

Abstract
The stress-related catecholamine hormones and the α- and β-adrenergic receptors (α- and β-AR) may affect carcinogenesis. The β-AR GRK/β-arrestin biased agonist carvedilol can induce β-AR-mediated transactivation of the epidermal growth factor receptor (EGFR). The initial purpose of this study was to determine whether carvedilol, through activation of EGFR, can promote cancer. Carvedilol failed to promote anchorage-independent growth of JB6 P+ cells, a skin cell model used to study tumor promotion. However, at non-toxic concentrations carvedilol dose-dependently inhibited EGF-induced malignant transformation of JB6 P+ cells suggesting that carvedilol has chemopreventive activity against skin cancer. Such effect was not observed for the β-AR agonist isoproterenol and the β-AR antagonist atenolol. Gene expression, receptor binding, and functional studies indicate that JB6 P+ cells only express β2-ARs. Carvedilol, but not atenolol, inhibited EGF-mediated activator protein-1 (AP-1) activation. A topical 7,12-dimethylbenz[α]anthracene (DMBA)-induced skin hyperplasia model in SENCAR mice was utilized to determine the in vivo cancer preventative activity of carvedilol. Both topical and oral carvedilol treatment inhibited DMBA-induced epidermal hyperplasia (P < 0.05) and reduced H-ras mutations; topical treatment being the most potent. However, in models of established cancer, carvedilol had modest to no inhibitory effect on tumor growth of human lung cancer A549 cells in vitro and in vivo. In conclusion, these results suggest that the cardiovascular drug carvedilol may be repurposed for skin cancer chemoprevention, but may not be an effective treatment of established tumors. More broadly, this study suggests that β-ARs may serve as a novel target for cancer prevention.
Keywords: Skin cancer, chemoprevention, carvedilol, β-blocker, DMBA
Introduction
Skin cancer accounts for nearly 40% of all diagnosed cancers in the U.S and is the most common cancer worldwide (1). Each year more than a million cases of skin cancer are diagnosed in the US, and over 10,000 deaths annually (1). The primary causative agent for skin cancer is the ultraviolet (UV) radiation from sunlight (1). UV radiation causes DNA damage and reactive oxygen species (ROS) production that contribute to the development of all three major types of skin cancer: basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and melanoma (2). Everyone is recommended to limit sun exposure and use sunscreens for primary and secondary prevention of skin cancer. However, despite these efforts, the incidence of skin cancer continues to increase.
Chemoprevention, defined as using natural or synthetic substances to decrease the risk of developing cancer, has become an important approach toward decreasing cancer morbidity and mortality (3). Although many agents have been, and are continually, examined for chemoprevention, none has been approved by the FDA as a preventive strategy for skin cancer due to limited efficacy and intolerable adverse effects (4). Thus, there is a need to identify novel molecular targets and chemopreventive agents which are efficacious and have no, or very low, toxicity to normal cells.
Studies demonstrate an association between psychosocial factors such as chronic stress or depression with cancer onset and progression (5). Such effects are partly mediated through activation of the sympathetic nervous system which results in the release of the catecholamine hormones norepinephrine (NE) and epinephrine (Epi). The effects of NE and Epi are mediated through α- and β-adrenergic receptors (AR). Both Epi and NE impact several key pathways for cancer progression and metastasis (6). In particular, β-AR signaling is implicated in multiple cellular processes in cancer development (7), leading a number of researchers to suggest that the commonly prescribed β-AR antagonist drugs (β-blockers) may inhibit cancer progression (8). Indeed, several epidemiological and clinical studies have examined the relationship between β-blocker usage and cancer progression. Results from these studies, although not always consistent, suggest that β-blockers may have a protective role in reducing the incidence of all cancer types including skin cancer (8). However, questions regarding the chemoprevention efficacy and mechanisms of β-blockers remain unanswered.
An additional issue in these epidemiological studies is that not all β-blockers are merely blockers; some are biased agonists that can induce β-AR-mediated transactivation of the epidermal growth factor receptor (EGFR) and downstream ERK activation (9, 10). Because EGFR signaling is well known to promote proliferation, migration, and invasion of various types of cancer (11), our initial hypothesis was that the biased agonist carvedilol may promote malignant cell transformation. Therefore, we first determined whether carvedilol would induce the transformation of the mouse epidermal JB6 clone Cl 41-5a cells (JB6 P+) cells. The JB6 P+ cells are non-cancerous cells that are known to be transformable and form colonies in soft agar after exposure to several tumor promoters, such as EGF (12). However, our initial data proved the hypothesis incorrect and suggested that carvedilol prevented EGF-mediated transformation of JB6 P+ cells.
Therefore, we further investigated the cancer preventative attributes of carvedilol. To our surprise the data presented in this study strongly suggests that carvedilol prevents malignant transformation in vitro and in vivo models of skin carcinogenesis. The results led us to conclude that carvedilol may be a novel chemopreventive agent that is not only safe, but also represents a novel chemopreventive approach. Although this study focuses on skin cancer, these data may form the basis of clinical trials of these agents on prevention of other types of cancer.
Materials and methods
Compounds
Carvedilol was purchased from TCI America (Portland, OR). 4-(3-t-butylamino-2-hydroxypropoxy)-[5,7-3H]benzimidazole-2-one (3H-CGP) with a specific activity of 41.7 Ci/mmol was purchased from Perkin Elmer (Waltham, MA). Isoproterenol, 3-isobutyl-1-methylxanthine (IBMX), and forskolin were purchased from Sigma (St. Louis, MO). Atenolol, nebivolol, ICI-118,551, L-748,337, xamoterol humifumerate, formoterol humifumerate and L-755,507 were obtained from Tocris (Bristol, United Kindom). EGF was purchased from Peprotech (Rocky Hill, NJ) and dissolved in sterile deionized water as 100 ug/mL stock and stored in −20°C freezer.
Cell culture
JB6 CI 41-5a sensitive to promotion of transformation (JB6 P+) mouse epidermal cells, were purchased from American Type Culture Collection (ATCC, Manassas, VA). No authentication was done by the authors. JB6 P+ were maintained in Eagle’s minimum essential medium (EMEM) containing 5% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin. A549 and HEK-293 cells were obtained from ATCC, cultured in RPMI 1640 and DMEM, respectively, supplemented with 10% FBS and 1% penicillin-streptomycin. All cells from cell culture experiments were incubated at 37°C in 5% CO2/95% air.
