

Abstract
Nephronophthisis-related ciliopathies (NPHP-RC) are recessive diseases characterized by renal dysplasia or degeneration. We here identify mutations of DCDC2 as causing a renal-hepatic ciliopathy. DCDC2 localizes to the ciliary axoneme and to mitotic spindle fibers in a cell-cycle-dependent manner. Knockdown of Dcdc2 in IMCD3 cells disrupts ciliogenesis, which is rescued by wild-type (WT) human DCDC2, but not by constructs that reflect human mutations. We show that DCDC2 interacts with DVL and DCDC2 overexpression inhibits β-catenin-dependent Wnt signaling in an effect additive to Wnt inhibitors. Mutations detected in human NPHP-RC lack these effects. A Wnt inhibitor likewise restores ciliogenesis in 3D IMCD3 cultures, emphasizing the importance of Wnt signaling for renal tubulogenesis. Knockdown of dcdc2 in zebrafish recapitulates NPHP-RC phenotypes, including renal cysts and hydrocephalus, which is rescued by a Wnt inhibitor and by WT, but not by mutant, DCDC2. We thus demonstrate a central role of Wnt signaling in the pathogenesis of NPHP-RC, suggesting an avenue for potential treatment of NPHP-RC.
Introduction
Cilia are microtubule-based structures that project from the cell surface of numerous mammalian cells and mediate key pathways of development such as Wnt and Shh signaling. Many ciliary proteins participate in functional protein subcomplexes,1 which localize to primary cilia, centrosomes, the mitotic spindle, or the abscission structure in a cell-cycle-dependent manner.2 Mutations in genes that encode these ciliary proteins lead to developmental and degenerative diseases, which show a broad phenotypic spectrum. This complex of disorders has collectively been termed “ciliopathies” due to their localization to primary cilia and centrosomes.3 Moreover, numerous cilia-mediated signaling pathways have been implicated in the pathogenesis of ciliopathies, such as canonical Wnt/β-catenin,4 Shh,5 and DNA damage response (DDR).6,7
In this context, the term nephronophthisis-related ciliopathies (NPHP-RC) summarizes a group of rare autosomal-recessive cystic kidney diseases including nephronophthisis (NPHP [MIM 256100]), Senior-Loken syndrome (SLS [MIM 266900]), Joubert syndrome (JBTS [MIM 213300]), and Meckel Gruber syndrome (MKS [MIM 249000]). NPHP accounts for the majority of genetically caused end-stage renal disease (ESRD) during the first three decades of life. The most prominent diagnostic hallmarks are increased echogenicity and the presence of corticomedullary cysts upon renal ultrasound. Histologically, NPHP is characterized by tubular atrophy, basement membrane disintegration, interstitial fibrosis, and cyst formation. About 15% of the affected individuals with NPHP-RC exhibit extrarenal organ involvement, in particular hepatobiliary ductal plate malformation, progressive retinal dystrophy, and cerebellar vermis hypoplasia/aplasia as the hallmark of JBTS. The most severe manifestation of the NPHP-RC spectrum is MKS, a perinatally lethal ciliopathy.
Similar to the vast pleiotropy, NPHP-RC show broadly heterogeneous genotypes with monogenic ciliopathies established to be caused by recessive mutations in more than 90 genes.3–5,8–15 We recently demonstrated in a large cohort of 1,056 families with NPHP-RC that in more than 50% of cases the underlying disease-causing mutation is unknown.16,17 In order to identify additional single gene causes of NHPH-RC, we here applied homozygosity mapping and whole-exome sequencing in 100 affected individuals of consanguineous parents or sibling cases fulfilling the diagnostic criteria of NPHP-RC. In combination with high-throughput exon sequencing in a cohort of 800 individuals with NPHP-RC, we identified recessive truncating mutations in DCDC2 as a hitherto unknown cause of renal-hepatic variant of NPHP-RC. We demonstrate that DCDC2 interacts with the mediator of Wnt signaling dishevelled, and that DCDC2 overexpression inhibits β-catenin-dependent Wnt signaling. Thus, we demonstrate a central role of Wnt signaling in the pathogenesis of NPHP-RC, suggesting an avenue for potential treatment of NPHP-RC.
Material and Methods
Research Subjects
Blood samples and pedigrees were obtained from individuals with diagnosed NPHP-RC and informed consent. Approval for human subject research was obtained from Institutional Review Boards of the University of Michigan and the Boston Children’s Hospital.
Linkage Analysis
For genome-wide homozygosity mapping GeneChip® Human Mapping 250k StyI Array (Affymetrix) was used. Regions of homozygosity were identified using GENEHUNTER 2.118,19 and ALLEGRO20 with a disease allele frequency of 0.0001 and marker allele frequencies of European descent.12,21
Whole-Exome Sequencing
Whole exome sequencing (WES) and variant burden analysis was performed as previously described22 using Agilent SureSelect human exome capture arrays (Life Technologies™) with next generation sequencing (NGS) on an Illumina™ sequencing platform. Sequence reads were mapped against the human reference genome (NCBI build 36/hg18) using CLC Genomics Workbench (version 4.7.2) software (CLC bio). Mutation calling (Tables S1 and S2) was performed by geneticists/cell biologists, who had knowledge of clinical phenotypes, pedigree structure, homozygosity mapping, and WES evaluation.
High-Throughput Mutation Analysis by Array-Based Multiplex PCR and NGS
For 48 DNA samples simultaneously, 672 amplicons (592 exons) of 32 candidate genes, including DCDC2, were sequenced in a cohort of 800 individuals with NPHP-RC using a PCR-based 48.48 Access Array microfluidic technology (Fluidigm) with consecutive NGS as previously described.16,17 Detected variants were confirmed by Sanger sequencing and evaluated for segregation. Additional 21 individuals with early onset liver fibrosis were Sanger sequenced for coding regions of DCDC2 (Table S3).
cDNA and Splice Mutation
RNA of A4435-21 and healthy control was purified from whole blood, cDNA was synthesized (Agilent Technologies) and Sanger sequenced, using primers flanking exon 4 in order to confirm skipping of exon 4 (Figure S1; Table S3).
cDNA Cloning
Human full-length (Hs-FL) DCDC2 cDNA was subcloned by PCR from Hs-FL cDNA (origene SC114336). Full-length and partial clones were subcloned into pRK5-N-Myc using the gateway system (Invitrogen). Mutations were introduced at position c.649A>T to represent p.Lys217∗ (Figure S2C) and at c.123_124 delGT to represent p.Ser42Glnfs∗72 (Figure S2A) using “QuikChange II XL Site-Directed Mutagenesis” (Agilent Technologies). Using the same technique the nucleotides 349 to 425 of exon 4 were deleted in order to represent the splice mutation c.349-2A>G (Figure S2B). MAPK8IP1 (JIP1) full-length was subcloned from Hs-FL cDNA (NM_005456, origen sc124125) into pDEST40 (gateway). PAFAH1B1 (LIS1) (NM_000430.3 [MIM 601545]), open reading frame was amplified from Hs-FL cDNA (clone ID HsCD00378475) and subcloned to pDEST40. Human Disheveled 1,2,3 (NM_004421.2, NM_004422.2, NM_004423.3) full-length clones and fragment of DVL3 were a gift from Vita Bryja, Masaryk University.
