

Abstract
The study of gene expression in mammalian single cells via genomic technologies now provides the possibility to investigate the patterns of allelic gene expression. We used single-cell RNA sequencing to detect the allele-specific mRNA level in 203 single human primary fibroblasts over 133,633 unique heterozygous single-nucleotide variants (hetSNVs). We observed that at the snapshot of analyses, each cell contained mostly transcripts from one allele from the majority of genes; indeed, 76.4% of the hetSNVs displayed stochastic monoallelic expression in single cells. Remarkably, adjacent hetSNVs exhibited a haplotype-consistent allelic ratio; in contrast, distant sites located in two different genes were independent of the haplotype structure. Moreover, the allele-specific expression in single cells correlated with the abundance of the cellular transcript. We observed that genes expressing both alleles in the majority of the single cells at a given time point were rare and enriched with highly expressed genes. The relative abundance of each allele in a cell was controlled by some regulatory mechanisms given that we observed related single-cell allelic profiles according to genes. Overall, these results have direct implications in cellular phenotypic variability.
Introduction
In diploid organisms, the mammalian transcription machinery has the choice of transcribing two alleles. Apart from well-known exceptions in which one allele is known to be exclusively expressed—such as in imprinted genes,1,2 X-linked genes,3,4 and genes with random “allelic exclusion”5–10—it is unclear whether ongoing transcription of active genes in individual cells occurs simultaneously from two alleles and whether the allele-specific mRNA level is uniform in all cells.
Studies performed on multiple selected genes in various cell types via RNA fluorescence in situ hybridization suggest that only a fraction of alleles are actively transcribed and associated with RNA polymerase II transcription factories.11–17 Rare are the genes displaying two detectable transcription spots in a large fraction of the cells. Recent single-cell studies have described pervasive random monoallelic expression of autosomal genes in mouse embryonic progenitors and cultured adult murine fibroblasts.18 Allele-biased expression at the single-cell level in 15 single cells from Epstein-Barr-virus-transformed human lymphoblastoid GM12878 cells was also recently reported.19
To investigate the extent of allele-specific transcription of autosomal human protein-coding genes, we used single-cell RNA sequencing (RNA-seq) technology to study 203 single cells from two different human primary fibroblast cell lines. By analyzing informative single-nucleotide variants (SNVs), we determined the relative mRNA abundance of each of the two alleles. For most of the actively transcribed genes, our results revealed that one allele was predominantly detected in a single cell at a particular point in time, whereas the second allele was at low levels or undetectable. We observed a stochastic process given that equal numbers of single cells expressed one or the other allele and a minority of single cells expressed both alleles. Interestingly, we detected only a few genes with an equal mRNA level from both alleles in all single cells. Detailed genomic characterization of these “single-cell biallelic” genes revealed that they express high levels of mRNA in a large number of cells. Our study allowed us to explore the highly dynamic and stochastic nature of allele-specific transcription of human autosomal genes.
Material and Methods
Samples
Human newborn primary fibroblast culture (female, UCF1014, GenCord sample collection) was established from umbilical cord tissue obtained from newborns of western European origin.20 Human fetal primary fibroblast culture (T2N) was derived from postmortem skin tissue obtained from “Twin 2 normal” fetuses (16 weeks of gestation, female); see Dahoun et al.21 for details. The study was approved by the ethics committee of the University Hospitals of Geneva, and written informed consent was obtained from both parents of each individual prior to the study.
Cell Growth
Cells were cultured in Dulbecco’s modified Eagle’s medium GlutaMAX (Life Technologies) supplemented with 10% fetal bovine serum (Life Technologies) and 1% penicillin-streptomycin-fungizone mix (Amimed, BioConcept) at 37°C in a 5% CO2 atmosphere. The day before the single-cell-capture experiment, cells were trypsinized (0.05% trypsin-EDTA, Life Technologies) and replated at a density of 0.3 × 106 cells per 100 mm dish.
Single-Cell Capture
Single-cell captures were performed on the C1 Single-Cell Auto Prep system (Fluidigm) with the cell load script 1772x/1773x. Trypsinized cells were counted and sized with the CASY 1 Cell Counter + Analyzer System (Shärfe System). The average size of human primary fibroblasts was 20 μm (15–25 μm). A total of 4,500–6,000 dissociated live cells were loaded into the assay well of a primed microfluidic array (C1 Single-Cell Auto Prep array for mRNA sequencing [mRNA-seq, 17–25 μm], 96 chambers, Fluidigm) according to the manufacturer’s protocol. After 30 min of capture procedure, we visualized one by one the 96 chambers by using an inverted phase contrast microscope to annotate the content of the chambers. Only the chambers containing one individual cell were selected. Chambers containing debris or damaged cells were excluded from this analysis.