Anchorage-independent growth assay in soft agar
In a 96-well tissue culture plate, 2,000 JB6 P+ cells or 200 A549 cells per well were mixed with 0.33% agar suspended on top of a layer of 0.5% agar. 4% Nobel agar (Sigma-Aldrich) was prepared in PBS, autoclaved and stored at 4°C. 0.5% and 0.33% agar were diluted from 4% stock using EMEM supplemented with 10% FBS and 1% penicillin/streptomycin. EGF (10 ng/mL) was used to promote the anchorage-independent growth of JB6 cells, but not added for A549 cells. Various concentrations of β-AR agonist or β-blockers were added together with EGF into the top and bottom layers of the agar. Plates are incubated at 37 °C, 5% CO2 for 7–10 days. Colonies with greater than ten cells were counted manually under a microscope. Similar assay was conducted in a 12-well plate.
RT-PCR
JB6 P+ (6 × 105 cells per well) were seeded in 6-well plates and once confluent RNA was extracted using RNAeasy Plus Kit (Qiagen). cDNA was synthesized using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). PCR was performed using mouse Adrb1, Adrb2 and Adrb3 primers. The primer sequences are available upon request. Jumpstart RedTaq Ready Mix (Sigma) was used for polymerase chain reaction (PCR). PCR programming was as follows: 94°C for 5 minutes for denaturation; 35 cycles of amplification of 94°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds; then 70°C for 7 minutes to finalize elongation. Product size was validated through gel electrophoresis and visualized using a Gel Logic 1500 Imaging System (Kodak).
Radiolabeled binding assays
As the assay is nearly identical to previously published studies (10); herein, only deviations will be detailed. JB6 P+ cells were seeded in 12-well plates pretreated with poly-D-lysine (Sigma) and allowed to grow to confluency. For saturation binding, increasing concentrations of 3H-CGP in EMEM was added to the wells. For the competition assays, a 5 nM 3H-CGP stock solution in EMEM was created and subdivided into separate tubes where the inhibitors were added, and in identical experiments 1 μM isoproterenol was added to each sample in order to determine non-specific binding. The assay was conducted on ice within a 4°C refrigerator. Nonspecific binding was subtracted from each point, and data were expressed as a percent of total surface receptors determined in the control.
Cyclic AMP (cAMP) assay
cAMP was measured as previously described (10, 13) with the following changes. JB6 P+ cells were grown in poly-D-Lysine coated 12-well plates. All cells were treated with 500 μM IBMX for 5 minutes, then 10 nM of the β1-AR-specific agonist xamoterol, 10 nM of the β2-AR-specific agonist formoterol, 5 nM of the β3-AR-specific agonist L-755,507, or 10 μM forskolin was added to the cells at 37°C for 15 minutes (n = 4 for each treatment). Additionally, a complete second set of samples were treated with 100 nM carvedilol.
Cell proliferation assay
96-well plates were seeded with 3,000 to 4,000 JB6 P+ cells per well and allowed to attach overnight. Cells were treated with test compounds for 72 hours and incubated at 37°C in 5% CO2/95% air. Cell viability was determined using Sulforhodamine B (SRB) assay of Sigma according to manufacturer’s protocol.
Luciferase reporter gene assay
HEK293 cells were transfected with pGL4.22-AP1 (gift, Dr. D. Sanchez) and pRL-TK-luc (Promega) at a 40:1 ratio using FuGENE HD Transfection Reagent (Roche Applied Science, Indianapolis, IN) according to the manufacturer’s instructions. Twenty-four hours after transfection, the cells were exposed to test agents for another 24 hours. Cell lysates were used for determining luciferase activities of both firefly and renilla by the dual luciferase reporter gene assay (Promega). Firefly luciferase activity was normalized to renilla luciferase activity.
Cell scratch-wound assay
A549 cells were seeded in 35mm dishes to create a confluent monolayer. The dishes were allowed to incubate overnight. On the following day, wounds were created by a straight scratch from a pipette tip in the center of the culture. The cells were then treated with DMSO or carvedilol at 1μM and 10μM concentrations. Gap distances were quantitatively evaluated using EVOS microscope (Life Technologies, Foster City, CA) immediately after the scratch and after 24-hour incubation.
Model of chemically induced murine skin tumorigenesis
All animal studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, and approved by the Western University of Health Sciences Institutional Animal Care and Use Committees. Five-week-old female SENCAR mice (National Cancer Insitute, Frederick, MD) (n=36) were divided into six groups and the backs of mice shaved. At 7-week of age, 100 nmol DMBA dissolved in 200 μl acetone was applied topically twice weekly for four weeks. Carvedilol was given at 5 weeks of age twice weekly topically in two doses (5 and 10 μM in 200 μL acetone) 30 min before DMBA exposure when co-administering, and orally by gavage in two doses (5 and 20 mg/kg in 1% methyl cellulose in PBS) 2 hours before DMBA exposure when co-administering. Two days after the last treatment mice were sacrificed, and samples of skin were excised and fixed immediately in formalin and embedded in paraffin blocks. The embedded tissues were cut into 3-micron thick sections and stained with H&E to determine the morphology.
Immunohistochemistry (IHC) analysis
Paraffin-embedded sections were baked at 60°C for 1 hour and deparaffinized in xylene and rehydrated through a graded alcohol series. The antigen was retrieved using citrate buffer (pH 6.0) for 20 min at 95°C. Briefly, sections were blocked by 10% normal goat serum for 2 hours followed by overnight incubation at 4°C with 1:1000 dilution of proliferation cell nuclear antigen (PCNA; Cell signaling technologies) primary antibody. Sections were then incubated for 2 hours with 1:5000 dilution of HRP secondary antibody followed by 5-minute incubation with DAB substrate (Vector labs; Burlingame, USA) counterstained with Mayer’s hematoxylin.
Competitive Allele-Specific TaqMan PCR (castPCR)
Genomic DNA was isolated from frozen skin tissues by DNAzol (MRC, Inc) according to the protocol provided by the manufacturer. CastPCR was performed with TaqMan Mutation Detection Assay designed to detect CAA → CTA transversion in codon 61 of the mouse H-ras gene (Applied Biosystems by Life Technologies) following manufacturer’s instruction. The castPCR was run on a GeneAmp 7300 Sequence Detection system (Applied Biosystems) using a universal mutation detection thermal-cycling protocol. The mutational status of a sample was determined by calculating the ΔCt value between the mutant allele assay and wild-type allele assay to obtain the percent mutation according to manufacturer’s instruction.