Coimmunoprecipitation
Coimmunoprecipitation experiments upon co-overexpression in NIH 3T3 and HEK293T cells were performed as described previously.23
Luciferase Reporter Gene Assay
The Wnt/β-catenin reporter assay has been performed as described.23 In brief, NIH 3T3 cells were transfected with pcDNA3/S33Y β-catenin, pTOPFLASH, pGL4.74[hRluc/TK] (Promega) and DCDC2 (WT/mutants) or the empty vectors. At 36 hr posttransfection, luciferase activities were measured using a Dual-Luciferase® Reporter Assay and GloMax™ 96 microplate luminometer (Promega) according manufacturer’s instruction. The luciferase activities were normalized to Renilla luciferase activities and protein concentration.
Antibodies
For immunofluorescence studies, the following primary antibodies were used: Mouse anti-DCDC2 (Abcam, ab 157186), goat-anti-DCDC2 (Santa Cruz, sc-50728), rabbit anti-Kif3a (Abcam ab11259), mouse anti-Jip-1 (Santa Cruz sc-25267), mouse anti-DVL3 (Santa Cruz sc-365581), mouse anti-SDCCAG8 (Abcam, ab67098), rabbit anti-Cep164 (Sigma, hpa037606), mouse anti-Pericentrin (Abcam, ab28144), rabbit anti-PCNT (Atlas Antibodies, 019887), rabbit anti-PCM-1 (Cell Signaling, 5259), and rabbit anti-IFT88 (ProteinTech, 13967-1-AP). For immunoblotting, the following primary antibodies were used: rabbit anti-DCDC2 (Sigma Aldrich D2945), and mouse anti-Jip-1 (Santa Cruz sc-25267).
Immunofluorescence and Confocal Microscopy in Cell Lines
Cells were prepared for immunofluorescence as previously described24, incubated in primary antibodies (see above) overnight at 4°C, and imaged using Leica SP5X system with an upright DM6000 microscope and A1R confocal microscope (Nikon Instruments).
Immunofluorescence and Confocal Microscopy on Tissues
Human and murine paraffin-embedded samples were obtained from Zyagen. Paraffin-embedded tissue sections of 5–7 μm were deparaffinized, rehydrated, stained after heat-induced antigen retrieval, and imaged on a LSM510 confocal microscope (Carl Zeiss Microimaging), and on an A1R confocal microscope (Nikon Instruments).
Knockdown of Dcdc2
Transfection of non-target siRNA was performed in parallel to targeted siRNA using Lipofectamine RNAimax (Invitrogen) following manufacturer’s instructions. Experiments were performed 48 hr after siRNA transfection. For all knockdown experiments, Dharmacon ON-TARGETplus siRNA 11 and 12 against murine Dcdc2a were used (Table S3). The knockdown efficiency was shown by immunoblot (Figure S12) and by qPCR (Figure S10).
Spheroid Assay
Spheroid assays were performed as previously described.6 In brief, IMCD3 cells were transfected with human DCDC2 cDNA constructs at day 1. After 24 hr, cells were transfected with siRNA against murine Dcdc2a. 24 hr after the second transfection, cells were resuspended in matrigel (BD Bioscience) and seeded on Lab TekII chambered coverglasses. After 72 hr, cells that had formed spheroids with visible cleared lumens were stained and imaged using a Zeiss LSM700 confocal microscope. In each spheroid, nuclei were counted, followed by cilia. The percentage of “ciliated nuclei” in each spheroid was used as a distinct value in the results. To detect the presence of cilia, we scored 200–2,000 cells per condition for the presence of cilia. GraphPad Prism 6.0 (GraphPad Software) was used to graph results and perform statistical tests using ANOVA statistical analysis. Experiments were repeated at least two times independently and data combined, graphs show mean value and SEM.
Zebrafish Embryos and Microinjections of Morpholinos and mRNA
Zebrafish embryos were staged 24 hr post fertilization (hpf) and 1-phenyl-2-thiourea (PTU, Sigma) was used to block pigmentation in embryos older than 24 hpf. A morpholinooligonucleotide (MO) against the translation start site of dcdc2b (AUGMO) (Table S3) was designed (Gene Tools, LLC), dissolved in 1× Danieau’s buffer and injected at varying concentrations (50, 100, 150, 200, and 250 μM) into WT zebrafish embryos at the one-cell stage. 5′-capped sense mRNA of human DCDC2 WT and the two mutant clones hDCDC2_Lys217∗ and hDCDC2_Ser42Glnfs∗72 were synthesized using SP6 mMessage mMachine Kit (Ambion) and was coinjected (approximately 100 pg) with dcdc2b MO for rescue experiments. Two additional MOs targeting splice sites were not efficient in dcdc2b knockdown as evidenced by RT PCR (data not shown). All zebrafish experiments were performed in accordance with ethical permits approved by Stockholm North Experimental Animal Committee (Dnr N29-12).
Treatment with WNT/β-Catenin Inhibitor iCRT14
Zebrafish embryos injected with AUGMO were exposed to 500 nM or 750 nM or 1 μM or 5 μM concentrations of iCRT14 at the beginning of gastrulation. Compound treated embryos were raised at 28.5°C in Petri dishes for 48 hr and screened for the rescue of ciliopathy phenotypes. As solvent control, 1% DMSO was used.
Whole-Mount In Situ Hybridization, Histology, and Immunohistochemistry
Digoxigenin-labeled antisense probe for foxa3 was synthesized as described previously.25 For in situ hybridization staged embryos were fixed in 4% PFA for 24 hr and processed.26 Immunohistochemistry and histology experiments on Zebrafish embryos have been performed as described.27,28
Transmission Electron Microscopy
For electron microscopy, 3.5-day-old control and morphant embryos were fixed in 2% glutaraldehyde solution containing 1% PFA in 0.1 M phosphate buffer (PB), pH 7.4. Sectioning was performed using a Leica EM UC 6 (Leica) and stained with uranyl acetate and lead citrate. Electron micrographs were examined and acquired using a Tecnai 10 transmission electron microscope with a veleta camera.
Statistical Analysis
Results in Figure 2 are presented as scattergrams with means and error bars for SD for the indicated number of values. Statistical analysis of continuous data was performed with a two-tailed Student’s t test. p < 0.05 was considered statistically significant.
Figure 2.
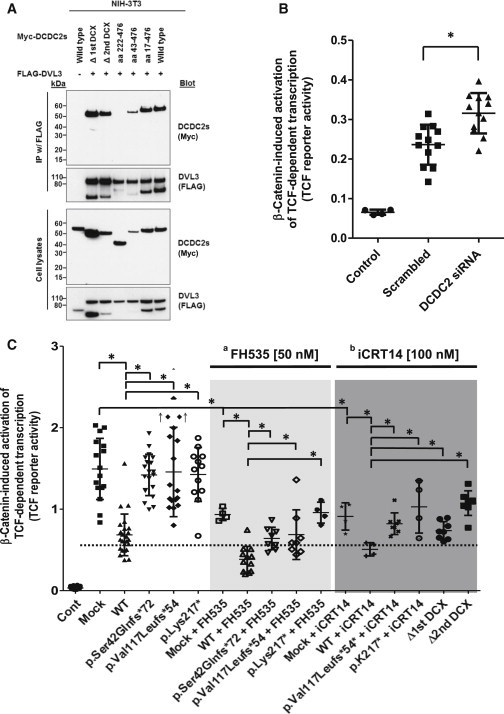
DCDC2 Interacts with Dishevelled and Inhibits β-Catenin-Dependent Wnt Signaling Synergistically with Wnt Inhibitors
(A) DCDC2 interacts with DVL3 when both are transiently overexpressed in NIH 3T3 cells. A construct (aa 222-476) lacking both doublecortin (DCX) domains (see Figure S2 for constructs) does not interact with DVL3, whereas the deletion of either DCX domain alone is not sufficient to abrogate the interaction.
