

Abstract
CODAS syndrome is a multi-system developmental disorder characterized by cerebral, ocular, dental, auricular, and skeletal anomalies. Using whole-exome and Sanger sequencing, we identified four LONP1 mutations inherited as homozygous or compound-heterozygous combinations among ten individuals with CODAS syndrome. The individuals come from three different ancestral backgrounds (Amish-Swiss from United States, n = 8; Mennonite-German from Canada, n = 1; mixed European from Canada, n = 1). LONP1 encodes Lon protease, a homohexameric enzyme that mediates protein quality control, respiratory-complex assembly, gene expression, and stress responses in mitochondria. All four pathogenic amino acid substitutions cluster within the AAA+ domain at residues near the ATP-binding pocket. In biochemical assays, pathogenic Lon proteins show substrate-specific defects in ATP-dependent proteolysis. When expressed recombinantly in cells, all altered Lon proteins localize to mitochondria. The Old Order Amish Lon variant (LONP1 c.2161C>G[p.Arg721Gly]) homo-oligomerizes poorly in vitro. Lymphoblastoid cell lines generated from affected children have (1) swollen mitochondria with electron-dense inclusions and abnormal inner-membrane morphology; (2) aggregated MT-CO2, the mtDNA-encoded subunit II of cytochrome c oxidase; and (3) reduced spare respiratory capacity, leading to impaired mitochondrial proteostasis and function. CODAS syndrome is a distinct, autosomal-recessive, developmental disorder associated with dysfunction of the mitochondrial Lon protease.
Introduction
Cerebral, ocular, dental, auricular, skeletal (CODAS) syndrome (MIM 600373) was first described in 1991 as a distinctive constellation of developmental delay, craniofacial anomalies, cataracts, ptosis, median nasal groove, delayed tooth eruption, anomalous cusp morphology, malformed helices, hearing loss, short stature, delayed epiphyseal ossification, metaphyseal hip dyplasia, and vertebral coronal clefts.1 CODAS is a rare disease; only three additional cases (from Brazil, Canada, and France) were reported between 1995 and 2010.2–4 Autosomal-recessive inheritance of CODAS syndrome was first suggested by Shebib et al.,1 who identified the phenotype within an endogamous Mennonite community, and it was corroborated by the observations of Innes et al.3
We used whole-exome and Sanger sequencing to study ten individuals with CODAS syndrome from three distinct ancestral backgrounds: Amish-Swiss from the United States (n = 8), Mennonite-German from Canada (n = 1), and mixed European (German, Scottish, Irish, English) from Canada (n = 1). Our analysis revealed four different mutations within LONP1 (RefSeq accession number NM_004793.3 [isoform 1]; [MIM 605490]). These mutations were either homozygous (LONP1 c.2161C>G [p.Arg721Gly] and LONP1 c.2026C>T [p.Pro676Ser]) or compound heterozygous (LONP1 c.1892C>A/c.2171C>T [p.Ser631Tyr/p.Ala724Val]) (Table 1). In humans, LONP1 encodes the ATP-dependent mitochondrial Lon protease, which belongs to the AAA+ superfamily of ATPases associated with various cellular activities. AAA+ proteins share a conserved ATPase module and mediate diverse cellular processes such as DNA replication, membrane fusion, signal transduction, and transcriptional regulation.5–8
Table 1.
Four Pathogenic LONP1 Alleles Among Ten Individuals with CODAS Syndrome
| Origin of Sample | n | Ancestry |
LONP1 Variants |
Previously Reported | |
|---|---|---|---|---|---|
| Allele 1a | Allele 2a | ||||
| Pennsylvania | 8 | Amish-Swiss | Arg721Gly | Arg721Gly | |
| Canada | 1 | Mennonite-German | Pro676Ser | Pro676Ser | Shebib et al., 19911 |
| Canada | 1 | Mixed European | Ser631Tyr | Ala724Val | |
In every case, genotyping showed allele segregation consistent with recessive inheritance.
Lon is an ATP-driven proteolytic machine and is highly conserved from archaea to mammals. In human mitochondria, Lon is the simplest of four energy-dependent proteolytic systems that include ClpXP, m-AAA, and i-AAA.9 Mutations of CLPP (MIM 601119), which encodes the proteolytic component of ClpXP, are associated with Perrault syndrome type 3 (MIM 614129),10 and mutations of SPG7, which encodes a subunit of m-AAA, are linked to hereditary spastic paraplegia (MIM 607259).11 In humans, Lon functions as a protease, chaperone, and DNA-binding protein. It contributes to mitochondrial proteostasis by eliminating misfolded and oxidatively damaged proteins12–14 and supports cell viability during proteotoxic, hypoxic, and endoplasmic-reticulum stress.15–17 Lon also degrades key regulatory proteins of mitochondrial metabolism, for example ALAS-1 and StAR, rate-limiting enzymes of heme and steroid hormone biosynthesis, respectively.18,19
Independent of its proteolytic activity, Lon influences function of the mitochondrial genome and respiratory chain. Its chaperone-like function promotes assembly of respiratory-chain complexes within the mitochondrial inner membrane,16,20,21 and knockout of Lon-encoding genes in yeast and mammals leads to mtDNA deletions and depletion, respectively.15,22,23 Lon binds single-stranded mtDNA in a sequence- and strand-specific manner24–27 and associates preferentially with the mtDNA control region where replication and transcription are initiated.24
Here, we establish a link between LONP1 and CODAS syndrome in humans and discuss potential mechanisms linking the dysfunction of mitochondrial Lon to diverse clinical manifestations.
Subjects and Methods
Subjects
We studied a total of ten individuals from two countries (United States, n = 8 and Canada, n = 2). All had the core developmental, ocular, dental, auricular, and skeletal features of CODAS syndrome.1–4 Detailed phenotype studies were conducted on four children homozygous for LONP1 c.2161C>G, a founder mutation among the Pennsylvania Amish (Figures 1 and 2). The study was approved by the institutional review board of Lancaster General Hospital. Parents consented to research on their affected children on their behalf and signed a separate consent to reproduce photographs. Affected children were evaluated by routine clinical methods, including bone radiographs, 1.5 T magnetic resonance imaging, slit-lamp and dilated indirect ophthalmoscopy, middle-ear tympanometry, distortion-product otoacoustic emissions, evoked auditory brainstem responses, and direct laryngoscopy.
Figure 1.
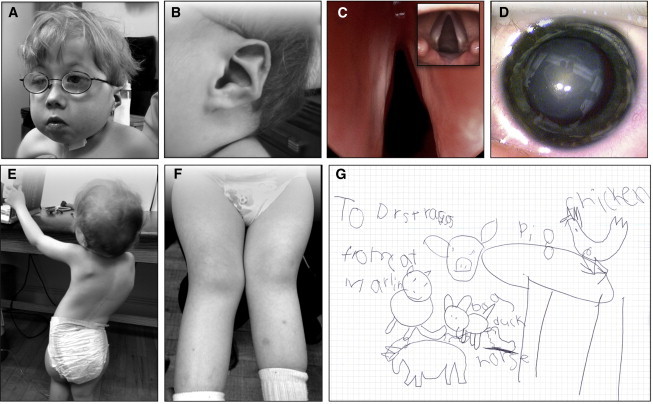
CODAS Phenotype
(A and B) A 5 year-old child who is homozygous for LONP1 c.2161C>G (p.Arg721Gly) and who has characteristic physical features of CODAS syndrome. Features include (A) broad skull and flattened midface; ptosis; grooved nasal tip; anteverted nares; and (B) helix hypoplasia (“crumpled” ears).
(C) Laryngoscopy of an affected child revealed airway narrowing secondary to paretic, atrophic vocal cords (inset: normal vocal cords for comparison).
(D) Dense bilateral nuclear cataracts develop rapidly between 2 and 6 months of age in all CODAS-syndrome-affected Amish individuals.
(E and F) Scoliosis and genu valgum progress with advancing age.
(G) Despite previous reports of developmental delay in children with CODAS syndrome, timely ophthalmologic and audiologic intervention appear to be critical developmental determinants; at age 5 years our oldest subject has a vocabulary of more than 100 words, reads at the first grade level, communicates effectively by signing, and has advanced drawing and writing skills.
Figure 2.
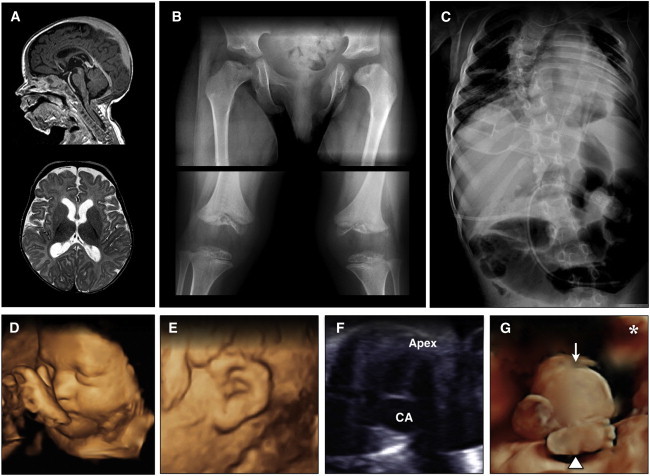
Radiographic Features of CODAS Syndrome
(A) Sagittal T1 (upper) and axial T2 (lower) images of an affected 4-year-old Amish child show mild diffuse cortical atrophy, an immature pattern of myelination, and hypoplasia of the corpus callosum.
(B) Skeletal radiographs show metaphyseal dysplasia most evident at hip joints (upper) and valgus knees with both hypoplasia and delayed ossification of epiphyses (lower).
(C) Severe scoliosis is evident early in childhood, and coronal clefts are observed at various levels of the vertebral column.
(D–G) Prenatal imaging of a 32-week-old fetus with CODAS syndrome revealed polyhydramnios, a two-vessel umbilical cord, midfacial hypoplasia, (D) crumpled helices, (E) a balanced atrioventricular canal, (F) common atrium, omphalocele, (G, arrow) and absence of left lower long bones, which were replaced by a relatively well-formed foot attached directly to the hip. (G, arrowhead) The asterisk in (G) marks the left palpebral fissure for orientation.
