1016/s0166-3542(01)00125-5. [DOI] [PubMed] [Google Scholar]
- Shao L., Rapp L.M., Weller S.K. Herpes simplex virus 1 alkaline nuclease is required for efficient egress of capsids from the nucleus. Virology. 1993;196:146–162. doi: 10.1006/viro.1993.1463. [DOI] [PubMed] [Google Scholar]
- Shuangsuo D., Zhengguo Z., Yunru C., Xin Z., Baofeng W., Lichao Y., Yan’an C. Inhibition of the replication of hepatitis B virus in vitro by emodin. Med. Sci. Monit.: Int. Med. J. Exp. Clin. Res. 2006;12 [PubMed] [Google Scholar]
- Sinclair J. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim. Biophys. Acta. 2010;1799:286–295. doi: 10.1016/j.bbagrm.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Sydiskis R.J., Owen D.G., Lohr J.L., Rosler K.H., Blomster R.N. Inactivation of enveloped viruses by anthraquinones extracted from plants. Antimicrob. Agents Chemother. 1991;35:2463–2466. doi: 10.1128/aac.35.12.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Shenk T. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J. Virol. 2005;79:10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller S.K., Seghatoleslami M.R., Shao L., Rowse D., Carmichael E.P. The herpes simplex virus type 1 alkaline nuclease is not essential for viral DNA synthesis: isolation and characterization of a lacZ insertion mutant. J. Gen. Virol. 1990;71(Pt 12):2941–2952. doi: 10.1099/0022-1317-71-12-2941. [DOI] [PubMed] [Google Scholar]
- Xiong H.R., Luo J., Hou W., Xiao H., Yang Z.Q. The effect of emodin, an anthraquinone derivative extracted from the roots of Rheum tanguticum, against herpes simplex virus in vitro and in vivo. J. Ethnopharmacol. 2011;133:718–723. doi: 10.1016/j.jep.2010.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim H., Lee Y.H., Lee C.H., Lee S.K. Emodin, an anthraquinone derivative isolated from the rhizomes of Rheum palmatum, selectively inhibits the activity of casein kinase II as a competitive inhibitor. Planta Med. 1999;65:9–13. doi: 10.1055/s-1999-13953. [DOI] [PubMed] [Google Scholar]