(B) Knockdown of DCDC2 in NIH 3T3 activates the β-catenin-dependent WNT pathway. Cells were transfected with an empty vector pcDNA3 (Control) or pcDNA3-S33Y-β-catenin together with scrambled or DCDC2 siRNA. The ratio between the luciferase activity obtained from the cotransfected TCF-responsive reporter and the control luciferase reporter gene construct (pGL4.74[hRluc/YK]) was calculated and designated as “TCF reporter activity.”
(C) Cells were transfected with an empty vector pcDNA3 (Control) or pcDNA3-S33Y-β-catenin (Mock) together with DCDC2 constructs. Left panel: Overexpression of WT, but not mutant, DCDC2 inhibits β-catenin-induced activation of TCF-dependent reporter gene. Light gray panel: The Wnt inhibitor FH535 suppresses TCF reporter activity. Overexpression of WT DCDC2, but not three mutant DCDC2 clones, inhibits TCF reporter activity additively to the effect of FH535. Dark gray panel: Likewise, the Wnt inhibitor iCRT14 suppresses TCF reporter activity. Overexpression of WT DCDC2, but not mutants, inhibits TCF reporter activity additively to the effect of iCRT14. Two cDNA clones lacking the first or second DCX domain, respectively (Δ1st DCX and Δ2nd DCX), also fail to suppress TCF reporter activity. A dotted horizontal line is drawn for easier interpretation of results. ↑ indicates two outliers at 2.4 and 2.7. aFH535 (middle light gray panel) is known to suppress β-catenin/TCF-mediated transcription and to inhibit β-catenin and GRIP-1 recruitment to PPARγ; biCRT14 (left dark gray panel) is known to modulate interaction between β-catenin and TCF and inhibit β-catenin/TCF-mediated transcription.
Bioinformatics
Genetic location is according to the February 2009 Human Genome Browser data.
Results
DCDC2 Mutations Cause a Renal-Hepatic Ciliopathy
To elucidate pathomechanisms of NPHP-RC, we sought additional causative mutations and performed homozygosity mapping21 and whole-exome sequencing24 in 100 consanguineous cases or sibling cases fulfilling the diagnostic criteria of NPHP-RC. In consanguineous individual A3547-22, who had hepatic fibrosis (Figure 1A) at age 11 months and end-stage renal disease (ESRD) from NPHP at age 14 years (Table 1), we obtained by homozygosity mapping eight candidate regions of homozygosity by descent (Figure 1B). Whole-exome sequencing yielded a homozygous truncating mutation (c.649A>T, p.Lys217∗) in DCDC2 (double cortin domain-containing 2) (Figures 1C–1E; Table 1). No additional homozygous truncating mutations were detected in any other gene within the mapped candidate regions (Tables S1 and S2). Direct inspection of sequence alignments did not yield a mutation in any of the >90 genes with a known connection to NPHP-RC. By high-throughput exon sequencing16,17 in a large worldwide cohort of 800 additional families with NPHP-RC, in whom mutations in known genes were excluded, we sequenced all exons of DCDC2. In less than ten of these individuals liver fibrosis was present. We detected in individual A4435-21, who had hepatic fibrosis requiring liver transplantation at 2 years of age, two compound heterozygous mutations: A frameshift mutation (c.123_124 delGT, p.Ser42Glnfs∗72) (Table 1; Figure 1E) and an obligatory splice site mutation (c.349-2A>G) that we show produces the frameshift product p.Val117Leufs∗54 (Table 1; Figure 1F; Figures S1 and S2). None of the affected individuals had retinal degeneration, cerebellar vermis hypoplasia, hydrocephalus, obesity, or bone disease. An additional 21 individuals with early onset liver fibrosis were screened for variants in the coding region of DCDC2 by Sanger sequencing (Table S3). No additional mutations were detected. We generated cDNA clones reflecting the human DCDC2 mutations (Figure S2). When re-evaluating the Dcdc2 knockout mouse model,29 we found that there was periportal fibrosis at age 11 months consistent with the phenotype seen in humans with DCDC2 mutations (Figure S3).
Figure 1.
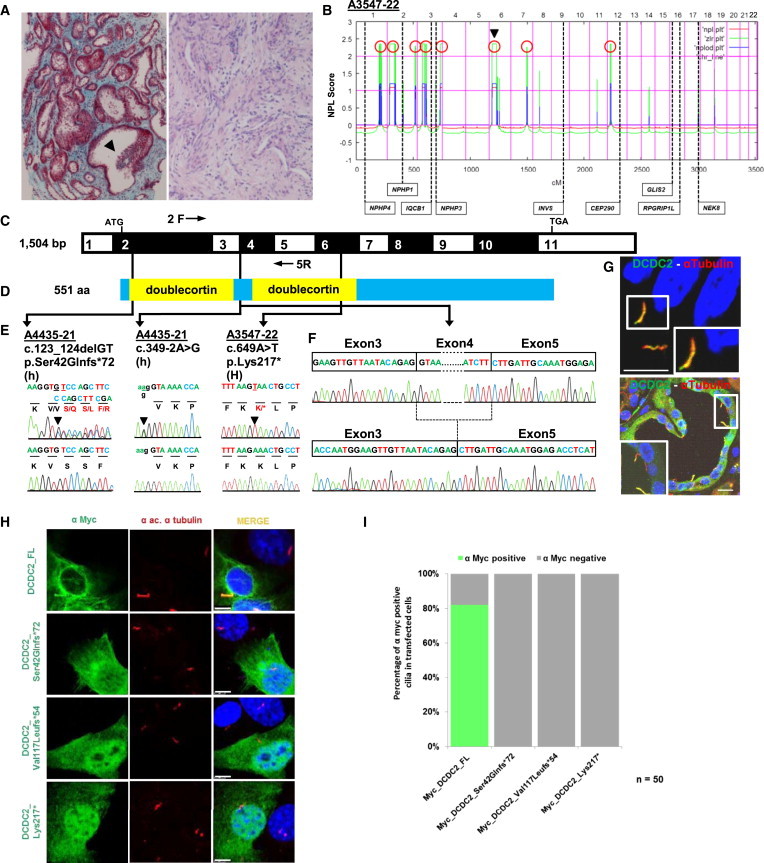
Homozygosity Mapping and WES Detect DCDC2 Mutations in Individuals with Renal-Hepatic Ciliopathy
(A) In A3547-22 with a renal-hepatic ciliopathy renal histology (left panel; Masson trichrome staining) reveals tubulointerstitial fibrosis and tubular dilation with epithelial luminal budding (arrowhead). Hepatic histology (right panel; H&E staining) of A3547-22 shows areas of florid fibrosis with destruction of bile ducts, focal ductular proliferation with cholestasis, and bile plugging.
(B) For individual A3547-22 nonparametric LOD (NPL) scores from whole-genome mapping are plotted across the human genome. The x axis shows Affymetrix 250K StyI array SNP positions on human chromosomes concatenated from pter (left) to qter (right). Genetic distance is given in cM. Eight maximum NPL peaks (red circles) indicate candidate regions of homozygosity by descent. Note that the DCDC2 locus (arrow head) is positioned within a maximum NPL peaks on chromosome 6p.
(C) Exon structure of human DCDC2 cDNA. Positions of start codon (ATG) and stop codon (TGA) are indicated.