Xenograft lung tumor growth
Sub-confluent A549 cells were trypsinized and then suspended in RPMI 1640. 1 × 106 cells in 100 μl RPMI 1640 was injected subcutaneously into the right and left flanks of eight-week-old female NOD SCID mice (Taconic, Hudson, NY) (n = 5 for each group). Mice were given vehicle (1% methylcellulose in PBS) or 600 μg carvedilol (estimated as 26 mg/kg) orally every day for seven weeks. Tumor volumes were regularly measured starting 19 days after implantation. The tumor volume was calculated according to the formulation: .
Statistical Analysis
Data are expressed as mean ± standard error, unless stated otherwise, and was analyzed using NCSS 2007 (Kaysville, UT) or Graph GraphPad Prism version 6.0 (La Jolla, CA); additionally Grubs outlier test was run to exclude single samples from a group that were significant outliers. The specific tests are detailed in the text and figure legends. For all statistical analysis, means were indicated to be statistically different when p < 0.05.
Results
Carvedilol inhibited EGF-induced epidermal cell transformation in JB6 P+ cells
This project was initiated to test the hypothesis that the GRK/β-arrestin biased agonist β-blockers, which are known to transactivate EGFR (9), would promote the transformation of JB6 P+ cells. Surprisingly, carvedilol alone had no effect on JB6 P+ colony formation (Figure 1A). A serendipitous mistake allowed monitoring of the effect of carvedilol on EGF-mediated transformation of JB6 P+ cells. Co-administration of EGF and carvedilol to JB6 P+ cells significantly reduced the numbers of colony growing in soft agar. These series of data invalidated our initial hypothesis and suggested that carvedilol is a chemopreventive agent; therefore, we designed experiments to further examine this effect and test the new hypothesis that carvedilol is a chemopreventive agent.
Figure 1. Effects of β-AR ligands on EGF-mediated neoplastic transformation of JB6 P+ cells.

Effects of EGF or carvedilol (Car) on JB6 P+ cell transformation (A). The β-AR agonist isoproterenol (Iso) (B), the β-blockers carvedilol (C) and atenolol (Aten) (D) showed different degree of inhibition of EGF-induced cell transformation. Groups with different Greek letters are statistically different (p < 0.05) as determined by ANOVA with Tukey-Kramer post hoc test.
To determine the effects of agonizing or antagonizing the β-ARs in skin cell transformation, we tested the β-AR agonist isoproterenol as well as the β-blockers carvedilol and atenolol on 10 ng/mL EGF-mediated neoplastic transformation of JB6 P+ cells (Figure 1B–D). Atenolol was chosen because it is the most commonly prescribed β-blocker and the most prevalent β-blocker in the aforementioned clinical studies. 0.1 μM and 1.0 μM isoproterenol, doses that are higher than those needed to activate β-ARs, did not alter EGF-mediated neoplastic transformation (Figure 1B). Treatment with carvedilol resulted in drastic inhibition of colony number (Figure 1C). Although atenolol had a statistically significant effect, the inhibition by atenolol was modest (Figure 1D). As this assay is dependent on cell growth, we conducted sulforhodamine B (SRB) colorimetric assays for cytotoxicity, and parallel colony formation assays (Figure 2). Treatment of JB6 P+ cells with carvedilol demonstrated a dose-dependent inhibition of EGF-mediated transformation (log IC50 = −6.365 ± 0.158 M) that was fully efficacious, while only toxic at 100 μM with no significant toxicity seen at any other concentrations (Figure 2A). These data indicate that the lack of EGF-mediated colony formation is not due to a general cytotoxic effect. On the other hand, atenolol demonstrated a dose-dependent inhibition of EGF-mediated transformation (log IC50 = −5.037 ± 0.266 M) that only reached 48% inhibition of colony formation, while displaying no toxicity (Figure 2B). Representative images of colonies in soft agar are shown in Figure 2C. Since the JB6 P+ transformation assay has a positive predictive value for in vivo efficacy of chemopreventive agents (14), these data suggest that carvedilol may have chemopreventive activity at non-toxic concentrations.
Figure 2. Comparison of the effects of carvedilol and atenolol on transformation and cytotoxicity of JB6 P+ cells.

The β-blockers carvedilol (A) or atenolol (B) were examined for their effects on the cell transformation (by soft agar assay, in black) and cytotoxicity (by SRB assay, in gray) and data were normalized to their respective controls and plotted together. (C) Representative images of colonies after 10 days in soft agar within 12-well plates at 4x magnification; scale bar equals 1 mm.
Only β2-adrenergic receptors are expressed in JB6 P+ cells
To determine whether the JB6 P+ cells express any of the β-AR receptors, the expression levels of β1-, β2- and β3-ARs were evaluated using RT-PCR analysis and ligand binding. RT-PCR indicates expression of only β2-ARs, while the positive control displayed the expected band for each receptor (Figure 3A). Radiolabeled receptor binding assays were performed using the cell impermeant β-AR non-specific ligand 3H-CGP that binds with the following affinities: 0.3, 0.79, and 152 nM for the β1-, β2-, and β3-ARs, respectively (15–17). First a saturation binding experiment was conducted; 3H-CGP Bmax was found to be 0.2024 ± 0.0437 DPM/Hoechst reached at 30 to 100 nM 3H-CGP (data not shown), which suggests JB6 P+ do not express β3-ARs due to the Bmax concentration being well below the reported affinity for the β3-AR. Competition assays conducted with 5 nM 3H-CGP, which will occupy approximately 25% of the specific binding sites, was examined by competition with highly specific inhibitors: nebivolol (β1) Ki = 0.9 nM (18), ICI-118,551 (β2) Kd = 0.55 nM (19), and L-748,337 (β3) Ki = 4.0 nM (20). Each antagonist was used up to 100-times the reported affinity to ensure competition (Figure 3B). Only ICI-118,551 significantly reduced CGP binding in a dose-dependent manner. The calculated Kd for ICI-118,551 is 7.537 nM. Although the calculated Kd appears high, accounting for only a quarter of the receptors occupied by CGP results in the 95% confidence interval to include 0.55 nM (CI: 0.18 to 19.83 nM).