Molecular Genetics
Genetic mapping of CODAS syndrome was performed as previously described.28–33 In brief, SNP genotyping was performed with the GeneChip Mapping 10K Assay Kits (Affymetrix). Data were analyzed in Microsoft Excel spreadsheets that were custom formatted at the Clinic for Special Children. SNP positions came from Affymetrix genome-annotation files, and genotype data came from the Affymetrix GeneChip Human Mapping 10K Xba 142 arrays. Data analyses were designed for rapid identification of genomic regions that were identically homozygous between affected individuals.
Exome sequencing was performed as previously described.34–36 A solution-hybrid-selection methodology was used for isolating exomic DNA, which was subjected to sequencing on the Illumina GA-II platform. In brief, DNA oligonucleotides, corresponding to 120 bp of target sequence flanked by 15 bp of universal primer sequence, were synthesized in parallel on an Agilent microarray, then were cleaved from the array. The oligonucleotides were amplified by PCR, then transcribed in vitro in the presence of biotinylated uridine-5′-triphosphate so that single-stranded RNA “bait” would be generated. Genomic DNA was sheared, ligated to Illumina sequencing adapters, and selected for lengths between 200 and 350 bp. This “pond” of DNA was hybridized with an excess of bait in the solution. The “catch” was pulled down by magnetic beads coated with streptavidin, eluted, and sequenced on the Illumina GA-II.
Massively parallel sequencing data were processed by the Sequencing Platform at the Broad Institute via two pipelines. The first, called “Picard,” utilized the reads and qualities produced by the Illumina software and produced a single BAM file representing each sample. The second pipeline, called Genome Sequencing Analysis, then performed post-processing and analysis of the data, including SNP identification, small-insertion and -deletion identification, local realignment of insertion- or deletion-containing reads, gene annotation, and filtering with common polymorphisms (1000 Genomes, dbSNP build 129). The details of the sequencing-data processing have been described elsewhere.37 For inter-exome analyses, variant call data were imported into a FileMaker Pro database, and rare and novel variants shared among affected individuals were queried.
Molecular Modeling
A homology model of the human mitochondrial Lon hexamer was generated from the crystal structure of B. subtilis Lon (PDB 3M6A).9,38 In brief, we generated a homology model of a single Lon subunit that lacks its amino-terminal mitochondrial-targeting sequence (MTS) (see Figures 3B–3D) by using three independent molecular modeling software tools: Prime (Schrodinger), Modeller (see Web Resources), and the I-TASSER online server at the University of Michigan. We obtained the final model of the Lon subunit by averaging three models. We obtained the Lon hexamer by superimposing the structure of one subunit onto the crystal structure of B. subtilis Lon (PDB 3M6A).
Figure 3.
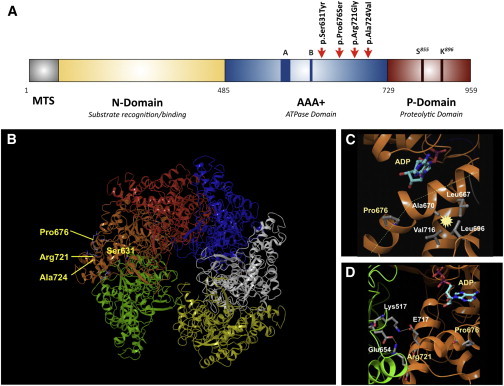
Location of Amino Acids That Are Mutated within Mitochondrial Lon in CODAS Syndrome
(A) Domain structure of the human Lon subunit. Mitochondrial targeting sequence (MTS), substrate recognition/binding (N) domain, ATPase domain (AAA+), and a protease domain (P). Red arrows indicate the position of pathogenic CODAS mutations.
(B) Homology model of human mitochondrial Lon. The model shows the position, within a single Lon subunit (shown in amber), of amino acids that are altered in CODAS syndrome.
(C and D) The positions of amino acids Pro676 and Arg721 (yellow), which are altered in proteins encoded by CODAS homozygous alleles. ADP is shown occupying the ATP- and ADP-binding pocket (a green dashed line represents a proline-induced kink in the helix; a yellow dotted line represents a salt bridge; starbursts represent hydrophobic interactions). (C) The Pro676-induced kink promotes hydrophobic interactions between Ala670, Leu667, Leu696, and Val716. (D) The positions of Arg721 as well as Pro676, Glu717, and the ATP- and ADP-binding site are located on the same Lon subunit (amber). Arg721 and Glu717 form salt bridges with Glu654 and Lys517, respectively, which are located on the adjacent Lon subunit (green).
cDNA Constructs
Human LONP1 cDNA (RefSeq NM_004793.3) that was amplified from ARPE-19 (human adherent retinal pigment epithelium-19) total RNA by RT-PCR (SuperScript II reverse transcriptase, Life Technologies; Phusion DNA polymerase, New England BioLabs)35 was cloned into pENTR/D-TOPO (Life Technologies), and CODAS mutations were introduced by site-directed mutagenesis (QuikChange II, Agilent) (Table S2). For mammalian expression of Lon proteins with C-terminal V5 or FLAG tags, the LONP1 cDNA was cloned in frame into pcDNA3.2/V5-DEST or pcDNA3.2/FLAG-DEST by recombination from pENTR/D-TOPO/LONP1 constructs with LR Clonase II (Life Technologies). For bacterial expression, cDNAs encoding Lon proteins that lacked the first 114 amino acids (containing the predicted MTS) were sub-cloned into pET24c(+) as well as into pProEX-1; the latter constructs encoded a His tag fused to the N terminus.25 All amino acid numberings were assigned on the basis of the full-length Lon precursor.
Purification of Recombinant Lon
Wild-type and pathogenic Lon variants were overexpressed in Rosetta (DE3) (Novagen) and purified as previously described.39–41 Lon constructs were cloned into either pET24c(+) (Figure 4) or ProEX (hexa-histidine tag26) (Figure S1). For Figure 4, Lon-positive fractions were pooled and concentrated, and protein concentrations were determined via the Bradford assay, for which BSA was used as a standard and UV absorbance was set at 280 nm, and using the equation ε280 = W(5500) + Y(1490) + C(125), where ε280 is the molar absorptivity at 280 nm and W, Y, and C are the numbers of tryptophan, tyrosine, and cysteine residues, respectively.42,43 Protein concentrations were further corrected on the basis of protein-band optical density normalized to LonWT in Coomassie-stained SDS-PAGE gels (Image J). Recombinant steroidogenic acute regulatory protein (StAR) and mitochondrial transcription factor A (TFAM) were purified as described previously.41,44
Figure 4.
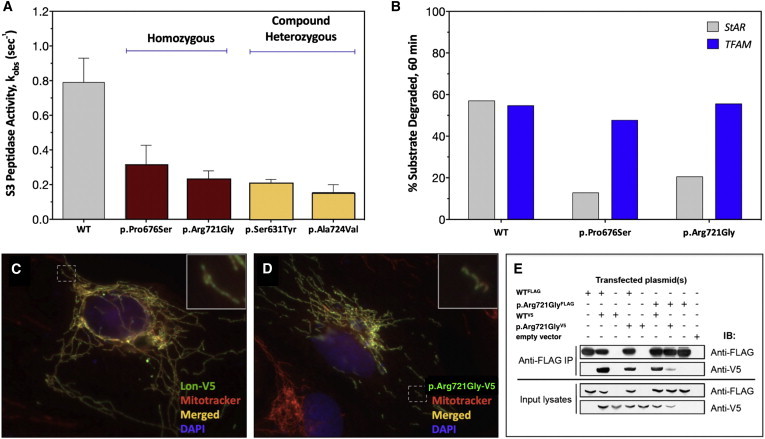
Enzymology, Cellular Expression, and Homo-oligomerization of Pathogenic Lon Proteins
(A) ATP-dependent degradation of the fluorogenic peptide substrate S3. The rates of S3 cleavage were determined with recombinant Lon proteins (250 nM monomer) and 0.5 mM of S3 (0.5-fold Km) in buffer. The degradation reactions were initiated by the addition of Mg-ATP at 37°C for 600–900 s. S3 cleavage was monitored as an increase in fluorescence at an excitation and emission of 320 nm and 420 nm, respectively (three or more replicates). Error bars represent SEM.
(B) StAR (5.6 μM) or TFAM (5 μM) were combined in buffer with Mg-ATP, and reactions were initiated by the addition of Lon (500 nM) at 37°C. Aliquots were removed at time points, reactions were terminated with 5× reducing sample buffer, and aliquots were analyzed by SDS-PAGE and Coomassie Brilliant Blue staining. ImageJ was used for determining the percent StAR and TFAM degraded after a 60 min incubation period with the same Lon variant (no replicates).
(C and D) Overexpression of (C) wild-type Lon-V5 and (D) p.Arg721Gly-V5 (green = V5 immunofluorescence) in ARPE-19 cells counterstained with Mitotracker Red (red) and DAPI (blue). Nontransfected cells display only red Mitotracker fluorescence.
(E) Coimmunoprecipitation (IP) of Lon-V5 by Lon-FLAG (co-overexpressed in HEK293T cell lysates) with anti-FLAG M2 antibody. Immunoblotting (IB) with anti-FLAG and V5 antibodies followed.
Enzymatic Assays
Peptidase and Protease Assays
Fmoc-protected amino acids, Boc-Abz, Fmoc-protected Lys-Wang resin, and HBTU (N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate, O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate) were purchased from Advanced ChemTech and NovaBiochem. Peptide substrate S1 was purchased from Genscript. Peptide synthesis of S2 was performed with Fmoc solid-phase synthesis methodologies as described previously.40 Peptidase activity was measured on a Fluoromax-4 spectrofluorimeter (Horiba Group). All reactions mixtures contained 50 mM HEPES (pH 8.0), 5 mM Mg(OAc)2, 1 mM DTT, 250 nM wild-type and mutant Lon proteins, and 0.5 mM S3. To avoid problems from the inner filter effect, we used S3, a mixed substrate consisting of 10% fluorescently labeled peptide S2 [Y(NO2)RGITCSGRQK(Abz)] and 90% non-fluorescent analog of the peptide S1 (YRGITCSGRQK(Ac)). After the mixture equilibrated at 37°C for 1 min, the reaction was initiated with 1 mM ATP. We measured the amount of hydrolyzed peptide by determining the maximum fluorescence generated per micromole of peptide after complete digestion by trypsin. The steady-state rate of the reaction was determined from the tangent of the linear portion of the time course. This rate was converted to an observed rate constant (kobs) by division of the rate by the enzyme concentration. At least three identical experiments were performed.