(D) Protein domain structure of DCDC2. The N terminus contains two doublecortin domains.
(E) Three homozygous (H) or compound-heterozygous (h) DCDC2 mutations detected in two families with a renal-hepatic ciliopathy (see Table 1). Family number (underlined), mutation, and predicted translational changes are indicated. Healthy control sequence is shown underneath.
(F) Sequenced RT-PCR product from cDNA of lymphocytes of A4435-21 with obligatory splice mutation (c.349-2A>G) using primers located in exons 2 (2F) and 5 (5R). Positions of primers are indicated in (C).
(G) Hepatic and renal localization of DCDC2. Coimmunofluorescence using anti-DCDC2 or anti-α-acetylated tubulin antibodies demonstrates DCDC2 at the ciliary axoneme in cilia of cholangiocytes (upper panel) and of renal epithelial cells (lower panel). Immunofluorescence was performed on human paraffin embedded sections. Nuclei are stained with DRAQ5. Scale bars are 20 μm.
(H) Upon overexpression in NIH 3T3 cells WT DCDC2 decorates the ciliary axoneme, whereas all three mutant clones reflecting mutations in NPHP-RC families lack ciliary localization.
(I) Quantitation of ciliary staining in 50 transfected cells per clone. Scale bars are 5 μM.
Table 1.
Mutations of DCDC2 in Two Families with Nephronophthisis-Related Ciliopathies
| Family-Individual | Ethnic Origin | Nucleotide Alterationa(Segregation) | Deduced protein Change | Exon/intron (state) | Parental Consanguinity | Kidney Phenotype | Liver Phenotype | Other (Clinical Characteristics) |
|---|---|---|---|---|---|---|---|---|
| A3547−22 | UK | c.649A>T (M: het, P: het) | p.Lys217∗ | 6 (Hom) | Yes | Increased echogenicity, severe interstitial fibrosis, tubular dilation with prominent epithelial luminal budding, ESRD at 14 yr. died at 16 yr. from esophageal bleeding | Hepatosplenomegaly, extensive florid fibrosis with destruction of bile ducts, bile focal duct proliferation with cholestasis | Left terminal ICA region aneurysm, foci of signal abnormality in subcortical and deep white matter |
| A4435−21 | Czech | c.123_124 delGT (M: het) c.349-2A>G (P: het) | p.Ser42Glnfs∗72b 3′ splice site (100% conserved) (p.Val117Leufs∗54)c | 2 (het) 4 (het) | No | No renal involvement current age 9 yr. | Hepatosplenomegaly, ductal plate malformation, hepatic fibrosis, scant cholestasis, LTX at 2 yr. | N/A |
CRF, chronic renal failure; ESRD, end-stage renal disease; het, heterozygous; Hom, homozygous; ICA, internal carotid artery; LTX, liver transplantation; M, maternal; N/A, not applicable; P, paternal; yr., years.
DCDC2: cDNA mutations are numbered according to human cDNA reference sequence NM_001195610.1.
The allele appears 12/8,242 in the EVS-Server database.
G is not among alternative nucleotides in the splice site consensus (3′ acceptor splice site position). The allele appears 3/67366 in the ExAC Browser database.
DCDC2 Colocalizes with Other NPHP-RC Proteins to the Cilia-Centrosome-Spindle Complex
To elucidate the role of DCDC2 in the pathogenesis of NPHP-RC, we examined its localization in epithelia of different tissues that are involved in NPHP-RC phenotypes, using indirect immunofluorescence studies. In kidney and liver, we observed that DCDC2 colocalizes with acetylated α-tubulin to the axoneme of primary cilia of human renal tubule cells and cholangiocytes in liver (Figure 1G) and to multiciliated ependymal cells and pia mater cells in mouse brain (Figure S4). However, DCDC2 did not localize to the basal body (Figure S4).
Because NPHP-RC proteins localize to primary cilia, centrosomes, the mitotic spindle, and the abscission structure in a cell-cycle-dependent manner,2 we performed colocalization studies for DCDC2 and other NPHP-RC proteins using confocal laser immunofluorescence microscopy in MDCK-II and hTERT-RPE1 cells (Figures S6 and S7). We found that within the different phases of mitosis DCDC2 fully or partially colocalizes with acetylated α-tubulin during metaphase and anaphase to the spindle microtubules, during late telophase/diakinesis to the abscission structure, and in ciliated cells during interphase to the ciliary axoneme (Figures S5–S7). Throughout those cell-cycle phases, DCDC2 is excluded from the basal body (in interphase) and the mitotic spindle poles (in metaphase and anaphase), as well as the midbody (in diakinesis) (Figures S5–S7), in contrast to other NPHP-RC proteins, which preferentially stain basal body and spindle poles, reciprocally omitting the axoneme and mitotic spindle fibers. These NPHP-RC proteins include SDCCAG8/NPHP10 (Figure S6) and CEP164/NPHP15 (Figure S7). In metaphase, DCDC2 is absent from the interpolar spindle domain of the mitotic spindle (Figure S5). Three proteins that had been described as interacting with DCDC2, DVL3 (this study), JIP1,30 and Kif3a31 labeled basal bodies, mitotic spindle poles, and midbody rather than the ciliary axoneme (Figure S8).
Overexpression in hTERT-RPE1 cells revealed upon immunofluorescence that mutations observed in individuals with NPHP-RC and constructs, from which either of the two doublecortin domains are deleted (Figure S2) fail to localize to the primary cilia (Figure S9). These results are in accordance to observations made in rat hippocampal neurons.31 Immunocytochemistry of cells overexpressing WT and the truncating mutants p.Lys217∗, p.Val117Leufs∗54, and p.Ser42Glnfs∗72 shows that the mutations abrogate localization of DCDC2 to the primary cilium in NIH 3T3 (Figure 1H) and RPE1 cells (Figure S9).
DCDC2 Interacts with DVL1-3, and Mutations Abrogate Wnt Signaling
Because we have previously shown that INVS4,32 and CEP164,6 defects in which are associated with NPHP-RC, interact with proteins that participate in Wnt signaling, we tested whether DCDC2 interacts with the disheveled proteins (DVL1, DVL2, or DVL3), which act as regulators of Wnt signaling.31,33
We performed coimmunoprecipitation studies in NIH 3T3 cells demonstrating that full-length DCDC2 interacts with all three dishevelled proteins, DVL1, DVL2, and DVL3. This interaction was not abrogated in cDNA constructs representing the mutations that we identified in individuals with NPHP-RC (Figure S11). However, a construct (aa 222-476) lacking both doublecortin (DCX) domains (see Figure S2) failed to interact with DVL3, whereas deletion of either DCX domain alone did not abrogate interaction with DVL3 (Figure 2A). (An overview on cDNA clones is given in Figure S2, where p.Ser42Glnfs∗72 represents the shortest variant of A4435-21).
As DVL regulates Wnt signaling, we examined signaling effects downstream of DCDC2 and found that siRNA knockdown of Dcdc2a in NIH 3T3 cells increases β-catenin-induced activation of T cell factor (TCF)-dependent transcription (Figure 2B). Reciprocally, overexpression of DCDC2 WT reduced β-catenin-induced activation of TCF-dependent transcription (Figure 2C, left panel). This effect was abrogated by all three mutations (Table 1, Figures 1C–1E) that we detected in individuals with NPHP-RC (Figure 2C, left panel). Two different Wnt inhibitors (FH535 and iCRT14) reduced β-catenin-induced activation of TCF-dependent transcription with an effect that was additive when combined with DCDC2 overexpression (Figure 2C, middle and right panels).