Figure 3. β-adrenoceptor expression in JB6 P+ cells.

(A) RT-PCR analysis revealed that JB6 P+ cells only express β2-ARs. The positive control (+Ctr) is Mouse Universal Reference cDNA. (B) Binding assays utilizing 3H-CGP and specific β-blockers: nebivolol (β1-AR), ICI-118,551 (β2-AR), and L-748,337 (β3-AR) indicate that only inhibition of the β2-AR showed significant (p < 0.05) displacement of CGP in a dose-dependent manner via an ANOVA with Tukey-Kramer post hoc test, indicated by an asterisk (*); n ≥ 6. (C) Functional assays examining cAMP accumulation utilizing specific agonists: xamoterol (β1-AR), formoterol (β2-AR), or L-755,507 (β3-AR) and carvedilol demonstrate that only stimulation of the β2-AR resulted in a statistically significant increase in cAMP compared to control (p < 0.05) via Kruskal-Wallis multiple-comparison test, indicated by an asterisk (*), which was reversed by carvedilol treatment.
To further confirm the RT-PCR and binding data, and demonstrate functionality of the β-ARs, the ability of specific β-AR agonists to increase cAMP levels was examined (Figure 3C). As a positive control, forskolin was utilized to verify the assay conditions; it stimulated cAMP production five-fold over control (Data not shown). The concentrations of each β-AR agonist were chosen based on their EC50 values for their specific receptor and to ensure there is minimal activation of the other two β-ARs. Only the β2-AR agonist formoterol statistically increased cAMP levels beyond the control values. Additionally, carvedilol prevented formoterol-mediated cAMP accumulation. These results support the previous conclusions that the β2-ARs are on the surface of JB6 cells and functional. Additionally, this data demonstrates that carvedilol functionally blocks β2-ARs.
Carvedilol inhibited EGF-induced AP-1 activity
To investigate carvedilol’s possible mechanisms of action its effect on EGF-mediated activation of AP-1 was evaluated; AP-1 is a major transcription factor involved in EGF-induced transformation of JB6 P+ cells (12). As shown in Figure 4A, 1 μM and greater doses of carvedilol significantly inhibited EGF-mediated AP-1 activity (P < 0.05). In contrast, atenolol, which showed slight anti-transformation activity (Figure 1D & 2B), showed no significant inhibition of AP-1 activity (P > 0.05) (Figure 4B). These results indicated that inhibition of AP-1 activity by carvedilol may explain part of the mechanism underlying the inhibitory activity against EGF-induced cell transformation.
Figure 4. Effects of carvedilol and atenolol on EGF-induced AP-1 transactivation.
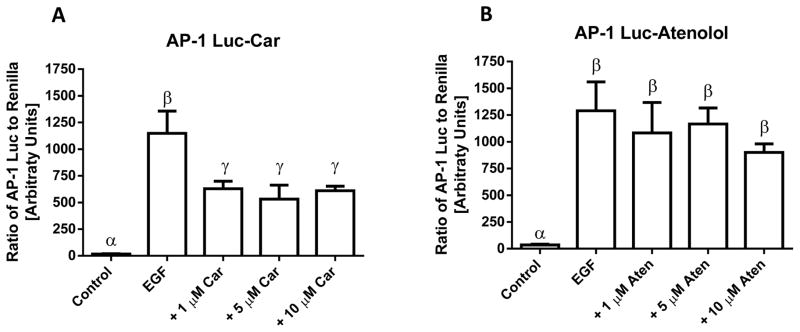
HEK-293 expressing an AP-1 luciferase reporter and renilla control were treated with vehicle control, EGF (10 ng/mL), EGF plus β-blockers carvedilol (A) or atenolol (B) for 24 h; data shown as means ± SE. Groups with different Greek letters are statistically different (p < 0.05) as determined by ANOVA with Tukey-Kramer post hoc test; n = 5.
Carvedilol inhibited DMBA-induced skin carcinogenesis in mice
To determine the in vivo chemopreventive activity of carvedilol a 4-week DMBA-induced skin hyperplasia assay in SENCAR mice was utilized. Carvedilol treatments included topically in two doses (5 and 10 μM), and orally in two doses (5 and 20 mg/kg), beginning two weeks before the first dose of DMBA. Representative samples of the epidermis (H&E staining) where DMBA was applied with and without carvedilol treatment shown in Figure 5A. Skin thickness was measured 20 times at various locations along the epidermis and averaged to obtain a single sample’s epidermal thickness (Figure 5B) DMBA treatment increased epidermal thickness 3-fold from 10.9 ± 1.5 μm in control to 36.5 ± 6.0 μm. There was no significant difference between the two topical or the two oral doses of carvedilol (data not shown). However, topical treatment proved to be slightly more efficacious as the epidermal thickness was only 19.9 ± 3.1 in the 5 μM topical group. Analogous to the H&E data, immunohistochemical analysis indicates an increased expression of PCNA in the basal layer of the epidermis of mouse skin after DMBA treatment (Figure 5A). Carvedilol treatment reduced the number of PCNA positive cells within the epidermis.
Figure 5. Effects of carvedilol on DMBA-induced epidermal hyperplasia and the mutation of H-ras in SENCAR mice.

(A) Representative microphotographs of the control and treated murine skin (H&E staining and PCNA expression). (B) Measurement of epidermal thickness; Groups with different Greek letters are statistically different (p < 0.05) as determined by ANOVA with Tukey-Kramer post hoc test. (C) H-ras CAA → CTA mutations from the epidermal samples shown in A and B. Groups with different Greek letters are statistically different (p < 0.05) as determined by ANOVA with Kruskal-Wallis Multiple-Comparison Z-Value with the Bonferroni Test; n ≥ 5.
One of the key events in tumor initiation in mouse skin is the mutation of H-ras at codon 61 (CAA → CTA) (21). Utilizing TaqMan Mutation Detection Assays based on castPCR to assess H-ras mutations, it was found that the control mice had no mutations while all the treatment groups harbored a small percentage of CAA → CTA mutation (Figure 5C). As in the previous figure (Figure 5B), 5 μM topical carvedilol showed the greatest reduction of H-ras mutation, yet there are no statistical differences between the treatment groups. Together, these experiments further support the notion that carvedilol is a chemopreventive agent.