Peptidase assays using the fluorescent dipeptide substrate rhodamine 110, bis-(CBZ-L-alanyl-L-alanine amide) (AA2-Rh110, Anaspec), were performed in quadruplicate (20 μl) in 384-well plates as previously described.39 Lon (800 nM monomer) or no-enzyme controls were incubated in a reaction buffer (150 mM NaCl, 50 mM HEPES [pH 8.0], and 10 mM MgCl2) containing AA2-Rh110 (6 μM) and ATP (2 mM) for 3 hr at 37°C, after which fluorescence was measured at an excitation and emission of 485 and 535 nm, respectively, with a Perkin Elmer Victor3 V. The relative fluorescence units (RFU) of the background (no-enzyme control) measurements were subtracted, and the resultant values were normalized to percent activity of the no-drug reactions. Data were fit to 4-parameter dose-response curves using GraphPad Prism 6, and the error bars represent the SD of four replicate reactions. At least three identical experiments were performed. StAR and TFAM purification and degradation assays were performed as previously described.19,41,44,45
ATPase Assay
The ATPase activity of wild-type and mutant Lon variants was measured in quadruplicate reactions (5 μl) in a 384-well plate via the ADP-Glo Assay (Promega) according to the manufacturer’s recommendations. All steps were performed at room temperature. Purified human Lon (400 nM monomer) was incubated in a reaction buffer (40 mM Tris-HCl [pH 7.5], 20 mM MgCl2, and 0.1 mg/ml BSA) with Ultra Pure ATP (1 mM; Promega) for 60 min. ADP-Glo™ Reagent (5 μl) was added for 40 min followed by Kinase Detection Substrate (10 μl) for 60 min, which was linear with [ADP]. Relative luminescence units (RLU) were measured on a Perkin Elmer Victor3 V, and background (no-enzyme control) RLU were subtracted. At least three identical experiments were performed.
Cell Culture
ARPE-19 cells, human embryonic kidney (HEK293T) cells, HeLa cells (ATCC, Manassas, VA), and mouse auditory sensory epithelium (UB/OC-2) cells (gift from Matthew Holley, University of Sheffield, UK) were cultured in DMEM:F12, DMEM, Eagle’s MEM, or MEM with GlutaMax and 50U/ml interferon-gamma (Life Technologies), respectively, each with 10% fetal bovine serum (FBS; Sigma or Life Technologies) at 37°C/5% CO2. Epstein-Barr virus (EBV)-transformed B-lymphoblastoid cell lines (LCLs) were generated from two Amish CODAS-syndrome-affected probands homozygous for LONP1 c.2161C>G (p.Arg721Gly) as well as from their respective heterozygous parents (Lineberger Comprehensive Cancer Center, University of North Carolina). LCLs were cultured in RPMI (ATCC formulation) with 15% FBS at 37°C and 5% CO2. Genotyping of LCLs was performed by Sanger sequencing (The Pennsylvania State University Nucleic Acid Facility). In brief, SuperScript III reverse transcriptase (Life Technologies) and oligo dT primers were used for reverse transcription of cDNA from LCL total RNA (Trizol, Life Technologies). cDNA was then amplified by the polymerase chain reaction (PCR) with LONP1-specific primers (Table S1) and purified for DNA sequencing with QIAGEN PCR purification spin columns. Lon knockdown in HeLa, PC3, and HEK293T cells was performed as previously described46 (and as also described in Supplemental Data).
Mitochondrial Localization
Wild-type and mutant Lon variants were overexpressed by transient transfection (FuGENE 6, Promega) in ARPE-19, HeLa, and UB/OC-2 cells cultured on glass coverslips. After 40–48 hr, cells were incubated with MitoTracker Red CMXRos (300 nM) at 37°C/5% CO2 for 30 min, washed with PBS (pH 7.2), and fixed in 4% paraformaldehyde in PBS for 15 min at 37°C. Cells were permeabilized in acetone at −20°C for 5 min, washed with PBS, and blocked in 1% bovine serum albumin (BSA) with 0.2% Triton X-100 in PBS for 10 min. Immunofluorescence detection of Lon-V5 fusion proteins was conducted as described elsewhere.35
Immunoblotting and Immunoprecipitation
Cells were lysed in the following buffers as indicated: in RIPA buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1.0% octylphenoxypolyethoxyethanol CA-630, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) supplemented with 1 mM EDTA, 2 mM NaF, 1 mM Na3VO4, and protease-inhibitor cocktail (Roche); Triton X-100 buffer (50 mM Tris, [pH 7.5], 300 mM NaCl, and 0.5% Triton X-100) supplemented with Halt protease- and phosphatase-inhibitor cocktail (ThermoScientific); or urea buffer (50 mM triethylammonium bicarbonate buffer [TEAB, Sigma] [pH 8.5], 8 M urea) supplemented with protease-inhibitor cocktail and phosphatase-inhibitor cocktail (Sigma). When the RIPA or Triton X-100 buffer was used, cells were incubated for 15 min on ice, then centrifuged at 14,000 rpm for 15 min at 4°C so that lysates would be cleared. For the urea buffer, cells were sonicated with microprobe for 15 s followed by a 30 s pulse, and this was repeated 3–5 times before centrifugation at 4°C cleared lysates.
The protein concentration of cell extracts was measured with the Bradford assay and then normalized.42 Immunoblotting and co-immunoprecipitation were performed as described elsewhere;35 washes following immunoprecipitation contained 5% Tween-20. The following antibodies were employed: rabbit anti-Lon (1:400, custom made and affinity purified as previously described)24 or rabbit anti-Lon (1:100, Novus, H00009361-D01P); rabbit anti-ClpX (1:3,000, custom made by Dr. Irene Lee); rabbit anti-aconitase (1:200, provided by Dr. Luke Szweda, Oklahoma Medical Research Foundation); mouse anti-ClpP (1:4,000, Abcam 56455); rabbit anti-Pink1 (1:500, Abcam 23707); rabbit anti-mt-cytochrome oxidase subunit II (1:5,000, Abcam 91317); OXPHOS Antibody Cocktail (1:500, Mitoscience 604); goat anti-actin (1:2,000, 1615; Santa Cruz); mouse anti-β-actin (Sigma, 1:1,000,000); rabbit anti-mt-cytochrome B (1:1,000, Aviva Systems Biology ARP50256_P050); mouse monoclonal anti-V5 (1:5,000, Life Technologies) or mouse anti-FLAG M2 (1:1,000 Sigma); and HRP-conjugated secondary antibodies (Cell Signaling Technologies or Santa Cruz Biotechnology). Immunoblots were developed by chemiluminescence.
Transmission Electron Microscopy
LCLs were prepared for transmission electron microscopy (TEM) as described previously.39,47 In brief, cells were pelleted by centrifugation at 1,000 rpm for 5 min, washed with PBS, re-pelleted, and re-suspended in 2.5% glutaraldehyde and 4% paraformaldehyde in 0.1 M PBS overnight at 4°C. LCLs were then post-fixed in 1% osmium tetroxide for 1 hr at 20°C and dehydrated with ethanol; infiltration with Spurr’s resin (Electron Microscopy Sciences) followed. Infiltrated LCLs were collected by centrifugation and embedded directly into BEEM capsules, which were gently centrifuged so that the cells would be driven to the bottom of the capsule. Ultrathin sections (70-90 nm) were cut with glass knives, mounted on naked copper grids (200 mesh), and stained with a saturated solution of uranyl acetate in 50% ethanol for 20 min, followed by Reynold’s lead citrate for 15 min. Differences in the frequency of abnormal mitochondrial morphology between proband and parent cells were analyzed by two-way ANOVA with Bonferroni post-hoc correction (SPSS, IBM).
Quantitative PCR (qPCR) Determination of mtDNA Copy Number
Genomic DNA (100 ng) from peripheral blood lymphocytes and LCL cells was used for amplification of both the mtDNA-encoded MT-CYB gene and the nuclear-DNA-encoded APP gene as a reference control. Genomic DNA was amplified in reactions (20 μl; Applied Biosystems; universal PCR master mix) with the following Taqman primers. Forward: APP, 5′-TTTTTGTGTGCTCTCCCAGGTCT-3′; and MT-CYB, 5′-GCCTGCCTGATCCTCCAAAT-3′). Reverse: APP 5′-TGGTCACTGGTTGGTTGGC-3′; and MT CYB, 5′-AAGGTAGCGGATGATTCAGCC-3′. TaqMan probes: APP, 5′-[6FAM]CCCTGAACTGCAGATCACCAATGTGGTAG[TAM]-3′; and MT-CYB, 5′-[6FAM]CACCAGACGCCTCAACCGCCTT[TAM]-3′ An AB 7500 RT-PCR system (Applied Biosystems) and standard qPCR conditions were employed: 95°C for 10 min, 40 cycles at 95°C for 15 s, and 60°C for 1 min. All reactions were performed in triplicate. Relative quantitation of the mtDNA copy number was calculated by the ΔΔCt method.
Mitochondrial Oxygen-Consumption Measurements
Proband and parent cells (1 × 105/well) in a 100 μl unbuffered assay medium (DMEM with 11 mM glucose, 1 mM pyruvate, and 2 mM glutamine) were plated in poly-D-lysine-coated Seahorse XF 24-well plates (50 μg/ml) and centrifuged so that cells were adhered to the plates. After the uniformity of cells plated in each well was ensured, plates were transferred to a 37°C incubator without CO2 for 30 min, 575 μl warm assay medium was added, and then plates were left in the incubator for another 20 min. Each plate was transferred to the XF24 Analyzer, and the oxygen consumption rate (OCR) was measured at baseline. Sequential injections were then performed with the following: oligomycin (1 μM), an inhibitor of complex V (the ATP synthase); FCCP (750 nM), an uncoupler that dissipates the membrane potential across the mitochondrial inner membrane; and rotenone (1 μM), an inhibitor of complex I (NADH dehydrogenase), which blocks the transfer of electrons from complex I to ubiquinone. The spare respiratory capacity (SRC), which is the maximum OCR after FCCP injection, was calculated as a percentage of baseline OCR. The mean, standard deviation, and statistical significant difference between proband and mother cells were calculated by Student’s t test in Microsoft Excel.