DCDC2 Interacts with JIP1
Previous studies showed that DCDC2 interacts with the protein JIP1 (mitogen-activated protein kinase 8 interacting protein 1).30,34 JIP1 plays a role in aggregating components of the MAP kinase module (MLK, MKK7, JNK), in facilitating JNK signal transduction35 activating JNK,36 thereby mediating MAPK activation. We therefore performed coimmunoprecipitation in HEK293T cells (Figure S12A). Whereas WT DCDC2 and the mutant p.Val117Leufs∗54 still interacted with full-length JIP1, the DCDC2 truncated mutant p.Ser42Glnfs∗72 that we had detected in individual A4435-21 lacked interaction with JIP1. (For DCDC2 constructs see Figure S2.) When phosphorylation of the JNK downstream targets c-Jun and ATF2 was assayed in NIH 3T3 cells, knockdown of DCDC2 in NIH 3T3 cells did not affect JNK pathway activation (Figure S12B). It was previously shown that lissencephaly-1 (LIS1) interacts with doublecortin (DCX), a protein that belongs with DCDC2 to a family of doublecortin domain-containing proteins.37 When performing coimmunoprecipitation to test for interaction of DCDC2 with LIS1 we did not detect an interaction between the two proteins (Figure S13).
Loss of Dcdc2 Function Disturbs Renal Epithelial Ciliation in 3D Cultures
In order to test the effects of DCDC2 on ciliation, we assessed the result of Dcdc2 siRNA knockdown in a 3D cell culture system. IMCD3 cells cultured in 3D matrigel mimic the normal processes of renal tubulogenesis by forming spheroids or small tubules composed of polarized cells, which exhibit a lumen and a ciliated apical membrane. Counting the number of cilia present on this apical membrane is an established model of ciliogenesis.38 Spheroids grown from cells treated with two different RNAi oligonucleotides targeted against Dcdc2 displayed significantly fewer cilia than spheroids grown from cells treated with a non-target control RNAi but did not display any severe architectural changes in lumen formation (Figure 3). In both cases, this ciliation defect could be rescued by concurrent transfection of WT human DCDC2 but not by any of the mutant DCDC2 clones Ser42Glnfs∗72 and Val117Leufs∗54 that were derived from the affected individuals (Figures 3A–3C). The third mutant (Lys217∗) was not efficiently expressed in the spheroids. The overexpression of empty vector, human WT DCDC2, or either of the mutants did not produce a significant difference in ciliation, excluding a dominant-negative or overexpression effect caused by these alleles (Figure 3; Figure S10). We conclude that DCDC2 is important for the generation and maintenance of renal cilia, but knockdown of DCDC2 does not recapitulate the spheroid defects commonly observed after the loss of function of other NPHP proteins.1,24
Figure 3.
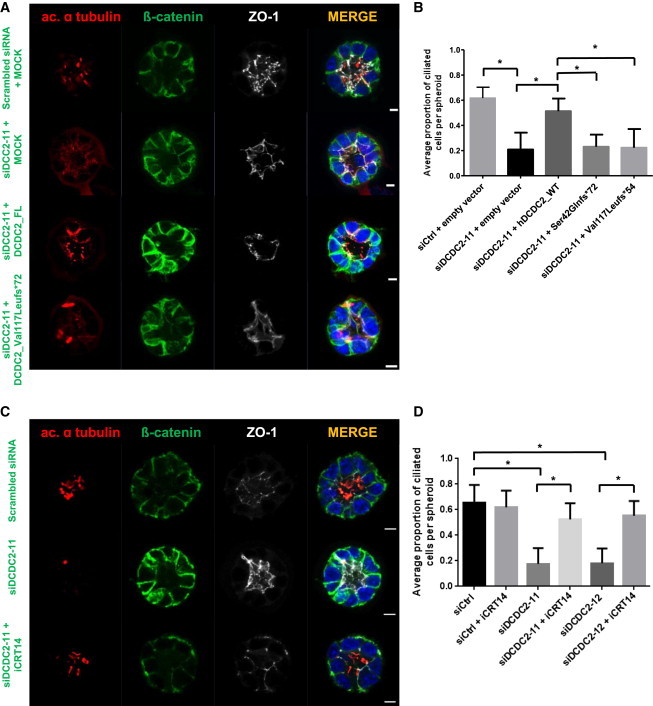
Lack of Dcdc2 Is Rescued by Full-Length DCDC2 and by Wnt Inhibition, but Not by Mutants Detected in NPHP-RC
(A) siRNA (siDcdc2-11) mediated knockdown of Dcdc2 in IMCD3 cells grown in 3D spheroid culture causes a ciliogenesis defect that is rescued by overexpression of human WT DCDC2, but not by mutants found in humans with NPHP-RC. For each condition >50 spheroids were evaluated for the percentage of ciliated cells and the experiment was repeated three times independently. Note that the red structures visible in the mutant-transfected spheroid are midbodies of dividing cells, not cilia. Scale bars are 5 μm.
(B) Ratio of ciliated cells per nuclei within each spheroid upon knockdown and after attempted rescue with WT DCDC2 and two mutants. ∗p < 0.001, as determined by ANOVA analysis. Experiments were repeated at least two times independently and the data combined. Graphs show mean value and standard error of the mean (SEM).
(C) The ciliogenesis defect was rescued by growing spheroids in medium treated with Wnt inhibitor iCRT14 (100 μM). Scale bars are 5 μm.
(D) Quantification for two siRNAs of rescue of ciliogenesis defect by iCRT14 treatment. ∗p < 0.001, as determined by ANOVA analysis. Experiments were repeated at least two times independently and the data combined. Graphs show mean value and SEM.
Because loss of Dcdc2 constitutively activates Wnt signaling (Figure 2B), we hypothesized that treating spheroids grown from renal cells depleted for Dcdc2 with Wnt inhibitors would restore the frequency of cilia. We grew IMCD3 spheroids in the presence of either 50 nM FH535 or 100 nM iCRT14 for 72 hr and scored for cilia per nucleus in 3D structures. The cells exposed to 50 nM FH535 failed to thrive and resulted in no viable spheroids (data not shown). However, the cells treated with 100 nM iCRT14 did generate well-polarized spheroids with lumens. The Wnt inhibitor treatment noticeably rescued the effects of Dcdc2 knockdown in the cells growing in 3D structures by restoring cilia numbers (Figures 3C and 3D). This suggests a role for Wnt signaling in the process of ciliation.
Dcdc2 Knockdown Replicates Ciliopathy Phenotypes in Zebrafish
To examine the renal cystic phenotype in an in vivo vertebrate model, we studied loss-of-function of dcdc2b in zebrafish using a morpholino targeted against the start codon (AUGMO) to suppress the translation of dcdc2b mRNA. The knockdown efficiency of the AUGMO was dose dependent, producing clear visible phenotypes in higher percentage of embryos at 200 μM. In comparison with WT control embryos, knockdown of dcdc2b resulted in typical ciliopathy-related defects such as ventrally curved body axis, hydrocephalus, kidney cysts, kinky tails, and occasionally pericardial edema (Figure 4). At 2 days postfertilization (dpf), 81% of the morphants showed body-axis defects with kinks or waviness in the tail (Figure 4A). Hydrocephalus was observed in 58% of morphants at 2 dpf, and kidney cysts were prominantly visible in 31% of the morphants at 3.5 dpf (Figures 4A–4C). In addition, dcdc2b morphants also showed malformation in otolith assembly (Figure S14). Compared to control embryos that normally had two otoliths, some of the dcdc2b morphants had one or two fused or three otoliths (Figure S14F). These results demonstrate that dcdc2b in zebrafish might have a vital role in cilia formation or function.