Carvedilol has a modest effect on tumor growth of human lung cancer A549 cells in vitro and in vivo
As most preclinical and clinical studies examined the effect of β-blockers on developed cancer, the anticancer effects of carvedilol on A549 cells, an established human lung cancer cell line, was examined. Soft agar colony formation assay of A549 cells allowed for testing the effect of carvedilol on anchorage-independent growth. Carvedilol showed an inhibitory effect at a high concentration (30 μM). The SRB assay was also performed in order to evaluate the cytotoxicity of carvedilol on A549 cells (anchorage-dependent) and plotted together with the soft agar assay data (Figure 6A). Carvedilol was only toxic at 100 μM. Such results are very different from the non-cancerous JB6 P+ cells (Figure 2A). Additionally, cell mobility assays were conducted on A549 cells. Cells treated with vehicle or 1 μM carvedilol significantly shifted to close the gap; whereas, cells treated with 10 μM carvedilol failed to close the gap (Figure 6B), indicating an inhibitory effect of carvedilol on cell mobility only at high concentrations. Next, the effect of oral administration of carvedilol on xenograft tumor growth model utilizing A549 cells was evaluated (Figure 6C). Repeated measures ANOVA showed a minor difference between the two treatment groups (P = 0.074255) and significant tumor growth over time (P < 0.000001). The data was significantly different on only two days: days 42 and 49. Throughout the experiment, the body weights of the two groups were similar (data not shown). This in vivo study suggests that carvedilol treatment has little effect on established tumors.
Figure 6. Effects of carvedilol on human lung tumor A549 growth and migration in vitro and in vivo.

(A) The β-blocker carvedilol was examined for effects on soft agar assay (in black) and SRB assay (in gray) and data were normalized to their respective controls and plotted together; asterisk (*) and cross (†) indicates the soft agar and SRB assay is statistically different than control, respectively, (p < 0.05) as determined by ANOVA with Tukey-Kramer post hoc test. (B) Cell migration of A549 cells was quantified by the gap distances before and after 24 hour treatment with carvedilol; asterisk (*) indicates that the 24 hour period was statistically different than the initial (p < 0.05) as determined by paired t-test, n = 3. (C) Effect of carvedilol on xenograft lung tumor growth in mice. Asterisk (*) indicates p-value is < 0.05 between the two points as determined via a RM-ANOVA with a Newman-Keuls multiple-comparison post hoc test; n ≥ 4.
Discussion
β-blockers are often prescribed for cardiovascular diseases (22). While activation of β-AR signaling has been linked to several types of cancer (7), the effects of β-blockers on cancer initiation and promotion are unclear. Although the most commonly used drugs in published studies focusing on the anticancer effects of β-blockers are atenolol and propranolol, in this study carvedilol, a third-generation β-blocker that is actually a GRK/β-arrestin biased agonist (9, 10, 22), was utilized. The cancer-related effects of carvedilol, one of the most tolerable β-blockers, has not been extensively studied. We were interested in carvedilol because it transactivates the EGFR (9), which is a molecular target for multiple types of solid tumors including skin cancer (11). Therefore, one important question was whether carvedilol can promote cancer through activation of the EGFR. The mouse JB6 P+ cell line is a characterized model for neoplastic transformation in response to EGF; EGF induces these cells to acquire anchorage-independent growth in soft agar culture (12). Therefore, JB6 P+ cells were treated with carvedilol to determine if carvedilol could induce transformation similar to EGF. To our surprise carvedilol treatment was indistinguishable from the vehicle control group indicating that carvedilol does not induce transformation of JB6 P+ cells, and thus is not a tumor promoter, dispelling our hypothesis and concerns (23). This conclusion is supported not only by the data presented in this study, but numerous clinical studies of this approved cardiovascular drug that has been on the market for more than 15 years without evidence of increasing cancer risk.
The JB6 P+ cell line is also a good system to evaluate the potency and mechanisms of chemopreventive agents. In addition, many advances in cancer biology have been extended from the JB6 model in culture to several in vivo models of human cancers of epithelial origin. A serendipitous error occurred where carvedilol was mixed with EGF in one experiment; surprisingly carvedilol proved to be an efficacious inhibitor of EGF-induced colony formation in JB6 P+ cells. This finding allowed for an interesting switch in the hypothesis of this study – carvedilol may be a chemopreventive agent.
To test this new hypothesis, a β-AR agonist (isoproterenol) and the most commonly prescribed β-blocker (atenolol) were also used to determine if the effects are somewhat unique to carvedilol (Figure 1). Since carvedilol is a biased agonist, inhibits alpha-adrenergic receptors, and generates nitric oxide (24); it is not surprising that neither isoproterenol nor atenolol replicates the results of carvedilol. However, atenolol did have partial activity at nearly 10-fold greater concentrations. The lesser potency can be explained by affinities for the receptor (see below); however, the decreased efficacy indicates that the effect goes beyond mere affinities and has a biological component. The nature of this component and whether it can be successfully targeted to prevent cancer is an ongoing investigation.
The presence of β-ARs on JB6 P+ cells prior to this study was unknown; thus, to determine if these effects could even begin to be ascribed to a β-AR, the receptors must be identified. As determined by RT-PCR, receptor binding, and functional studies with highly specific agonists, only the β2-AR is present on JB6 P+ cells (Figures 3).
Carvedilol signals through the β2-AR utilizing the GRK/β-arrestin pathway resulting in the transactivation of the EGFR and activation of ERK1/2 (22). Although it remains to be determined whether carvedilol is able to stimulate EGFR transactivation in the JB6 P+ cell line, there is little reason to believe it does not activate the EGFR as it has been shown to signal through the β2-AR in mouse cells (10). If indeed carvedilol transactivates the EGFR in these cells, then these data suggest, in a bioassay, that transactivation of EGFR via the β2-AR/β-arrestin pathway is not identical to direct activation of EGFR with its natural ligand EGF, as previously described in intestinal epithelial cells (25).
As stated, the lack of β1-ARs explains the reduced potency of atenolol (Figures 1 & 2) as it has approximately 6-fold greater selectivity for the β1-AR (26); however, at 100 μM atenolol is expected to saturate β2-ARs. Thus, the effects cannot be ascribed solely to a change in potency. Supporting this conclusion, the effects of micromolar concentrations of atenolol had no effect on EGF-mediated activation of AP-1 (Figure 4). At these concentrations, atenolol does have a statistically differentiable effect on colony formation with no toxicity. However, carvedilol significantly attenuated EGF-mediated activation of AP-1 (Figure 4). These data indicate that the cellular effects of carvedilol differ from atenolol. This may be due to GRK/β-arrestin biased agonism or other pleiotropic effects ascribed to carvedilol (27, 28). Since AP-1 is a mediator of transformation (12), one potential mechanism of carvedilol-mediated inhibition of EGF-induced colony formation is through blocking the activity of AP-1.