Results
Molecular Genetic Studies
We initiated a genetic mapping study involving two Amish children who were affected with CODAS syndrome. Affymetrix GeneChip Mapping 10K DNA microarrays were initially used for genotyping, but no significant regions of homozygosity were detected. Exome sequencing was performed, and variants were filtered so that those with minor allele frequencies (MAFs) > 1% in either 1000 Genomes or the Exome Variant Server or with MAF > 10% in our own internal Amish variant database were excluded. Analysis of variants detected in CODAS-affected individuals revealed a single shared homozygous allele, LONP1 c.2161C>G (p.Arg721Gly), located at physical position chr19:5,694,557 (NCBI GRCh37, hg19) The gene mapped to a region of chromosome 19 with poor coverage on the Affymetrix 10K array, hence our inability to conclusively map the phenotype.
The presence of LONP1 c.2161C>G was confirmed by Sanger sequencing, and we verified proper segregation by genotyping parents and siblings. Using an unlabeled probe to genotype 576 Amish controls by high-resolution melt analysis, we found a surprisingly high population allele frequency of 5.9%. We next acquired additional CODAS-syndrome DNA samples accompanied by clinical data from collaborators in Canada and designed primers to amplify and sequence all 18 exons of LONP1. The first sample was from a Mennonite child from Manitoba, Canada, who was the subject of a previous publication.1 She was found to be homozygous for LONP1 c.2026C>T (p.Pro676Ser). The second sample was from a child of mixed European ancestry (German, Scottish, Irish, and English) from Ontario, and she was found to be compound heterozygous for LONP1 c.1892C>A (p.Ser631Tyr) and c.2171C>T (p.Ala724Val)(Table 1).
Phenotype Associated with CODAS Syndrome
Eight children from the Old Order Amish community of Pennsylvania were homozygous for LONP1 c.2161C>G (Table 2). Three died of laryngeal obstruction in the first days of life, and one with a tracheostomy died from pneumonia during early infancy. Four remaining children (ages 0.6–14.2 years, two female) required tracheostomies to survive infancy; only one child was subsequently decannulated. All three surviving children who underwent sedated laryngoscopies had paretic, atrophic vocal cords, glottic narrowing, chronic sialorrhea, and swallowing dysfunction. Three of four surviving affected children were nourished exclusively by gastrostomy tube.
Table 2.
Phenotype of CODAS-Syndrome-Affected Children Homozygous for LONP1 c.2161C>G (p.Arg721Gly)
|
Agea |
14.2b |
4.6 |
1.2 |
0.6 |
|---|---|---|---|---|
| Gender | F | M | F | M |
| Cerebral | ||||
| Hypotonia and motor delay | + | + | + | + |
| Intellectual disability | + | + | n/a | n/a |
| Epilepsy | + | − | − | − |
| Ocular | ||||
| Ptosis | + | + | + | + |
| Cataractsc | + | + | + | + |
| Dental | ||||
| Delayed tooth eruption | + | + | n/a | n/a |
| Enamel dysplasia | + | + | n/a | n/a |
| Auricular | ||||
| Scapha and helix dysplasia (“Crumpled” Ears) | + | + | + | + |
| Conductive hearing loss | n/a | + | + | + |
| Sensorineural hearing loss | + | + | − | − |
| Skeletal | ||||
| Short stature | + | + | + | + |
| Metaphyseal dysplasia | + | + | + | + |
| Epiphyseal hypoplasia and delayed ossification | + | + | + | + |
| Scoliosis | + | + | + | + |
| Genu valgus | + | + | + | + |
| Pes valgus | + | + | n/a | n/a |
| Vertebral coronal clefts | + | + | n/a | n/a |
| Dens hypoplasia | + | + | n/a | n/a |
| Other | ||||
| Grooved nasal tip | + | + | + | + |
| Anteverted nares | + | + | + | + |
| Tracheostomy | + | + | + | + |
| Vocal-cord paresis | − | + | + | + |
| Endocardial cushion (atrial septum defect)d | − | + | + | − |
| Gastresophageal reflux | − | + | − | + |
| Gastrostomy tube feeding | − | + | + | + |
| Omphalocele | − | − | − | + |
| Imperforate anus | + | − | − | + |
| Rectovaginal fistula | + | − | − | − |
| Cryptorchidism | n/a | + | n/a | − |
| Tongue hemiatrophy | − | + | − | + |
Abbreviations are as follows: F, female; M, male; n/a, not applicable because age or clinical data were not available.
Limited clinical data are available for four of eight children who had CODAS syndrome and died in the perinatal period as a result of respiratory complications (these children are not represented in Table 1). Two had omphalocele.
Died at 14.2 years of age as a result of accidental trauma.
Dense nuclear cataracts develop rapidly between 2 and 6 months of age.
Two surviving children with CODAS syndrome have atrial septal defects; two who died perinatally had atrioventricular canal defects. The total incidence of congenital heart disease was 50%.
LONP1 c.2161C>G homozygotes had physical features characteristic of CODAS syndrome.1–4 These included broad skull and flattened midface, helix hypoplasia (“crumpled” ears), ptosis, grooved nasal tip, anteverted nares, and with advancing age, short stature, scoliosis, genu valgus, and pes valgus (Figures 1A–1F). Teeth erupted late with cusp-tip extensions. Dense bilateral nuclear cataracts developed rapidly between 2 and 6 months of age. Audiological testing showed impaired tympanic membrane mobility (type B pattern), normal otoacoustic emissions and neural synchrony, and low-frequency conductive hearing loss. The two oldest individuals had a mixed pattern involving mild-to-moderate sensory hearing loss in the 2–4 kHz range.
All affected children had hypotonia, developmental delay, and variable intellectual disability, some of which was remediable. One child with delayed ambulation was able to walk independently when knee-ankle-foot orthoses were applied at age 4 years. This same child had early audiological intervention (myringotomy tubes and amplification). By age 5, he had a vocabulary of more than 100 words, could read at the first grade level, could communicate effectively by signing, and had advanced drawing and writing skills (Figure 1G).
Neuroimaging of one affected child (age 4 years) was notable for prominent cortical sulci, symmetric ventriculomegaly, subcortical hypomyelination, and a thin corpus callosum (Figure 2A). Skeletal radiographs showed metaphyseal dysplasia (most evident at hip joints) and both hypoplasia and delayed ossification of epiphyses (Figure 2B). Two children had cervical radiographs that showed dens hypoplasia and, in one case, synostosis between the odontoid and C2. Scoliosis was evident within the first few years of life, and coronal clefts could be observed at various levels of the vertebral column (Figure 2C).
Two surviving LONP1 c.2161C>G homozygotes had an imperforate anus, in one case associated with omphalocele and in the other with a rectovaginal fistula. Omphalocele was also observed in two CODAS-affected siblings who died during infancy. One of two affected males had unilateral cryptorchidism. Two affected children had striking hemiatrophy of the tongue, presumably because of hypoplasia or aplasia of the ipsilateral hypoglossal nerve; both also had vocal cord paresis and chronic sialorrhea.
Given the diverse roles of Lon in mitochondrial homeostasis, we screened affected individuals for biochemical indices of mitochondrial dysfunction. Plasma lactate (1.4 ± 0.9 mM; reference range 1.9 ± 0.7 mM) and alanine (332 ± 150 μM; reference range 435 ± 121 μM)48 were normal in children with CODAS syndrome, as were all urine-tricarboxylic-acid-cycle intermediates, including citrate, alpha-ketoglutarate, succinate, fumarate, and malate49 (data not shown).
Prenatal Presentation of CODAS Syndrome
One unborn female Amish infant with CODAS syndrome had a prenatal ultrasound at 32 weeks’ gestational age. Estimated weight (1,516 g; 1st centile) and femur length (6.0 cm, 10th centile) were low; estimated head circumference was normal (32.1 cm; 93rd centile). The study was notable for the presence of polyhydramnios, a two-vessel umbilical cord, midfacial hypoplasia, crumpled helices, retrognathia, a balanced atrioventricular canal, omphalocele (containing intestine, stomach, and part of the liver), and mild thoracic scoliosis and the absence of left lower long bones, which were replaced by a relatively well-formed foot attached directly to the hip (Figures 2D–2G). She was stillborn at 39 weeks’ gestation.
Location of Amino Acids Altered in CODAS Syndrome within the Homology Model of Lon
The Lon holoenzyme is composed of six identical subunits that each consist of four domains: an MTS, a substrate recognition and binding (N) domain, an ATPase (AAA+) domain, and a proteolytic (P) domain (Figure 3A). The amino terminal MTS targets the newly synthesized cytosolic polypeptide to the mitochondrial matrix, where removal of the MTS produces mature Lon. The AAA+ domain contains Walker boxes A and B that mediate ATP binding and hydrolysis, and the P domain active site contains a Ser855-Lys896 dyad required for peptide-bond hydrolysis.50
Using the crystal structure of B. subtilis Lon as a template, we established a homology model of human mitochondrial Lon, which has a homohexameric ring-shaped arrangement.9 All four Lon variants identified in CODAS syndrome resulted in amino acid substitutions within the AAA+ domain. Ser631, Arg721, and Ala724 are located at the interface between Lon subunits and mediate subunit interactions. Pro676 is at the ATP-binding site (Figures 3A and 3B).
Replacements of Pro676 with serine (p.Pro676Ser) and Arg721with glycine (p.Arg721Gly) are pathogenic in the homozygous state (Table 1), allowing homo-oligomeric composition of the holoenzyme to be inferred. Pro676 is on an alpha-helix near the ATP-binding pocket and is involved in subdomain interactions (Figure 3C); it generates an alpha-helix kink that facilitates hydrophobic interactions between Leu667, Ala670, Leu696, and Val716. These interactions most likely maintain the geometry of the ATP-binding pocket. Arg721 contributes to intersubunit interactions by forming a salt bridge with Glu654 on the neighboring subunit (Figure 3D). Arg721 is near Glu717, which also forms a salt bridge with Lys517 in the AAA+ domain of the neighboring subunit (Figure 3D). The p.Arg721Gly substitution most likely weakens intersubunit interactions and reduces stability of the holoenzyme.