Figure 4.
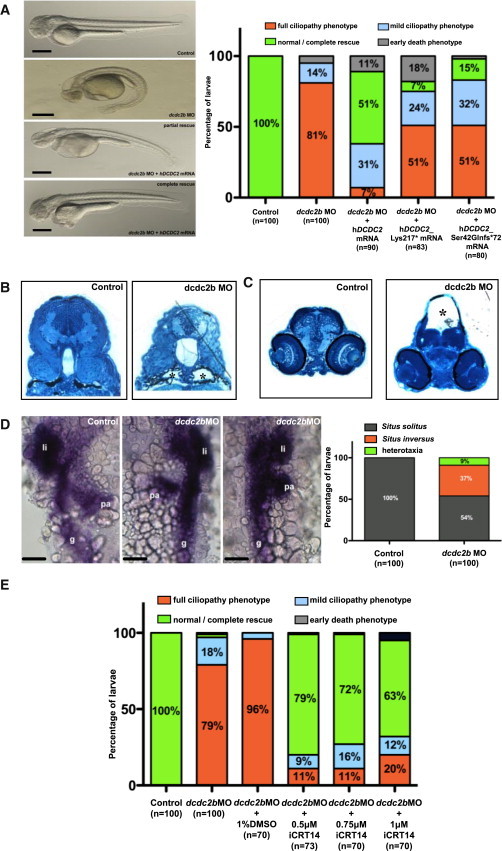
MO Knockdown of dcdc2b Replicates Ciliopathy Phenotypes in Zebrafish that Cannot Be Rescued by cDNA Clones Representing Human Ciliopathy Mutants
(A) Zebrafish embryos injected with AUGMO at one-cell stage produced defects characteristic of cilia dysfunction. Lateral view of 2-day-old control and morphant embryos. dcdc2b morphant developed ventrally bent body axis, hydrocephalus, tail kinks, and pericardial edema (“full ciliopathy phenotype”). Morphologically visible ciliopathy phenotypes in dcdc2b morphants were completely or partially rescued (lacking at least one phenotype) by coinjection of 5′capped mRNA of WT human DCDC2. Coinjection of AUGMO with capped mRNA of either of the two human DCDC2 mutant clones hDCDC2_Lys217∗ or hDCDC2_Ser42Glnfs∗72 mostly failed to rescue ciliary defects. Scale bar is 100 μm.
(B) Histological sections of pronephros from control and morphant embryo at 3.5 dpf. dcdc2b morphants clearly showed dilation of the pronephric duct (asterisks) compared to control embryos.
(C) Transverse brain sections shows hydrocephalus (asterisk) in dcdc2b morphants as compared to control brain sections (methylene blue and silver stain).
(D) Left-right asymmetry defects in liver, gut, and pancreas as visualized by in situ hybridization for the expression of foxa3 with quantitation by histogram (li, liver; pa, pancreas; g, gut). Scale bar is 50 μm.
(E) Treatment with β-catenin inhibitor iCRT14 within the dose range of 0.5–1 μM rescued dcdc2b knockdown ciliopathy phenotypes in the morphants.
Histograms are representation of two or three independent experiments.
We further observed that suppression of dcdc2b caused laterality defects in liver, gut, and pancreas, which was visualized by whole-mount in situ hybridization for the expression of foxa3 (Figure 4D). Interestingly, immunohistochemical staining with an anti-acetylated tubulin antibody revealed that knockdown of dcdc2b does not alter motile cilia length in zebrafish kidney or spinal cord (Figure S14A–S14D). Electronmicrographs of the motile pronephric cilia of dcdc2b morphants showed no structural defects in comparison to WT pronephric cilia (Figure S14E).
To confirm the specificity of the phenotype and to determine whether the biological function of DCDC2 is evolutionarily conserved between human and zebrafish, we performed an mRNA rescue experiment by coinjecting human WT DCDC2 mRNA along with dcdc2b MO into one-cell-stage embryos. Injection of WT human DCDC2 mRNA rescued morphologically visible ciliary phenotypes in 51% of the morphants, suggesting a similar role for dcdc2b as for DCDC2 in cilia function (Figure 4A). We further tested the pathogenicity of the DCDC2 mutations identified in individuals diagnosed for NPHP-RC by a similar rescue approach where dcdc2b MO was coinjected with capped mRNA of the human mutation representing clones hDCDC2_Lys217∗ or hDCDC2_Ser42Glnfs∗72 into one-cell-stage embryos. Both mutations were unable to completely rescue the phenotype of dcdc2b morphants, confirming that these mutations indeed affect the function of the protein (Figure 4A).
Zebrafish Ciliopathy Phenotype Is Rescued by a β-Catenin Inhibitor
Since knockdown or overexpression of Dcdc2 in NIH 3T3 cells altered the Wnt/β-catenin pathway, we hypothesized that the ciliopathy phenotype observed in zebrafish dcdc2b morphants might be a result of derailed Wnt signaling pathway. To test this hypothesis, we exposed the morphants to 0.5 μM, 0.75 μM, and 1 μM of a potent β-catenin inhibitor, iCRT14 at the beginning of gastrulation. After 48 hr period of exposure, the embryos were scored for body curvature, hydrocephalus, tail kinks, and pericardial edema. Interestingly, iCRT14 was able to effectively rescue the phenotype in higher percentage of embryos at doses ranging from 0.5 to 1 μM (Figure 4E). However, concentrations higher than 1 μM were toxic to the embryos (data not shown). Our in vivo and in vitro results strongly suggest a role of Wnt signaling pathway in the pathogenesis of NPHP-RC.
Discussion
We here identified mutations of DCDC2 as a previously unknown cause of a renal-hepatic ciliopathy in humans that is characterized by severe early-onset liver fibrosis within the first year of life. DCDC2 localized to the ciliary axoneme and to mitotic spindle fibers in a cell-cycle-dependent manner. We showed that DCDC2 interacts with DVL, and DCDC2 overexpression inhibited β-catenin-dependent Wnt signaling in an effect additive to Wnt inhibitors. Mutations detected in human NPHP-RC lacked these effects. A Wnt inhibitor restored ciliogenesis in 3D IMCD3 cultures, emphasizing the importance of Wnt signaling for renal tubulogenesis. The fact that knockdown of DCDC2 reduces the number of cilia in cell culture, but not in dcdc2b zebrafish morphants might be explained by the biological differences between primary cilia of IMCD3 cells and multiciliated, motile cilia of zebrafish larval pronephric duct. Finally, knockdown of dcdc2b in zebrafish recapitulated NPHP-RC phenotypes, which were rescued by a Wnt inhibitor and by WT but not by mutant DCDC2. We thus demonstrate a central role of Wnt signaling in the pathogenesis of NPHP-RC, suggesting an avenue for potential treatment of NPHP-RC.
Broad pleiotropy is a common feature of NPHP-RC. In this context, the absence of renal involvement in individual A4435-21 at 9 years of age does not exclude that the affected individual might develop renal involvement later in life, because individual A3547-22 developed chronic kidney disease at 14 years of age with no signs of renal involvement earlier in life. Due to the significant overlap of the observed phenotype with other forms of NPHP-RC we introduce the term “NPHP19” for this variant of NPHP-RC.