We furthered the experiments with two different in vivo models. First we examined a cancer prevention model utilizing DMBA treated SENCAR mice (Figure 5). As expected DMBA resulted in hyperplasia of the exposed epidermis, which is a hallmark of early stage neoplastic transformation in this model (29). In accordance with the JB6 P+ model, carvedilol given either topically or orally reduced DMBA-induced epidermal thickness; however, topical administration of carvedilol proved to be the most effective. Topical administration can be logically thought to be more efficacious due to direct application of the drug to the area exposed to DMBA. This becomes intriguing when one considers that most skin cancer arises from excessive UV exposure, and there are many topical treatments for the prevention of sunburns and treatment of sunburns. Thus, one could formulate a cancer preventative agent, such as carvedilol, so that it can be added to these existing products and hopefully reduce the prevalence of skin cancer. Additionally, such an agent can be added to moisturizing creams and other regularly used non-sun exposure ointments to provide a skin cancer preventative affect. Furthermore, use of a topical β-blocker would likely mitigate cardiovascular effects of the drug, but this remains to be tested. Oral administration more closely resembles the current clinical use of β-blockers; thus, retrospective studies that focus on carvedilol may reveal a reduced incidence of skin, and/or other, cancers compared to the untreated population. There are studies indicating that carvedilol prevents cisplatin- and adriamycin-induced toxicity (30–32). However, epidemiological studies examining β-blockers and cancer that differentiate between β-blockers do not have enough carvedilol patients to make sound conclusions (33). Hopefully, this study will initiate retrospective clinical studies focused on carvedilol.
Secondly, we examined the role of carvedilol in the growth of an established tumor cell line, A549 lung cancer cells, in vitro and within a xenograft model (Figure 6). A549 lung cancer cells express β2-ARs, although with disputed functionality (24, 34). The overall effect of carvedilol was negligible and often not statistically different than the control. The subtle effects observed on cell growth and mobility may partially explain the modest in vivo effects of carvedilol in the xenograft model. Collectively, the A549 studies suggest that carvedilol is not very effective against existing tumors. Previous studies support this conclusion via indicating that carvedilol alone only transiently slows tumor growth, but synergizes with conventional antitumor drugs (35, 36). Collectively, the data suggest that the direct anticancer properties of carvedilol are rather weak when used alone, and thus is best used as an adjuvant therapy (37–41).
Taken together, the drastic difference seen in the prevention study (Figure 5) compared to the treatment study (Figure 6) supports the hypothesis that the mechanism(s) of cancer prevention is different from the mechanisms of cancer treatment. Identification of the mechanism via which carvedilol attenuates carcinogenesis in this model may allow for the generation of novel chemopreventive agents. There are two likely mechanisms that are not mutually exclusive and may coordinate to make carvedilol superior to atenolol in our studies: GRK/β-arrestin biased agonism plus inhibition of classical cAMP signaling at the β2-AR and antioxidant properties of the drug.
The most likely mechanism of such effect involves the direct targeting of the β2-AR. Previous studies provide evidence that catecholamines may affect cancer development, and several epidemiological and clinical studies have examined the association between β-blocker uses and cancer progression (42, 43). In particular, β2-AR signaling occurs in normal human keratinocytes and is implicated in multiple cellular processes underlying skin cancer development (8, 44). Moreover, there are several studies that described a functional connection between EGFRs and β-ARs. β2-AR and EGFR were found to comprise a positive feedback loop in human cancer cells (6), and EGF-induced cancer cell proliferation requires the transactivation of β-ARs (45). Thus, inhibition of the β2-AR should help reduce cancer, but this clearly is not the complete mechanism due to the relative lack of effect of atenolol.
The second plausible mechanism may be related to the antioxidant and ROS scavenging properties of carvedilol (46). The antioxidant property is possibly due to its unique structure properties: the carbazole moiety, which is not present in propranolol and atenolol (46). Alternatively, or in combination, carvedilol-mediated signaling resulting in activation of the nuclear factor E2-related factor 2 (Nrf2)-antioxidant response element (ARE) (30) could reduce ROS as it is an important cellular stress response involved in controlling ROS (47). ROS can activate AP-1 in JB6 P+ cells (12), and AP-1 is involved in the process of cell transformation. Thus, the antioxidant effect of carvedilol may contribute to its cancer preventive activity.
Collectively, these studies suggest that carvedilol can prevent skin cancer. Until further examination of this phenomenon, the mechanism cannot be fully elucidated, and there is a possibility that novel cancer preventive mechanisms not discussed, or known, may underline the presented observations. Compared to other reported potential chemopreventive agents, carvedilol stands out because it is clinically-employed and well tolerated by patients. Based on these features and the present findings, carvedilol is a very promising candidate for future clinical trials of skin cancer prevention.
Acknowledgments
Financial support
This work was supported by Western University of Health Sciences and by NIH grant DK079307.
We thank Dr. David Sanchez at Western University of Health Sciences for kindly providing pGL4.22-AP1.
Footnotes
Conflict of Interest statement
The author(s) declare that they have no conflict interests.