Recombinant CODAS Proteins Show Substrate-Specific Defects in Enzymatic Activity
Individual CODAS variants were purified as recombinant proteins from bacteria, and their respective peptidase, protease, and ATPase activities were compared with those of wild-type Lon. We measured ATP-dependent peptidase activity by using the fluorogenic decapeptide reporter substrate S3.51 Relative to wild-type protein, all four CODAS variants (p.Ser631Tyr, p.Pro676Ser, p.Arg721Gly, and p.Ala724Val) showed decreased energy-dependent peptidase activity; cleavage of S3 ranged from 19%–39% of wild-type activity (Figure 4A).
For the Amish p.Arg721Gly variant, we also determined ATP-independent peptidase activity by using the fluorescent dipeptide reporter AA2-Rh110, which is cleaved by wild-type Lon.39 In the absence of ATP, p.Arg721Gly and wild-type Lon showed significantly increased cleavage of AA2-Rh110. However, in the presence of ATP, peptidase activity of wild-type Lon was augmented 4-fold in comparison to minimal augmentation of p.Arg721Gly, and direct determination of ATPase function showed that p.Arg721Gly was 80% less active than wild-type Lon (data not shown).
Finally, we examined ATP-dependent proteolysis of p.Pro676Ser and p.Arg721Gly by using two purified recombinant natural Lon substrates: StAR19,44,45 and TFAM41,52 (Figures 4B and S1). Compared to wild-type Lon, which degraded 50%–60% StAR, p.Pro676Ser and p.Arg721Gly degraded only 10%–20% StAR during a 60 min incubation. In contrast, TFAM degradation by both p.Pro676Ser and p.Arg721Gly was comparable to that of the wild-type (50%–55% of TFAM was degraded during 60 min incubation)(Figure 4B). Control experiments confirmed that TFAM degradation was dependent on both ATP and Lon (Figures S1C and S1D). The efficiency with which p.Pro676Ser and p.Arg721Gly degrade different proteins is probably influenced by the substrate’s secondary or tertiary structure (i.e., whether the substrate is tightly or loosely folded). For example, folded and unfolded TFAM have been shown to be in equilibrium with one another.53 These results suggest that in CODAS syndrome, Lon-mediated degradation of some but not all protein substrates is impaired.
Impaired Homo-oligomerization of Transiently Expressed R721G
To characterize the behavior of CODAS variants in cultured cells, we engineered fusion proteins with C-terminal epitope tags to distinguish them from endogenous Lon. When transiently expressed in ARPE-19, UB/OC-2, or HeLa cells, CODAS and wild-type Lon were expressed at similar levels (Figures 4, S2, and S3). All four CODAS variants localized to mitochondria, as determined by double labeling with immunofluorescence and Mitotracker Red (Figures 4, S2, and S3).
To determine whether p.Arg721Gly was able to oligomerize with wild-type or other p.Arg721Gly Lon, we performed co-immunoprecipitation experiments by using recombinant proteins with different epitope tags (e.g., FLAG and V5 epitopes) (Figure 4E), and we transiently co-expressed p.Arg721GlyFLAG or wild-typeFLAG with either p.Arg721GlyV5 or wild-typeV5 in HEK293T cells. Both wild-typeFLAG and p.Arg721GlyFLAG co-precipitated wild-typeV5; p.Arg721GlyFLAG co-precipitated significantly less wild-typeV5 (Figure 4E). Co-immunoprecipitation of p.Arg721GlyV5 by p.Arg721GlyFLAG was barely detected, suggesting impaired homo-oligomeric assembly of p.Arg721Gly or disassembly of an unstable complex during the experimental procedure.
Normal mtDNA Copy Number and Lon Substrate Levels in Immortalized CODAS Lymphoblast Cell Lines
We generated EBV-immortalized B- LCLs from the peripheral blood of two Amish family trios, each consisting of a homozygous p.Arg721Gly/p.Arg721Gly proband and his or her unaffected heterozygous p.Arg721Gly/wt parents. No difference in LONP1 transcript abundance was observed by RT-PCR in proband versus parent cells (Figure S4). Immunoblot analysis of whole-cell extracts demonstrated that Lon protein abundance was virtually identical in cell lines from the proband, mother, and father (Figure 5A). There was no compensatory upregulation of ClpX or ClpP, subunits of another mitochondrial AAA+ protease, and we found no change in the abundance of mitochondrial aconitase or TFAM (Figure 5A), which are natural Lon substrates under conditions of oxidative stress or mtDNA depletion, respectively.13,41 Recent work suggests that Lon is involved in degrading the PTEN-induced putative kinase 1 (PINK1).54,55 However, an increase in PINK1 abundance was not detected in proband or parent cells, not even in the presence of proteasome inhibitors MG262 or epoxomicin (data not shown), which stabilize PINK1 in vitro.56,57
Figure 5.
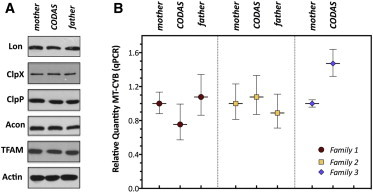
Lon Protein Levels and mtDNA Copy Numbers in CODAS-Syndrome-Affected Probands Are Similar to Those of Their Unaffected Heterozygous Parents
(A) Proteins were extracted from LCLs generated from CODAS-syndrome-affected probands and their parents. Triton X-100 lysis buffer was used, and 40 μg of the extract was immunoblotted for the proteins as shown.
(B) Quantitative PCR (qPCR) was performed so that the relative mtDNA copy number in CODAS-syndrome-affected probands and their respective mothers and fathers could be determined with genomic DNA isolated from the peripheral blood lymphocytes from the family members as shown. qPCR was performed with Taqman probes and primers for the mtDNA-encoded MT-CYB gene and the nuclear DNA-encoded APP gene as a reference. The relative quantity of mtDNA copy number was calculated by the ΔΔCt method. Error bars represent the maximum and minimum relative quantitation.
In lower and higher eukaryotes, knocking out Lon leads to reduced mtDNA copy-number or mtDNA deletions.22,23,58,59 We performed mtDNA copy-number analysis and mtDNA sequencing by using total genomic DNA isolated from peripheral blood lymphocytes obtained from three homozygous p.Arg721Gly probands and their unaffected heterozygous parents. Quantitative PCR showed no consistent difference of mtDNA copy number between probands and parents (Figure 5B). Similar results were obtained with genomic DNA isolated from proband and parental LCLs (Figure S5). Using genomic DNA isolated from peripheral blood lymphocytes for deep sequencing of mtDNA exposed no deletions or significant differences between mtDNA sequences of each proband and that individual’s mother (Table S1).
Abnormal Mitochondrial Morphology in CODAS Lymphoblast Cell Lines
Transmission electron microscopy of proband LCLs revealed enlarged mitochondria with swollen intra- or intercristal compartments, uniform vesicular structures, and electron-dense intramitochondrial inclusions (Figures 6A and 6C), suggestive of abnormal inner-membrane topology.60,61 By contrast, parental LCLs showed typical mitochondrial morphology with well-organized cristae and only occasional swollen cristal spaces (Figures 6B and 6D, and S6). Approximately 43 ± 3.4% (SEM) of mitochondria (n = 59) examined from two CODAS probands displayed abnormal mitochondrial morphology (p < 0.001), whereas only 5.2 ± 3.7% of paternal (n = 48), 14.1 ± 3.3% of maternal (n = 63), and 10.6 ± 5.1% (n = 326) of homozygous-normal LCL mitochondria were abnormal. There was no significant difference between maternal and paternal mitochondria (p = 0.232).
Figure 6.
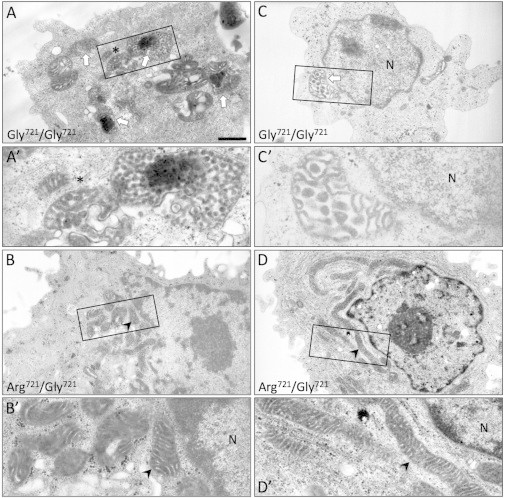
Abnormal Mitochondrial Ultrastructure in CODAS Cells
(A–D) Transmission electron micrographs were generated from cell lines of (A and C) two CODAS-syndrome-affected children homozygous for p.Arg721Gly (Gly721/Gly721) and (B and D, respectively) their heterozygous unaffected mothers (Arg721/Gly721). (A and C) In CODAS cells, 43% of mitochondria (n = 59 cells, p < 0.001) display abnormal morphology, including swollen intracristal space and/or vesiculated cristae and electron-dense aggregations (white arrows). The mitochondrion below the asterisk in (A′) appears to be in transition toward the vesiculated state when it is compared to the normal mitochondrion to the left of the asterisk. (B and D) The mothers’ cells display normal mitochondrial morphology (e.g., arrowheads); 14% show swelling of the intracristal space (n = 63 cells, p < 0.001) (N = nucleus). Scale bars represent 1 μm in (A–D) and 0.3 μm in magnified regions (A′–D′). The fathers’ cells show normal mitochondrial morphology, and only 5.2% show cristal-space swelling (n = 48) (Figure S6).
Reduced MT-CO2 and SRC in CODAS Mitochondria
Lon has been shown to mediate protein quality control and remodeling of cytochrome c oxidase, which is complex IV of the mitochondrial respiratory chain.16,17,21 We examined the abundance of various protein subunits belonging to respiratory chain complexes I-V and detected a specific and substantial reduction of mtDNA-encoded subunit II of cytochrome c oxidase (MT-CO2) in proband cells (Figure 7A). In contrast, proband and parental LCLs had similar abundances of other mtDNA-encoded (MT-CYB, MT-CO1) and nuclear-encoded (NDUFB8, SDHB, UQCRC2, ATP5A1) respiratory chain subunits.