cDNA Synthesis and Pre-amplification of Single Cells
We performed all cDNA preparations on the C1 single-cell array for mRNA-seq with the C1 Single-Cell Auto Prep system (Fluidigm). We used the SMARTer Ultra Low RNA Kit for Illumina Sequencing (Clontech) for the cell lysis and cDNA synthesis according to the manufacturer’s procedure. As recommended, we used the oligo(dT) 3′SMART CDS primer IIA to select for polyA+ RNA in a single-cell sample. No RNA extraction was performed; cDNA synthesis was coupled to the cell-lysis procedure. cDNA from single cells was pre-amplified with the Advantage 2 PCR Kit (Clontech) according to the manufacturer’s protocol. We used two different scripts: the standard mRNA-seq prep script (1772x/1773x, Fluidigm) and a modified version with 12 cycles for the PCR step instead of 22 cycles. The standard mRNA-seq prep script (22 cycles) was as follows: lysis at 72°C (3 min), 4°C (10 min), and 25°C (1 min); reverse transcription at 42°C (90 min) and 70°C (10 min); and PCR at 95°C (1 min), five cycles at 95°C (20 s), 58°C (4 min), and 68°C (6 min), nine cycles at 95°C (20 s), 64°C (30 s), and 68°C (6 min), seven cycles at 95°C (30 s), 64°C (30 s), and 68°C (7 min), and a hold at 72°C (10 min). The modified version with 12 cycles for the PCR step was as follows: lysis at 72°C (3 min), 4°C (10 min), and 25°C (1 min); reverse transcription at 42°C (90 min) and 70°C (10 min); and PCR at 95°C (1 min), two cycles at 95°C (20 s), 58°C (4 min), and 68°C (6 min), six cycles at 95°C (20 s), 64°C (30 s), and 68°C (6 min), four cycles at 95°C (30 s), 64°c (30 s), and 68°C (7 min), and a hold at 72°C (10 min). We harvested the 96 pre-amplified cDNAs from the C1 single-cell array (volume ∼ 13 μl) and quantified the cDNA by using the Qubit dsDNA BR Assay Kit (Invitrogen). We assessed cDNA quality on the 2100 Bioanalyzer (Agilent) with the high-sensitivity DNA chips (Agilent). See Table S1 (available online) for details.
Total RNA Extraction from Bulk Cell Samples
On the day of the single-cell capture, 1.5 × 106 cells from the same culture were collected and stored in TRIzol reagent (Invitrogen) at −80°C. Total RNA was isolated according to the manufacturer’s protocol.
mRNA-Seq Library Preparation
Single Cells
We used the Nextera XT DNA Kit (Illumina) to prepare 223 mRNA-seq libraries for 183 UCF1014 single cells and 40 T2N single cells with 0.3 ng of pre-amplified cDNA according to the manufacturer’s instructions. For cDNA samples below the threshold of Qubit detection, the starting material for the library preparation was 1.25 μl. Additionally, we included six samples from empty chambers and one sample of water instead of cDNA material. For those samples, we took 1.25 μl. In total, 230 samples were library prepared. For this paper, we retained a total of 203 single-cell samples (163 UCF1014 and 40 T2N) for allele-specific expression (ASE) analysis (see “Gene Quantification and De Novo Assembly” below).
Pool of Single Cells
We prepared two RNA-seq libraries with 1 ng of pooled cDNA as described for the single cells. For the pre-amplified cDNA (12 cycles), we took 8 μl of 78 single-cell cDNAs that we pooled. Because of the low concentration, we precipitated the cDNA pool with NaAc (3M [pH 5.2]) and 100% EtOH and then washed it with 70% EtOH. For the pre-amplified cDNA (22 cycles), we took 2 μl of 78 single-cell cDNAs. No precipitation step was necessary because the cDNA concentration of this pool was sufficient enough for library preparation.
Bulk TruSeq
We prepared two libraries with 500 ng of total RNA by using the TruSeq RNA Kit (Illumina) according to the manufacturer’s instructions.
Bulk Nextera
We reverse transcribed 10 ng of total RNA to cDNA by using the SMARTer Ultra Low RNA Kit for Illumina Sequencing (Clontech). The PCR amplification step was conducted with the Advantage 2 PCR Kit (Clontech) with 12 PCR cycles according to the manufacturer’s instructions. Two libraries were prepared with 1 ng of pre-amplified cDNA with the Nextera XT DNA Kit (Illumina) according to the manufacturer’s instructions.
Whole-Genome Sequencing
Library Preparation
Cells were harvested on the day of the single-cell capture. Genomic DNA was extracted with the QIAamp DNA Blood Mini Kit (QIAGEN) according to the manufacturer’s instructions, including for the RNase treatment. Purified genomic DNA (100 ng) was electrophoresed on a 0.8% agarose gel for quality assessment. We quantified the genomic DNA concentration by using the Qubit dsDNA BR Assay Kit (Invitrogen). Genomic DNA libraries were prepared with the TruSeq DNA Kit (Illumina). The starting amount of material was 1 μg of genomic DNA sheared with Covaris S2 to fragments 300–400 bp in size.