By immunofluorescence studies, we show that DCDC2 localizes to the ciliary axoneme and to mitotic spindle fibers. This localization pattern is clearly distinct from other NPHP-RC proteins, such as CEP164 and SDCCAG8 (Figures S6 and S7), which preferentially stain the basal body and the mitotic spindle poles. Interestingly, the interaction partners of DCDC2, DVL3 and JIP1,34 and KIF3a31 show a localization pattern that resembles CEP164 and SDCCAG8. The finding that there was no tight subcellular colocalization for these proteins could be due to the fact that the proteins bind to tubulin as suggested by Figure S8. The specific subcellular compartments to which NPHP-RC proteins localize are very small and often extremely dynamic during different phases of cell cycle, making colocalization experiments difficult to interpret. This holds true for many of the >90 NPHP-RC that are known so far.
Our in vitro studies in cell lines, spheroids, and our in vivo studies in zebrafish clearly demonstrate a role of increased canonical Wnt signaling in the pathogenesis of NPHP-RC. Loss-of-function mutations observed in individuals with NPHP-RC fail to reduce canonical Wnt signaling in vitro. The zebrafish phenotype observed upon knockdown of dcdc2b is reminiscent of the one seen in the zebrafish apc−/− mutant, a model of constitutively activated Wnt.39 The fact that the phenotype of dcdc2b zebrafish morphants could be mitigated by treatment with Wnt inhibitors further supports the significance of dysregulated canonical Wnt signaling in this model.
Consistent with the observed human phenotype, our histological studies of Dcdc2−/− mice reveal periportal liver fibrosis and demonstrate biliary duct proliferation and extensive collagen deposition surrounding the portal tracts compared to WT control (Figure S3). It has been frequently reported that Wnt signaling regulates tubule formation (reviewed in 40). Interestingly, the role of Wnt/β-catenin signaling during the development of liver fibrosis was recently analyzed.41 It was shown that abnormal activation of Wnt/β-catenin signaling promotes tissue fibrogenesis while downregulation of Wnt signaling suppresses the activity of hepatic stellate cells and the collagen synthesis. Our data suggest that loss of DCDC2 function does not profoundly affect renal tubulogenesis but renders the tubules more responsive to Wnt modulation in the 3D polarized tubule state. Activated Wnt signaling has so far been implicated in human NPHP-RC in the presence of INVS mutations.4 Our demonstration that Wnt inhibitors reverse the failure of DCDC2 mutants to reduce canonical Wnt signaling opens a potential route toward treatment for certain forms of NPHP-RC.
Acknowledgments
We are grateful to families and study individuals for their contribution. We would like to thank Milan Elleder and Helena Hůlkova (Institute for Inherited Metabolic Disorders) for histological preparation of liver biopsy specimen. We thank the zebrafish core facility, Karolinska Institutet for providing zebrafish embryos. We thank Kjell Hultenby, Eva Blomen, and Sally Cheung for technical support. We thank the Live Cell Imaging unit/Nikon Center of Excellence, Department of Biosciences and Nutrition, Karolinska Institutet for their support. This research was supported by grants from the National Institutes of Health to F.H. (DK1069274, DK1068306, DK064614), to P.C.H. (DK090728, DK059597), to R.A. (DK099434), and by the CIHR to L.P. (MOP130507). H.Y.G. is supported by the NephCure Foundation and by the ASN Foundation for Kidney Research. T.H. was supported by General University Hospital program RVO-VFN 64165/2012. This work was in part supported by grants to J.K. from Knut and Alice Wallenberg Foundation, the Swedish Research Council, the Centre for Biosciences, the Centre for Innovative Medicine, and Jonasson donation to the School of Technology and Health, Kungliga Tekniska Högskolan, Swedish Brain Foundation (Hjärnfonden) and Swedish Brain Foundations postdoc fellowship award to G.C., from the European Union Framework Programmes 241955 “SYSCILIA” and 305608 “EURenOmics” as well as the Dutch Kidney Foundation grants CP11.18 “KOUNCIL”/13A3D103 to R.H.G., and from the Deutsche Forschungs–gemeinschaft to K.Z. (ZE 205/14-1). F.H. is an Investigator of the Howard Hughes Medical Institute, a Doris Duke Distinguished Clinical Scientist, and the Warren E. Grupe Professor.
Contributor Information
Juha Kere, Email: juha.kere@ki.se.
Friedhelm Hildebrandt, Email: friedhelm.hildebrandt@childrens.harvard.edu.
Supplemental Data
Web Resources
ExAC Browser Beta, http://exac.broadinstitute.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Bioinformatics, http://genome.ucsc.edu/
References
- 1.Sang L., Miller J.J., Corbit K.C., Giles R.H., Brauer M.J., Otto E.A., Baye L.M., Wen X., Scales S.J., Kwong M. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hildebrandt F., Benzing T., Katsanis N. Ciliopathies. N. Engl. J. Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hildebrandt F., Otto E., Rensing C., Nothwang H.G., Vollmer M., Adolphs J., Hanusch H., Brandis M. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat. Genet. 1997;17:149–153. doi: 10.1038/ng1097-149. [DOI] [PubMed] [Google Scholar]
- 4.Otto E.A., Schermer B., Obara T., O’Toole J.F., Hiller K.S., Mueller A.M., Ruf R.G., Hoefele J., Beekmann F., Landau D. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet. 2003;34:413–420. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Attanasio M., Uhlenhaut N.H., Sousa V.H., O’Toole J.F., Otto E., Anlag K., Klugmann C., Treier A.C., Helou J., Sayer J.A. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat. Genet. 2007;39:1018–1024. doi: 10.1038/ng2072. [DOI] [PubMed] [Google Scholar]
- 6.Chaki M., Airik R., Ghosh A.K., Giles R.H., Chen R., Slaats G.G., Wang H., Hurd T.W., Zhou W., Cluckey A. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150:533–548. doi: 10.1016/j.cell.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou W., Otto E.A., Cluckey A., Airik R., Hurd T.W., Chaki M., Diaz K., Lach F.P., Bennett G.R., Gee H.Y. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet. 2012;44:910–915. doi: 10.1038/ng.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olbrich H., Fliegauf M., Hoefele J., Kispert A., Otto E., Volz A., Wolf M.T., Sasmaz G., Trauer U., Reinhardt R. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat. Genet. 2003;34:455–459. doi: 10.1038/ng1216. [DOI] [PubMed] [Google Scholar]
- 9.Otto E., Hoefele J., Ruf R., Mueller A.M., Hiller K.S., Wolf M.T., Schuermann M.J., Becker A., Birkenhäger R., Sudbrak R. A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am. J. Hum. Genet. 2002;71:1161–1167. doi: 10.1086/344395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mollet G., Salomon R., Gribouval O., Silbermann F., Bacq D., Landthaler G., Milford D., Nayir A., Rizzoni G., Antignac C., Saunier S. The gene mutated in juvenile nephronophthisis type 4 encodes a novel protein that interacts with nephrocystin. Nat. Genet. 2002;32:300–305. doi: 10.1038/ng996. [DOI] [PubMed] [Google Scholar]
- 11.Otto E., Loeys B., Khanna H., Hellemans J., Sudbrak R., Fan S., Muerb U., O’Toole J.F., Helou J., Attanasio M. A novel ciliary IQ domain protein, NPHP5, is mutated in Senior-Loken syndrome (nephronophthisis with retinitis pigmentosa), and interacts with RPGR and calmodulin. Nat. Genet. 2005;37:282–288. doi: 10.1038/ng1520. [DOI] [PubMed] [Google Scholar]