Figure 7.
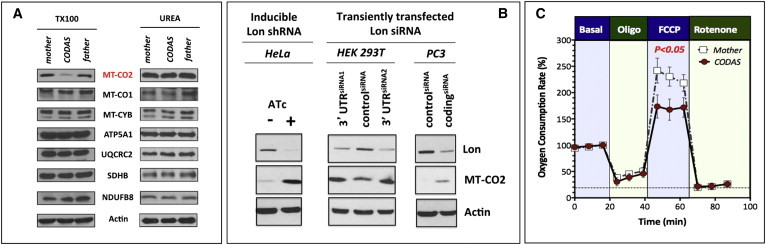
Alterations in the Structure and Function of the Electron Transport Chain in Proband LCLs
(A) For extraction of cellular proteins, lysis buffer containing 0.1% Triton X-100 (TX100) or 8 M urea (UREA) was used. Immunoblotting was performed for proteins as shown. MT-CO2 abundances were selectively reduced in CODAS cells but were recovered in the presence of 8 M urea.
(B) MT-CO2 is a protein substrate of mitochondrial Lon. A HeLa cell line with a stably integrated anhydrotetracycline (ATc)-inducible shRNA targeting Lon was grown in the presence or absence of ATc (500 ng/ml) for 4 days. HEK293T cells and PC3 cells were transiently transfected with a siRNA targeting the 3′ untranslated regionof Lon or a control siRNA and cultured for 3 days. Cell extracts (25 μg) were immunoblotted for Lon, MT-CO2, or actin as the loading control.
(C) Basal normalized oxygen-consumption rate was established at 18 min (representing 100%), followed by addition of oligomycin (Oligo, 1 μM); FCCP (750 nM); and finally rotenone (1 μM). A Student’s t test showed that cells from CODAS-syndrome-affected probands had significantly (p < 0.05) lower spare mitochondrial respiratory capacity in the presence of the uncoupler FCCP. Error bars represent ±SD.
One possible explanation for reduced MT-CO2 abundance in CODAS cell lines was its poor solubility in the protein extraction buffer employed, which contained 0.5% Triton X-100. To address this, we used cell-lysis buffer with 8 M urea, which effectively solubilizes most aggregated proteins and inclusion bodies. When cells were extracted with this strong denaturant, similar abundances of MT-CO2 were observed in proband and parental LCLs. Urea did not alter solubility of other mtDNA- or nuclear-encoded respiratory-chain subunits (Figure 7A).
Insolubility of MT-CO2 might be caused by the failure of p.Arg721Gly to properly degrade and/or assemble this subunit into complex IV. To determine whether MT-CO2 is a natural substrate of Lon, we knocked down the protease in different cell lines. MT-CO2 is highly hydrophobic and difficult to purify; we therefore used a cellular system rather than directly testing purified Lon activity against that of MT-CO2. Inducible or transient knockdown of Lon was associated with increased MT-CO2 abundance in HeLa, HEK293T, and PC3 cells (Figure 7B). Taken together, these results suggest that MT-CO2 is a natural Lon substrate, and that p.Arg721Gly’s failure either to degrade or to properly assemble this protein in CODAS cells results in its aggregation within mitochondria.
After observing the reduced solubility of MT-CO2, we investigated the efficiency of mitochondrial respiration. Normalized basal mitochondrial oxygen consumption and ATP-linked respiration were similar in proband and parental cells, but CODAS cells had significantly lower SRC when mitochondrial membrane potential was dissipated by the uncoupler FCCP (Figure 7C).
Discussion
Pathophysiology of CODAS Syndrome
Mitochondrial Lon is a multi-functional enzyme with diverse roles that include (1) elimination of misfolded and oxidatively damaged proteins,12–14,20,62 (2) selective degradation of key rate-determining mitochondrial proteins in response to metabolic flux and cell stress,16–18,58,63 (3) chaperone-like assembly of protein complexes within the respiratory chain,21,64 and (4) regulation of mitochondrial gene expression.46 Homozygous loss of Lon in mice results in embryonic death associated with poor growth and reduced mtDNA copy number, indicating the vital importance of this protein in mammals.15 In contrast, heterozygous mice mature similarly to wild-type.15 These findings, combined with our enzymatic studies, support the idea that allele combinations giving rise to CODAS syndrome must result in alteration and not complete abrogation of Lon function.
We speculate that the complex pathophysiology of CODAS syndrome most likely reflects dysregulation of multiple Lon-dependent proteins and pathways. There are clinical similarities between CODAS syndrome and other mitochondrial disorders, including hypotonia, motor delay, ptosis, and sensorineural hearing loss.48,65–67 However, many classic clinical and biochemical hallmarks48,67,68 of mitochondrial dysfunction were not observed in CODAS children, and several CODAS manifestations, including postnatal cataracts, skeletal and dental abnormalities, and conductive hearing loss, are atypical of mitochondrial disease. Such complications might reflect Lon-dependent processes that are currently unknown. Moreover, tissue-specific defects could arise because of differential histological dependence on Lon as a protease, a chaperone, and/or a DNA-binding protein. For example, some tissues might be more dependent on Lon to degrade oxidatively damaged proteins, whereas others might depend critically on as-yet-unknown Lon-mediated pathways. The reduced SRC of CODAS LCLs suggests limited capacity to boost ATP production in response to high energy demands, such as occurs during increased cellular stress and work load. Further investigations using tissues with metabolic demands higher than those of lymphoblastoid cells are needed if we are to delineate the physiological relevance of mechanistic defects in mitochondrial function and morphology associated with CODAS syndrome.
Pathogenic Amino Acid Changes in CODAS Syndrome Cluster in the AAA+ Domain
Our structural analysis of homozygous CODAS variants is consistent with observed alterations of their enzyme activity in vitro. Proline676 is located within an α helix near the ATP-binding pocket and generates a kink in this α helix69 (Figure 3C). Removal of the kink at Pro676 might alter key hydrophobic interactions with four other residues—Ala670 (on the same helix as Pro676), Leu667, Leu696, and Val716—that likely help to shape the active site (Figure 3C). The p.Pro676Ser substitution could disrupt this kink, changing active site geometry or flexibility of the AAA+ module (Figure 3C) and decreasing the protein’s capacity for ATP binding and hydrolysis. Accordingly, we found reduced ATP-dependent peptidase (Figure 4A) and substrate-specific protease (Figure 4B) activities of the p.Pro676Ser variant.
Arg721 also makes an important intersubunit contact by forming a salt bridge with Glu654 from the AAA+ module of the neighboring subunit (Figure 3D). This salt bridge, together with one formed between Glu717 and Lys517, appears to be critical for stabilizing the Lon complex. p.Arg721Gly might thus destabilize the Lon complex and explain reduced homo-oligomerization, ATPase activity, and peptidase activity of this variant (Figure 4).
Functional Consequences of Homozygous Lon p.Arg721Gly in Human Cells
Our studies using recombinant protein and CODAS LCLs indicate that the p.Arg721Gly pathogenic variant has altered activity. Purified p.Arg721Gly has reduced ATPase and peptidase activities and degrades some natural substrates (e.g., TFAM) but not others (e.g., StAR) (Figure 4B). Substrate-restricted protease activity might reflect poor conformational resilience or stability of the p.Arg721Gly holoenzyme, as suggested by co-immunoprecipitation experiments (Figure 4E).
In this study, we show that MT-CO2 is upregulated when Lon is knocked down in three different cell types, suggesting that MT-CO2 is a Lon substrate. In CODAS cells, we observed insolubility and aggregation of MT-CO2 (Figure 7A) but not MT-CO1, which is similarly hydrophobic. Previous work shows that wild-type Lon plays a role in the assembly of the cytochrome c oxidase complex and in its remodeling the during adaptive switching between normoxic and hypoxic conditions.16,17,21 Work in yeast and mammalian cells suggests that Lon promotes cytochrome c oxidase assembly through a chaperone function independent of its protease activity.16,21
The failure of p.Arg721Gly to degrade or chaperone MT-CO2 (and possibly other inner-membrane proteins yet to be identified) might promote protein aggregation and explain the morphological and bioenergetic changes observed in CODAS cell mitochondria (Figures 6 and 7). In this study, we examined immortalized B-LCLs generated from CODAS-syndrome-affected probands and their parents. Because this cell type is not principally affected in CODAS syndrome, future studies are aimed at employing primary cells and tissue samples as well as cell lines engineered with specific CODAS LONP1 mutations.
Natural History and Treatment of CODAS Syndrome
The high allele frequency (MAF = 5.9%, carrier frequency = 11.8%) of LONP1 c.2161C>G among the Amish was unexpected. Of all the known pathogenic alleles in the Lancaster County Amish population, the LONP1 variant has the second-highest carrier frequency, eclipsed only by that of a founder mutation for Ellis-van Creveld syndrome (EVC; MIM 225500; RefSeq NM_153717.2); c.1886+5G>T; carrier frequency = 16.2%). The eight known Amish CODAS-syndrome-affected individuals comprise six sibships. In two sibships, three affected children died shortly after birth. One affected fetus was stillborn. Given the high allele frequency and perinatal mortality, the Amish CODAS variant might result in a high rate of fetal demise. This could explain an apparent discrepancy between the relatively low birth incidence of CODAS syndrome and the high allele frequency of LONP1 c.2161C>G in the Amish population.
Accurate prenatal diagnosis of CODAS syndrome has important clinical implications (Figure 2). Newborns with CODAS syndrome are likely to struggle through the neonatal transition. For syndrome variants (e.g., p.Arg721Gly) associated with neonatal vocal-cord paresis, the ability to anticipate and prevent perinatal asphyxiation can be life saving. Three of eight (38%) Amish CODAS-syndrome-affected individuals died of airway complications shortly after birth, and one was stillborn. Surviving children required intubation, mechanical respiratory support, and tracheostomy. Incomplete cardiac septation occurred in 50% of p.Arg721Gly homozygotes and manifested in two cases (25%) as a complete atrioventricular defect. Thus, supportive cardiopulmonary care for babies with a suspected or confirmed diagnosis of CODAS syndrome requires tertiary-level subspecialty services and must begin immediately after birth. A careful, staged approach to subsequent management can allow for relatively good cognitive outcome and for independence in affected individuals.