Sequencing
Libraries were sequenced on two lanes for UCF1014 samples and on three lanes for T2N samples. In brief, we used the Burrows-Wheeler Aligner (v.0.5.9-r16) to align the sequencing reads to the human reference genome (UCSC Genome Browser GRCh37/hg19). We used SAMtools v.0.1.18 to remove paired-end duplicate reads and pile up the remaining reads. SNVs were called with BCFtools v.0.1.17.
RNA-Seq
Libraries were sequenced on an Illumina HiSeq2000 machine as paired-end 100 bp reads. Demultiplexed fastq files were obtained with the Illumina CASAVA v.1.8.2 software and processed by our in-house pipeline running at the Vital-IT High Performance Computing Center of the Swiss Institute of Bioinformatics.
The two bulk TruSeq libraries were run on one lane. The two bulk Nextera libraries were run on one lane. The two pools of single cells were run on one lane. The single-cell libraries were multiplexed 12 or 16 libraries per lane (see Table S1).
Spike-In Experiment
The spike-in mixture contained 92 External RNA Control Consortium (ERCC) synthesized RNAs (Ambion, Life Technologies). This mixture was added in the lysis buffer during a single-cell-capture preparation with a final dilution of 1:40,000. Both ERCC spike-ins and single-cell mRNA-seq libraries were sequenced simultaneously for 12 samples (50 bp, paired end). The absolute number of spike-in molecules was calculated according to the known concentration of each spike-in. Given that we knew the volume of the chamber (9–10 nl) and the dilution (40,000×) of the spike-in molecules, we derived the expected number of spike-in molecules per chamber.
RPSM Calculation
RPSM stands for reads at a single-nucleotide position per sequencing read length (in kb) and per million mapped reads. The formula for RPSM is (106 × A) / (B × C), where A is the number of mappable reads at a nucleotide position, B is the total number of mappable reads of the sample, and C is the sequencing read length (in kb; C = 0.199).
Read Mapping for RNA-Seq Samples
We employed the RNA pipeline from gemtools v.1.6.2 to map RNA-seq reads. For alignment to the human reference genome sequence (GRCh37/hg19, including the herpes virus sequence), we used the GEM mapping suite22 to first map and subsequently split map all reads that did not map entirely. The mapping pipeline and settings can be found on the GitHub website (see Web Resources).
The GEM output format was converted to BAM format with the following mapping quality scores and flags. (For reference, MAPQ is the Phred-scaled mapping quality score, XT is the mapper-defined tag, U is the number of unique matches, R is a perfect tie, and NM is the number of total mismatches [read 1 + read 2]. See further details of flag information in the SAMtools documentation in the Web Resources.)
-
1.
Matches that are unique and do not have any subdominant match: 251 ≥ MAPQ ≥ 255, XT = U
-
2.
Matches that are unique and have subdominant matches but a different score: 175 ≥ MAPQ ≥ 181, XT = U
-
3.
Matches that are putatively unique (not unique but distinguishable by score): 119 ≥ MAPQ ≥ 127, XT = U
-
4.
Matches that are a perfect tie: 78 ≥ MAPQ ≥ 90, XT = R
Furthermore, the NM flag contains the number of total mismatches (read 1 + read 2). In the analysis, we used reads in categories 1 and 2 (MAPQ ≥ 150).
ASE Analysis
ASE analysis was performed as in Lappalainen et al.23 In brief, we considered heterozygous sites obtained from whole-genome sequencing with DNA reads supporting both alleles. We used a minimum site quality call of 200. We excluded sites susceptible to allelic mapping bias, namely (1) sites with 50 bp mappability < 1 according to the UCSC mappability track (implying that the 50 bp flanking region of the site is not unique in the genome), and (2) sites where overlapping simulated RNA-seq reads showed a >5% mapping difference between those that carried the reference allele and those that carried the non-reference allele (see the methods in Lappalainen et al.23).
In all analyses, we only used uniquely mapped RNA-seq reads (GEM mapping quality > 150) and sites with base quality > 20 and support from at least 16 reads. Using information from SAMtools (v.0.1.19) mpileup,24 we obtained for each site and each sample the number of reads mapping in the reference, the number of alternative alleles, and the sum of both. Each site was then annotated with the overlapped genomic feature in GENCODE annotation v.15 or the novel exons from the de novo assemblies of each sample. For each site, the number of single cells (and non-single cells) where the site was assessed was also counted. The distribution of allelic ratios for all samples is reported in Figure S9.