References
- 1.Ricotti C, Bouzari N, Agadi A, Cockerell CJ. Malignant skin neoplasms. The Medical clinics of North America. 2009;93:1241–64. doi: 10.1016/j.mcna.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 2.Gupta S, Mukhtar H. Chemoprevention of skin cancer: current status and future prospects. Cancer Metastasis Rev. 2002;21:363–80. doi: 10.1023/a:1021275330385. [DOI] [PubMed] [Google Scholar]
- 3.Rao CV, Reddy BS. NSAIDs and chemoprevention. Curr Cancer Drug Targets. 2004;4:29–42. doi: 10.2174/1568009043481632. [DOI] [PubMed] [Google Scholar]
- 4.Cuzick J, Otto F, Baron JA, Brown PH, Burn J, Greenwald P, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 2009;10:501–7. doi: 10.1016/S1470-2045(09)70035-X. [DOI] [PubMed] [Google Scholar]
- 5.Chida Y, Hamer M, Wardle J, Steptoe A. Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat Clin Pract Oncol. 2008;5:466–75. doi: 10.1038/ncponc1134. [DOI] [PubMed] [Google Scholar]
- 6.Shi M, Liu D, Duan H, Qian L, Wang L, Niu L, et al. The beta2-adrenergic receptor and Her2 comprise a positive feedback loop in human breast cancer cells. Breast Cancer Res Treat. 2011;125:351–62. doi: 10.1007/s10549-010-0822-2. [DOI] [PubMed] [Google Scholar]
- 7.Armaiz-Pena GN, Allen JK, Cruz A, Stone RL, Nick AM, Lin YG, et al. Src activation by beta-adrenoreceptors is a key switch for tumour metastasis. Nature communications. 2013;4:1403. doi: 10.1038/ncomms2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang EV, Eubank TD. The impact of adrenergic signaling in skin cancer progression: possible repurposing of beta-blockers for treatment of skin cancer. Cancer biomarkers: section A of Disease markers. 2013;13:155–60. doi: 10.3233/CBM-130325. [DOI] [PubMed] [Google Scholar]
- 9.Kim IM, Tilley DG, Chen J, Salazar NC, Whalen EJ, Violin JD, et al. Beta-blockers alprenolol and carvedilol stimulate beta-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci U S A. 2008;105:14555–60. doi: 10.1073/pnas.0804745105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erickson CE, Gul R, Blessing CP, Nguyen J, Liu T, Pulakat L, et al. The beta-blocker Nebivolol Is a GRK/beta-arrestin biased agonist. PLoS One. 2013;8:e71980. doi: 10.1371/journal.pone.0071980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciardiello F, Tortora G. A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res. 2001;7:2958–70. [PubMed] [Google Scholar]
- 12.Dhar A, Young MR, Colburn NH. The role of AP-1, NF-kappaB and ROS/NOS in skin carcinogenesis: the JB6 model is predictive. Mol Cell Biochem. 2002;234–235:185–93. [PubMed] [Google Scholar]
- 13.Ren J, Mi Z, Jackson EK. Assessment of nerve stimulation-induced release of purines from mouse kidneys by tandem mass spectrometry. J Pharmacol Exp Ther. 2008;325:920–6. doi: 10.1124/jpet.108.137752. [DOI] [PubMed] [Google Scholar]
- 14.Steele VE, Sharma S, Mehta R, Elmore E, Redpath L, Rudd C, et al. Use of in vitro assays to predict the efficacy of chemopreventive agents in whole animals. J Cell Biochem Suppl. 1996;26:29–53. doi: 10.1002/jcb.240630704. [DOI] [PubMed] [Google Scholar]
- 15.Drvota V, Wei H, Haggblad J, Carlsson B, Sylven C. Beta-adrenergic receptor function in cultured AT-1 cardiomyocytes. Biochem Biophys Res Commun. 1995;207:13–9. doi: 10.1006/bbrc.1995.1146. [DOI] [PubMed] [Google Scholar]
- 16.Anakwe OO, Murphy PR, Moger WH. Characterization of beta-adrenergic binding sites on rodent Leydig cells. Biol Reprod. 1985;33:815–26. doi: 10.1095/biolreprod33.4.815. [DOI] [PubMed] [Google Scholar]
- 17.Strosberg AD, Pietri-Rouxel F. Function and regulation of the beta 3-adrenoceptor. Trends Pharmacol Sci. 1996;17:373–81. doi: 10.1016/s0165-6147(96)80011-3. [DOI] [PubMed] [Google Scholar]
- 18.Pauwels PJ, Gommeren W, Van Lommen G, Janssen PA, Leysen JE. The receptor binding profile of the new antihypertensive agent nebivolol and its stereoisomers compared with various beta-adrenergic blockers. Mol Pharmacol. 1988;34:843–51. [PubMed] [Google Scholar]
- 19.Bilski AJ, Halliday SE, Fitzgerald JD, Wale JL. The pharmacology of a beta 2-selective adrenoceptor antagonist (ICI 118,551) J Cardiovasc Pharmacol. 1983;5:430–7. doi: 10.1097/00005344-198305000-00013. [DOI] [PubMed] [Google Scholar]
- 20.Candelore MR, Deng L, Tota L, Guan XM, Amend A, Liu Y, et al. Potent and selective human beta(3)-adrenergic receptor antagonists. J Pharmacol Exp Ther. 1999;290:649–55. [PubMed] [Google Scholar]
- 21.Chakravarti D, Mailander P, Franzen J, Higginbotham S, Cavalieri EL, Rogan EG. Detection of dibenzo[a,l]pyrene-induced H-ras codon 61 mutant genes in preneoplastic SENCAR mouse skin using a new PCR-RFLP method. Oncogene. 1998;16:3203–10. doi: 10.1038/sj.onc.1201853. [DOI] [PubMed] [Google Scholar]
- 22.Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, et al. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci U S A. 2007;104:16657–62. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andresen BT. A pharmacological primer of biased agonism. Endocrine, metabolic & immune disorders drug targets. 2011;11:92–8. doi: 10.2174/187153011795564179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vanhoutte PM, Gao Y. Beta blockers, nitric oxide, and cardiovascular disease. Curr Opin Pharmacol. 2013;13:265–73. doi: 10.1016/j.coph.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 25.McCole DF, Truong A, Bunz M, Barrett KE. Consequences of direct versus indirect activation of epidermal growth factor receptor in intestinal epithelial cells are dictated by protein-tyrosine phosphatase 1B. J Biol Chem. 2007;282:13303–15. doi: 10.1074/jbc.M700424200. [DOI] [PubMed] [Google Scholar]