Acknowledgments
We thank Kimberly Jacob, Prashant Patel, Ana Quiroz, and Jennie Silver for technical assistance and D. Streck from the Molecular Genetics Laboratory at the Rutgers New Jersey Medical School for next-generation mtDNA sequencing. R.N.J., E.G.P., and K.A.S. were supported by Howard Hughes Medical Institute undergraduate science education awards 52006294 and 52007538. R.N.J. was also supported by the Center for Research on Women and Newborn Health and by ConnectCare3. I.L. was supported by National Science Foundation grant CHE-1213175. C.K.S. was supported in part by National Institutes of Health grants R01GM084039 and 1R21NS067668 and also by the NJ Health Science Foundation. The Clinic for Special Children is supported by charitable donations from the communities it serves.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
I-Tasser Online Protein Structure and Function Predictions, Zhang Laboratory, University of Michigan, http://zhanglab.ccmb.med.umich.edu/I-TASSER/
Modeller, https://salilab.org/modeller/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
RefSeq: NCBI Reference Sequence Database, http://www.ncbi.nlm.nih.gov/refseq/
Sali Laboratory, University of California San Francisco, http://salilab.org/index.html
SAMtools, http://samtools.sourceforge.net/
References
- 1.Shebib S.M., Reed M.H., Shuckett E.P., Cross H.G., Perry J.B., Chudley A.E. Newly recognized syndrome of cerebral, ocular, dental, auricular, skeletal anomalies: CODAS syndrome—a case report. Am. J. Med. Genet. 1991;40:88–93. doi: 10.1002/ajmg.1320400118. [DOI] [PubMed] [Google Scholar]
- 2.de Almeida J.C., Vargas F.R., Barbosa-Neto J.G., Llerena J.C., Jr. CODAS syndrome: a new distinct MCA/MR syndrome with radiological changes of spondyloepiphyseal dysplasia. Another case report. Am. J. Med. Genet. 1995;55:19–20. doi: 10.1002/ajmg.1320550107. [DOI] [PubMed] [Google Scholar]
- 3.Innes A.M., Chudley A.E., Reed M.H., Shuckett E.P., Hildes-Ripstein G.E., Greenberg C.R. Third case of cerebral, ocular, dental, auricular, skeletal anomalies (CODAS) syndrome, further delineating a new malformation syndrome: first report of an affected male and review of literature. Am. J. Med. Genet. 2001;102:44–47. doi: 10.1002/1096-8628(20010722)102:1<44::aid-ajmg1410>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 4.Marlin S., Ducou Le Pointe H., Le Merrer M., Portnoi M.F., Chantot S., Jonard L., Mantel-Guiochon A., Siffroi J.P., Garabedian E.N., Denoyelle F. Fourth case of cerebral, ocular, dental, auricular, skeletal syndrome (CODAS), description of new features and molecular analysis. Am. J. Med. Genet. A. 2010;152A:1510–1514. doi: 10.1002/ajmg.a.33242. [DOI] [PubMed] [Google Scholar]
- 5.Lee I., Suzuki C.K. Functional mechanics of the ATP-dependent Lon protease- lessons from endogenous protein and synthetic peptide substrates. Biochim. Biophys. Acta. 2008;1784:727–735. doi: 10.1016/j.bbapap.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanson P.I., Whiteheart S.W. AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 7.Neuwald A.F., Aravind L., Spouge J.L., Koonin E.V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 8.Truscott K.N., Lowth B.R., Strack P.R., Dougan D.A. Diverse functions of mitochondrial AAA+ proteins: protein activation, disaggregation, and degradation. Biochem. Cell Biol. 2010;88:97–108. doi: 10.1139/o09-167. [DOI] [PubMed] [Google Scholar]
- 9.Venkatesh S., Lee J., Singh K., Lee I., Suzuki C.K. Multitasking in the mitochondrion by the ATP-dependent Lon protease. Biochim. Biophys. Acta. 2012;1823:56–66. doi: 10.1016/j.bbamcr.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jenkinson E.M., Rehman A.U., Walsh T., Clayton-Smith J., Lee K., Morell R.J., Drummond M.C., Khan S.N., Naeem M.A., Rauf B., University of Washington Center for Mendelian Genomics Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 2013;92:605–613. doi: 10.1016/j.ajhg.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casari G., De Fusco M., Ciarmatori S., Zeviani M., Mora M., Fernandez P., De Michele G., Filla A., Cocozza S., Marconi R. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–983. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 12.Bota D.A., Van Remmen H., Davies K.J. Modulation of Lon protease activity and aconitase turnover during aging and oxidative stress. FEBS Lett. 2002;532:103–106. doi: 10.1016/s0014-5793(02)03638-4. [DOI] [PubMed] [Google Scholar]
- 13.Bota D.A., Davies K.J. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol. 2002;4:674–680. doi: 10.1038/ncb836. [DOI] [PubMed] [Google Scholar]
- 14.Ngo J.K., Pomatto L.C., Davies K.J. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 2013;1:258–264. doi: 10.1016/j.redox.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quiros P.M., Espanol Y., Acin-Perez R., Rodriguez F., Barcena C., Watanabe K., Calvo E., Loureiro M., Fernandez-Garcia M.S., Fueyo A. ATP-dependent Lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell Rep. 2014;8:542–556. doi: 10.1016/j.celrep.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 16.Hori O., Ichinoda F., Tamatani T., Yamaguchi A., Sato N., Ozawa K., Kitao Y., Miyazaki M., Harding H.P., Ron D. Transmission of cell stress from endoplasmic reticulum to mitochondria: enhanced expression of Lon protease. J. Cell Biol. 2002;157:1151–1160. doi: 10.1083/jcb.200108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukuda R., Zhang H., Kim J.W., Shimoda L., Dang C.V., Semenza G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 18.Tian Q., Li T., Hou W., Zheng J., Schrum L.W., Bonkovsky H.L. Lon peptidase 1 (LONP1)-dependent breakdown of mitochondrial 5-aminolevulinic acid synthase protein by heme in human liver cells. J. Biol. Chem. 2011;286:26424–26430. doi: 10.1074/jbc.M110.215772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Granot Z., Kobiler O., Melamed-Book N., Eimerl S., Bahat A., Lu B., Braun S., Maurizi M.R., Suzuki C.K., Oppenheim A.B., Orly J. Turnover of mitochondrial steroidogenic acute regulatory (StAR) protein by Lon protease: the unexpected effect of proteasome inhibitors. Mol. Endocrinol. 2007;21:2164–2177. doi: 10.1210/me.2005-0458. [DOI] [PubMed] [Google Scholar]
- 20.Wohlever M.L., Baker T.A., Sauer R.T. Roles of the N domain of the AAA+ Lon protease in substrate recognition, allosteric regulation and chaperone activity. Mol. Microbiol. 2014;91:66–78. doi: 10.1111/mmi.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rep M., van Dijl J.M., Suda K., Schatz G., Grivell L.A., Suzuki C.K. Promotion of mitochondrial membrane complex assembly by a proteolytically inactive yeast Lon. Science. 1996;274:103–106. doi: 10.1126/science.274.5284.103. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki C.K., Suda K., Wang N., Schatz G. Requirement for the yeast gene LON in intramitochondrial proteolysis and maintenance of respiration. Science. 1994;264:273–276. doi: 10.1126/science.8146662. [DOI] [PubMed] [Google Scholar]
- 23.Van Dyck L., Pearce D.A., Sherman F. PIM1 encodes a mitochondrial ATP-dependent protease that is required for mitochondrial function in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1994;269:238–242. [PubMed] [Google Scholar]
- 24.Lu B., Liu T., Crosby J.A., Thomas-Wohlever J., Lee I., Suzuki C.K. The ATP-dependent Lon protease of Mus musculus is a DNA-binding protein that is functionally conserved between yeast and mammals. Gene. 2003;306:45–55. doi: 10.1016/s0378-1119(03)00403-7. [DOI] [PubMed] [Google Scholar]
- 25.Fu G.K., Markovitz D.M. The human LON protease binds to mitochondrial promoters in a single-stranded, site-specific, strand-specific manner. Biochemistry. 1998;37:1905–1909. doi: 10.1021/bi970928c. [DOI] [PubMed] [Google Scholar]
- 26.Liu T., Lu B., Lee I., Ondrovicová G., Kutejová E., Suzuki C.K. DNA and RNA binding by the mitochondrial lon protease is regulated by nucleotide and protein substrate. J. Biol. Chem. 2004;279:13902–13910. doi: 10.1074/jbc.M309642200. [DOI] [PubMed] [Google Scholar]
- 27.Chen S.H., Suzuki C.K., Wu S.H. Thermodynamic characterization of specific interactions between the human Lon protease and G-quartet DNA. Nucleic Acids Res. 2008;36:1273–1287. doi: 10.1093/nar/gkm1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lohr N.J., Molleston J.P., Strauss K.A., Torres-Martinez W., Sherman E.A., Squires R.H., Rider N.L., Chikwava K.R., Cummings O.W., Morton D.H., Puffenberger E.G. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am. J. Hum. Genet. 2010;86:447–453. doi: 10.1016/j.ajhg.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puffenberger E.G., Hu-Lince D., Parod J.M., Craig D.W., Dobrin S.E., Conway A.R., Donarum E.A., Strauss K.A., Dunckley T., Cardenas J.F. Mapping of sudden infant death with dysgenesis of the testes syndrome (SIDDT) by a SNP genome scan and identification of TSPYL loss of function. Proc. Natl. Acad. Sci. USA. 2004;101:11689–11694. doi: 10.1073/pnas.0401194101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puffenberger E.G., Strauss K.A., Ramsey K.E., Craig D.W., Stephan D.A., Robinson D.L., Hendrickson C.L., Gottlieb S., Ramsay D.A., Siu V.M. Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7-kilobase deletion in LYK5. Brain. 2007;130:1929–1941. doi: 10.1093/brain/awm100. [DOI] [PubMed] [Google Scholar]