- 12.Sayer J.A., Otto E.A., O’Toole J.F., Nurnberg G., Kennedy M.A., Becker C., Hennies H.C., Helou J., Attanasio M., Fausett B.V. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 13.Valente E.M., Silhavy J.L., Brancati F., Barrano G., Krishnaswami S.R., Castori M., Lancaster M.A., Boltshauser E., Boccone L., Al-Gazali L., International Joubert Syndrome Related Disorders Study Group Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 2006;38:623–625. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- 14.Delous M., Baala L., Salomon R., Laclef C., Vierkotten J., Tory K., Golzio C., Lacoste T., Besse L., Ozilou C. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 15.Otto E.A., Trapp M.L., Schultheiss U.T., Helou J., Quarmby L.M., Hildebrandt F. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J. Am. Soc. Nephrol. 2008;19:587–592. doi: 10.1681/ASN.2007040490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halbritter J., Diaz K., Chaki M., Porath J.D., Tarrier B., Fu C., Innis J.L., Allen S.J., Lyons R.H., Stefanidis C.J. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J. Med. Genet. 2012;49:756–767. doi: 10.1136/jmedgenet-2012-100973. [DOI] [PubMed] [Google Scholar]
- 17.Halbritter J., Porath J.D., Diaz K.A., Braun D.A., Kohl S., Chaki M., Allen S.J., Soliman N.A., Hildebrandt F., Otto E.A., GPN Study Group Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum. Genet. 2013;132:865–884. doi: 10.1007/s00439-013-1297-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kruglyak L., Daly M.J., Reeve-Daly M.P., Lander E.S. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am. J. Hum. Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 19.Strauch K., Fimmers R., Kurz T., Deichmann K.A., Wienker T.F., Baur M.P. Parametric and nonparametric multipoint linkage analysis with imprinting and two-locus-trait models: application to mite sensitization. Am. J. Hum. Genet. 2000;66:1945–1957. doi: 10.1086/302911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 21.Hildebrandt F., Heeringa S.F., Rüschendorf F., Attanasio M., Nürnberg G., Becker C., Seelow D., Huebner N., Chernin G., Vlangos C.N. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet. 2009;5:e1000353. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boyden L.M., Choi M., Choate K.A., Nelson-Williams C.J., Farhi A., Toka H.R., Tikhonova I.R., Bjornson R., Mane S.M., Colussi G. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482:98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zariwala M.A., Gee H.Y., Kurkowiak M., Al-Mutairi D.A., Leigh M.W., Hurd T.W., Hjeij R., Dell S.D., Chaki M., Dougherty G.W. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am. J. Hum. Genet. 2013;93:336–345. doi: 10.1016/j.ajhg.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otto E.A., Hurd T.W., Airik R., Chaki M., Zhou W., Stoetzel C., Patil S.B., Levy S., Ghosh A.K., Murga-Zamalloa C.A. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet. 2010;42:840–850. doi: 10.1038/ng.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Odenthal J., Nüsslein-Volhard C. fork head domain genes in zebrafish. Dev. Genes Evol. 1998;208:245–258. doi: 10.1007/s004270050179. [DOI] [PubMed] [Google Scholar]
- 26.Thisse B., Heyer V., Lux A., Alunni V., Degrave A., Seiliez I., Kirchner J., Parkhill J.P., Thisse C. Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods Cell Biol. 2004;77:505–519. doi: 10.1016/s0091-679x(04)77027-2. [DOI] [PubMed] [Google Scholar]
- 27.Chandrasekar G., Vesterlund L., Hultenby K., Tapia-Páez I., Kere J. The zebrafish orthologue of the dyslexia candidate gene DYX1C1 is essential for cilia growth and function. PLoS ONE. 2013;8:e63123. doi: 10.1371/journal.pone.0063123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou W., Hildebrandt F. Molecular cloning and expression of phospholipase C epsilon 1 in zebrafish. Gene Expr. Patterns. 2009;9:282–288. doi: 10.1016/j.gep.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 29.Truong D.T., Che A., Rendall A.R., Szalkowski C.E., LoTurco J.J., Galaburda A.M., Holly Fitch R. Mutation of Dcdc2 in mice leads to impairments in auditory processing and memory ability. Genes Brain Behav. 2014;13:802–811. doi: 10.1111/gbb.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shmueli A., Gdalyahu A., Sapoznik S., Sapir T., Tsukada M., Reiner O. Site-specific dephosphorylation of doublecortin (DCX) by protein phosphatase 1 (PP1) Mol. Cell. Neurosci. 2006;32:15–26. doi: 10.1016/j.mcn.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 31.Massinen S., Hokkanen M.E., Matsson H., Tammimies K., Tapia-Páez I., Dahlström-Heuser V., Kuja-Panula J., Burghoorn J., Jeppsson K.E., Swoboda P. Increased expression of the dyslexia candidate gene DCDC2 affects length and signaling of primary cilia in neurons. PLoS ONE. 2011;6:e20580. doi: 10.1371/journal.pone.0020580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watnick T., Germino G. From cilia to cyst. Nat. Genet. 2003;34:355–356. doi: 10.1038/ng0803-355. [DOI] [PubMed] [Google Scholar]
- 33.Giles R.H., van Es J.H., Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta. 2003;1653:1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 34.Coquelle F.M., Levy T., Bergmann S., Wolf S.G., Bar-El D., Sapir T., Brody Y., Orr I., Barkai N., Eichele G., Reiner O. Common and divergent roles for members of the mouse DCX superfamily. Cell Cycle. 2006;5:976–983. doi: 10.4161/cc.5.9.2715. [DOI] [PubMed] [Google Scholar]
- 35.Yasuda J., Whitmarsh A.J., Cavanagh J., Sharma M., Davis R.J. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol. Cell. Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaeschke A., Czech M.P., Davis R.J. An essential role of the JIP1 scaffold protein for JNK activation in adipose tissue. Genes Dev. 2004;18:1976–1980. doi: 10.1101/gad.1216504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caspi M., Atlas R., Kantor A., Sapir T., Reiner O. Interaction between LIS1 and doublecortin, two lissencephaly gene products. Hum. Mol. Genet. 2000;9:2205–2213. doi: 10.1093/oxfordjournals.hmg.a018911. [DOI] [PubMed] [Google Scholar]
- 38.Renkema K.Y., Stokman M.F., Giles R.H., Knoers N.V. Next-generation sequencing for research and diagnostics in kidney disease. Nat. Rev. Nephrol. 2014;10:433–444. doi: 10.1038/nrneph.2014.95. [DOI] [PubMed] [Google Scholar]
- 39.Hurlstone A.F., Haramis A.P., Wienholds E., Begthel H., Korving J., Van Eeden F., Cuppen E., Zivkovic D., Plasterk R.H., Clevers H. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature. 2003;425:633–637. doi: 10.1038/nature02028. [DOI] [PubMed] [Google Scholar]
- 40.Miller R.K., McCrea P.D. Wnt to build a tube: contributions of Wnt signaling to epithelial tubulogenesis. Developmental dynamics: an official publication of the American Association of Anatomists. 2010;239:77–93. doi: 10.1002/dvdy.22059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ge W.S., Wang Y.J., Wu J.X., Fan J.G., Chen Y.W., Zhu L. Beta-catenin is overexpressed in hepatic fibrosis and blockage of Wnt/beta-catenin signaling inhibits hepatic stellate cell activation. Molecular medicine reports. 2014;9:2145–2151. doi: 10.3892/mmr.2014.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.