- 26.Smith C, Teitler M. Beta-blocker selectivity at cloned human beta 1- and beta 2-adrenergic receptors. Cardiovascular drugs and therapy/sponsored by the International Society of Cardiovascular Pharmacotherapy. 1999;13:123–6. doi: 10.1023/a:1007784109255. [DOI] [PubMed] [Google Scholar]
- 27.Rossig L, Haendeler J, Mallat Z, Hugel B, Freyssinet JM, Tedgui A, et al. Congestive heart failure induces endothelial cell apoptosis: protective role of carvedilol. J Am Coll Cardiol. 2000;36:2081–9. doi: 10.1016/s0735-1097(00)01002-0. [DOI] [PubMed] [Google Scholar]
- 28.Le DE, Pascotto M, Leong-Poi H, Sari I, Micari A, Kaul S. Anti-inflammatory and pro-angiogenic effects of beta blockers in a canine model of chronic ischemic cardiomyopathy: comparison between carvedilol and metoprolol. Basic research in cardiology. 2013;108:384. doi: 10.1007/s00395-013-0384-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kowalczyk MC, Kowalczyk P, Tolstykh O, Hanausek M, Walaszek Z, Slaga TJ. Synergistic effects of combined phytochemicals and skin cancer prevention in SENCAR mice. Cancer Prev Res (Phila) 2010;3:170–8. doi: 10.1158/1940-6207.CAPR-09-0196. [DOI] [PubMed] [Google Scholar]
- 30.Ouyang Y, Chen Z, Tan M, Liu A, Chen M, Liu J, et al. Carvedilol, a third-generation beta-blocker prevents oxidative stress-induced neuronal death and activates Nrf2/ARE pathway in HT22 cells. Biochem Biophys Res Commun. 2013;441:917–22. doi: 10.1016/j.bbrc.2013.10.160. [DOI] [PubMed] [Google Scholar]
- 31.Rodrigues MA, Rodrigues JL, Martins NM, Barbosa F, Curti C, Santos NA, et al. Carvedilol protects against the renal mitochondrial toxicity induced by cisplatin in rats. Mitochondrion. 2010;10:46–53. doi: 10.1016/j.mito.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 32.El-Shitany NA, Tolba OA, El-Shanshory MR, El-Hawary EE. Protective effect of carvedilol on adriamycin-induced left ventricular dysfunction in children with acute lymphoblastic leukemia. Journal of cardiac failure. 2012;18:607–13. doi: 10.1016/j.cardfail.2012.06.416. [DOI] [PubMed] [Google Scholar]
- 33.Hicks BM, Murray LJ, Powe DG, Hughes CM, Cardwell CR. beta-Blocker usage and colorectal cancer mortality: a nested case-control study in the UK Clinical Practice Research Datalink cohort. Ann Oncol. 2013;24:3100–6. doi: 10.1093/annonc/mdt381. [DOI] [PubMed] [Google Scholar]
- 34.Nakane T, Szentendrei T, Stern L, Virmani M, Seely J, Kunos G. Effects of IL-1 and cortisol on beta-adrenergic receptors, cell proliferation, and differentiation in cultured human A549 lung tumor cells. J Immunol. 1990;145:260–6. [PubMed] [Google Scholar]
- 35.Pasquier E, Street J, Pouchy C, Carre M, Gifford AJ, Murray J, et al. beta-blockers increase response to chemotherapy via direct antitumour and anti-angiogenic mechanisms in neuroblastoma. Br J Cancer. 2013;108:2485–94. doi: 10.1038/bjc.2013.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erguven M, Yazihan N, Aktas E, Sabanci A, Li CJ, Oktem G, et al. Carvedilol in glioma treatment alone and with imatinib in vitro. Int J Oncol. 2010;36:857–66. doi: 10.3892/ijo_00000563. [DOI] [PubMed] [Google Scholar]
- 37.Palm D, Lang K, Niggemann B, Drell TL, Masur K, Zaenker KS, et al. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int J Cancer. 2006;118:2744–9. doi: 10.1002/ijc.21723. [DOI] [PubMed] [Google Scholar]
- 38.Cakir Y, Plummer HK, III, Tithof PK, Schuller HM. Beta-adrenergic and arachidonic acid-mediated growth regulation of human breast cancer cell lines. Int J Oncol. 2002;21:153–7. [PubMed] [Google Scholar]
- 39.Guo K, Ma Q, Wang L, Hu H, Li J, Zhang D, et al. Norepinephrine-induced invasion by pancreatic cancer cells is inhibited by propranolol. Oncol Rep. 2009;22:825–30. doi: 10.3892/or_00000505. [DOI] [PubMed] [Google Scholar]
- 40.Shi M, Liu D, Duan H, Qian L, Wang L, Niu L, et al. The beta2-adrenergic receptor and Her2 comprise a positive feedback loop in human breast cancer cells. Breast Cancer Res Treat. 2010 doi: 10.1007/s10549-010-0822-2. [DOI] [PubMed] [Google Scholar]
- 41.Stanojkovic TP, Zizak Z, Mihailovic-Stanojevic N, Petrovic T, Juranic Z. Inhibition of proliferation on some neoplastic cell lines-act of carvedilol and captopril. J Exp Clin Cancer Res. 2005;24:387–95. [PubMed] [Google Scholar]
- 42.Bangalore S, Kumar S, Kjeldsen SE, Makani H, Grossman E, Wetterslev J, et al. Antihypertensive drugs and risk of cancer: network meta-analyses and trial sequential analyses of 324,168 participants from randomised trials. Lancet Oncol. 2011;12:65–82. doi: 10.1016/S1470-2045(10)70260-6. [DOI] [PubMed] [Google Scholar]
- 43.Algazi M, Plu-Bureau G, Flahault A, Dondon MG, Le MG. Could treatments with beta-blockers be associated with a reduction in cancer risk? Rev Epidemiol Sante Publique. 2004;52:53–65. doi: 10.1016/s0398-7620(04)99022-0. [DOI] [PubMed] [Google Scholar]
- 44.Luthy IA, Bruzzone A, Pinero CP, Castillo LF, Chiesa IJ, Vazquez SM, et al. Adrenoceptors: non conventional target for breast cancer? Curr Med Chem. 2009;16:1850–62. doi: 10.2174/092986709788186048. [DOI] [PubMed] [Google Scholar]
- 45.Liu X, Wu WK, Yu L, Li ZJ, Sung JJ, Zhang ST, et al. Epidermal growth factor-induced esophageal cancer cell proliferation requires transactivation of beta-adrenoceptors. J Pharmacol Exp Ther. 2008;326:69–75. doi: 10.1124/jpet.107.134528. [DOI] [PubMed] [Google Scholar]
- 46.Yue TL, Cheng HY, Lysko PG, McKenna PJ, Feuerstein R, Gu JL, et al. Carvedilol, a new vasodilator and beta adrenoceptor antagonist, is an antioxidant and free radical scavenger. J Pharmacol Exp Ther. 1992;263:92–8. [PubMed] [Google Scholar]
- 47.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]