- 31.Sherman E.A., Strauss K.A., Tortorelli S., Bennett M.J., Knerr I., Morton D.H., Puffenberger E.G. Genetic mapping of glutaric aciduria, type 3, to chromosome 7 and identification of mutations in c7orf10. Am. J. Hum. Genet. 2008;83:604–609. doi: 10.1016/j.ajhg.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strauss K.A., Puffenberger E.G., Bunin N., Rider N.L., Morton M.C., Eastman J.T., 3rd, Morton D.H. Clinical application of DNA microarrays: molecular diagnosis and HLA matching of an Amish child with severe combined immune deficiency. Clin. Immunol. 2008;128:31–38. doi: 10.1016/j.clim.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 33.Strauss K.A., Puffenberger E.G., Craig D.W., Panganiban C.B., Lee A.M., Hu-Lince D., Stephan D.A., Morton D.H. Genome-wide SNP arrays as a diagnostic tool: clinical description, genetic mapping, and molecular characterization of Salla disease in an Old Order Mennonite population. Am. J. Med. Genet. A. 2005;138A:262–267. doi: 10.1002/ajmg.a.30961. [DOI] [PubMed] [Google Scholar]
- 34.Gnirke A., Melnikov A., Maguire J., Rogov P., LeProust E.M., Brockman W., Fennell T., Giannoukos G., Fisher S., Russ C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puffenberger E.G., Jinks R.N., Sougnez C., Cibulskis K., Willert R.A., Achilly N.P., Cassidy R.P., Fiorentini C.J., Heiken K.F., Lawrence J.J. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS ONE. 2012;7:e28936. doi: 10.1371/journal.pone.0028936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puffenberger E.G., Jinks R.N., Wang H., Xin B., Fiorentini C., Sherman E.A., Degrazio D., Shaw C., Sougnez C., Cibulskis K. A homozygous missense mutation in HERC2 associated with global developmental delay and autism spectrum disorder. Hum. Mutat. 2012;33:1639–1646. doi: 10.1002/humu.22237. [DOI] [PubMed] [Google Scholar]
- 37.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duman R.E., Löwe J. Crystal structures of Bacillus subtilis Lon protease. J. Mol. Biol. 2010;401:653–670. doi: 10.1016/j.jmb.2010.06.030. [DOI] [PubMed] [Google Scholar]
- 39.Bernstein S.H., Venkatesh S., Li M., Lee J., Lu B., Hilchey S.P., Morse K.M., Metcalfe H.M., Skalska J., Andreeff M. The mitochondrial ATP-dependent Lon protease: a novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood. 2012;119:3321–3329. doi: 10.1182/blood-2011-02-340075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee I., Berdis A.J. Adenosine triphosphate-dependent degradation of a fluorescent lambda N substrate mimic by Lon protease. Anal. Biochem. 2001;291:74–83. doi: 10.1006/abio.2001.4988. [DOI] [PubMed] [Google Scholar]
- 41.Lu B., Lee J., Nie X., Li M., Morozov Y.I., Venkatesh S., Bogenhagen D.F., Temiakov D., Suzuki C.K. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol. Cell. 2013;49:121–132. doi: 10.1016/j.molcel.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 43.Pace C.N., Vajdos F., Fee L., Grimsley G., Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ondrovicová G., Liu T., Singh K., Tian B., Li H., Gakh O., Perecko D., Janata J., Granot Z., Orly J. Cleavage site selection within a folded substrate by the ATP-dependent lon protease. J. Biol. Chem. 2005;280:25103–25110. doi: 10.1074/jbc.M502796200. [DOI] [PubMed] [Google Scholar]
- 45.Fishovitz J., Li M., Frase H., Hudak J., Craig S., Ko K., Berdis A.J., Suzuki C.K., Lee I. Active-site-directed chemical tools for profiling mitochondrial Lon protease. ACS Chem. Biol. 2011;6:781–788. doi: 10.1021/cb100408w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu B., Yadav S., Shah P.G., Liu T., Tian B., Pukszta S., Villaluna N., Kutejová E., Newlon C.S., Santos J.H., Suzuki C.K. Roles for the human ATP-dependent Lon protease in mitochondrial DNA maintenance. J. Biol. Chem. 2007;282:17363–17374. doi: 10.1074/jbc.M611540200. [DOI] [PubMed] [Google Scholar]
- 47.di Sant’Agnese P.A., De Mesy-Jensen K.L. Diagnostic electron microscopy on reembedded (“popped off”) areas of large Spurr epoxy sections. Ultrastruct. Pathol. 1984;6:247–253. doi: 10.3109/01913128409018580. [DOI] [PubMed] [Google Scholar]
- 48.DiMauro S., Schon E.A., Carelli V., Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. 2013;9:429–444. doi: 10.1038/nrneurol.2013.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barshop B.A. Metabolomic approaches to mitochondrial disease: correlation of urine organic acids. Mitochondrion. 2004;4:521–527. doi: 10.1016/j.mito.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 50.Botos I., Melnikov E.E., Cherry S., Tropea J.E., Khalatova A.G., Rasulova F., Dauter Z., Maurizi M.R., Rotanova T.V., Wlodawer A., Gustchina A. The catalytic domain of Escherichia coli Lon protease has a unique fold and a Ser-Lys dyad in the active site. J. Biol. Chem. 2004;279:8140–8148. doi: 10.1074/jbc.M312243200. [DOI] [PubMed] [Google Scholar]
- 51.Thomas-Wohlever J., Lee I. Kinetic characterization of the peptidase activity of Escherichia coli Lon reveals the mechanistic similarities in ATP-dependent hydrolysis of peptide and protein substrates. Biochemistry. 2002;41:9418–9425. doi: 10.1021/bi0255470. [DOI] [PubMed] [Google Scholar]
- 52.Matsushima Y., Goto Y., Kaguni L.S. Mitochondrial Lon protease regulates mitochondrial DNA copy number and transcription by selective degradation of mitochondrial transcription factor A (TFAM) Proc. Natl. Acad. Sci. USA. 2010;107:18410–18415. doi: 10.1073/pnas.1008924107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong T.S., Rajagopalan S., Freund S.M., Rutherford T.J., Andreeva A., Townsley F.M., Petrovich M., Fersht A.R. Biophysical characterizations of human mitochondrial transcription factor A and its binding to tumor suppressor p53. Nucleic Acids Res. 2009;37:6765–6783. doi: 10.1093/nar/gkp750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin S.M., Youle R.J. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013;9:1750–1757. doi: 10.4161/auto.26122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomas R.E., Andrews L.A., Burman J.L., Lin W.Y., Pallanck L.J. PINK1-Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS Genet. 2014;10:e1004279. doi: 10.1371/journal.pgen.1004279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Um J.W., Stichel-Gunkel C., Lübbert H., Lee G., Chung K.C. Molecular interaction between parkin and PINK1 in mammalian neuronal cells. Mol. Cell. Neurosci. 2009;40:421–432. doi: 10.1016/j.mcn.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 57.Lin W., Kang U.J. Characterization of PINK1 processing, stability, and subcellular localization. J. Neurochem. 2008;106:464–474. doi: 10.1111/j.1471-4159.2008.05398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quirós P.M., Español Y., Acín-Pérez R., Rodríguez F., Bárcena C., Watanabe K., Calvo E., Loureiro M., Fernández-García M.S., Fueyo A. ATP-dependent Lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell Rep. 2014;8:542–556. doi: 10.1016/j.celrep.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 59.Guha S., López-Maury L., Shaw M., Bähler J., Norbury C.J., Agashe V.R. Transcriptional and cellular responses to defective mitochondrial proteolysis in fission yeast. J. Mol. Biol. 2011;408:222–237. doi: 10.1016/j.jmb.2011.02.044. [DOI] [PubMed] [Google Scholar]
- 60.Mannella C.A. Structure and dynamics of the mitochondrial inner membrane cristae. Biochim. Biophys. Acta. 2006;1763:542–548. doi: 10.1016/j.bbamcr.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 61.Scorrano L., Ashiya M., Buttle K., Weiler S., Oakes S.A., Mannella C.A., Korsmeyer S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 62.Smakowska E., Czarna M., Janska H. Mitochondrial ATP-dependent proteases in protection against accumulation of carbonylated proteins. Mitochondrion. 2014;19(Pt B):245–251. doi: 10.1016/j.mito.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 63.Teng H., Wu B., Zhao K., Yang G., Wu L., Wang R. Oxygen-sensitive mitochondrial accumulation of cystathionine β-synthase mediated by Lon protease. Proc. Natl. Acad. Sci. USA. 2013;110:12679–12684. doi: 10.1073/pnas.1308487110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Solheim C., Li L., Hatzopoulos P., Millar A.H. Loss of Lon1 in Arabidopsis changes the mitochondrial proteome leading to altered metabolite profiles and growth retardation without an accumulation of oxidative damage. Plant Physiol. 2012;160:1187–1203. doi: 10.1104/pp.112.203711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Di Fonzo A., Ronchi D., Lodi T., Fassone E., Tigano M., Lamperti C., Corti S., Bordoni A., Fortunato F., Nizzardo M. The mitochondrial disulfide relay system protein GFER is mutated in autosomal-recessive myopathy with cataract and combined respiratory-chain deficiency. Am. J. Hum. Genet. 2009;84:594–604. doi: 10.1016/j.ajhg.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hirano M., Nishigaki Y., Martí R. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a disease of two genomes. Neurologist. 2004;10:8–17. doi: 10.1097/01.nrl.0000106919.06469.04. [DOI] [PubMed] [Google Scholar]
- 67.Koopman W.J., Distelmaier F., Smeitink J.A., Willems P.H. OXPHOS mutations and neurodegeneration. EMBO J. 2013;32:9–29. doi: 10.1038/emboj.2012.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Breuer M.E., Koopman W.J., Koene S., Nooteboom M., Rodenburg R.J., Willems P.H., Smeitink J.A. The role of mitochondrial OXPHOS dysfunction in the development of neurologic diseases. Neurobiol. Dis. 2013;51:27–34. doi: 10.1016/j.nbd.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 69.Wilman H.R., Shi J., Deane C.M. Helix kinks are equally prevalent in soluble and membrane proteins. Proteins. 2014;82:1960–1970. doi: 10.1002/prot.24550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.