Gene Quantification and De Novo Assembly
We used the software Cufflinks (v.2.1.1)25,26 with default parameters and GENCODE v.12 as a reference annotation.27 On the basis of Cufflinks transcript (170,086) quantifications, we selected for further analysis single cells that passed the arbitrary threshold of 12,000 transcripts expressed at FPKM (fragments per kilobase of exon per million reads mapped) > 0.3. We retained 163 UCF1014 single-cell samples expressing an average of 15,807 transcripts (the remaining samples expressed an average of 4,998 transcripts). Additionally, for each sample we performed de novo assembly to identify novel transcripts (Figure S4) without using the reference annotation. We then used the program cuffcompare to compare the assembled transcripts with the GENCODE reference annotation (v.15). Finally, for the four bulk RNA samples, we merged the four assemblies into a merged bulk RNA assembly. We compared each single-cell de novo assembly against the merged bulk RNA assembly to identify novel single-cell-specific transcripts. The program intersectBed from bedtools28 was used for this last comparison.
Results
Human newborn primary fibroblasts (UCF1014, GenCord) and human fetal primary fibroblasts (T2N) were cultured, and hundreds of individual cells were captured with the C1 microfluidic system (Fluidigm). We conducted independent experiments that differed by the day of the cell capture and the number of PCR cycles used to pre-amplify the cDNA from individual cells (Figure 1; see Material and Methods). We performed 22 PCR cycles (standard) and 12 PCR cycles in order to test for PCR amplification bias. We sequenced RNA libraries from 163 UCF1014 single cells (98 bp, paired end) to an average depth of 36 million reads (22 PCR cycles) and 16 million reads (12 PCR cycles) (Table S1; Figures S1 and S3). In addition, 40 single cells from T2N were RNA sequenced to a similar depth and used as a replicate sample. We also sequenced four bulk RNA samples generated from 1.5 million cells and two in-vitro-pooled cDNA samples obtained from 78 single-cell cDNAs each (Figure 1). The four bulk RNA samples were prepared from the two different primary fibroblast lines (UCF1014 and T2N) according to two different library procedures: (1) SMARTer Ultra Low RNA Kit (Clontech) for cDNA synthesis followed by Nextera library construction (Illumina) and (2) TruSeq RNA Sample Preparation Kits (Illumina). Each in-vitro-pooled cDNA sample was made from 78 pre-amplified cDNAs (22 or 12 PCR cycles) obtained from 78 different single cells (Figure 1). The total number of reads obtained for the bulk RNA and pooled samples was on average 136 and 251 million reads, respectively (Table S1; Figure S2).
Figure 1.
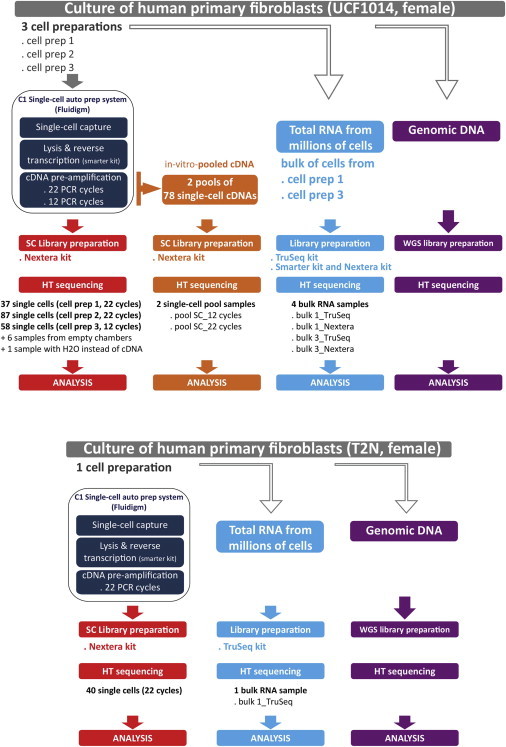
General Outline of the Experimental Workflow
To investigate the ASE at the single-cell level, we performed whole-genome sequencing of UCF1014 samples (26-fold coverage on average; Figure 1) in order to detect the majority of the heterozygous sites. After rigorous filtering, we calculated allelic ratios as the number of reads mapped to the reference allele divided by the total number of reads covering heterozygous SNVs. The main limitation of a single-cell RNA-seq approach is the accurate detection and quantification of two alleles for weakly transcribed genes.18 Thus, we devised a metric for the analysis by implementing a normalized read number at a nucleotide position, namely RPSM (see Material and Methods). This measure is preferable to RPKM (reads per kilobase of exon per million reads mapped) or FPKM measures because it is not dependent on the coverage of the transcript and it accurately reflects the abundance of the transcript at a specific nucleotide position and still allows comparison across samples.
Across 163 single cells from UCF1014, we analyzed 83,576 unique hetSNVs with a coverage ≥ 16 reads at the SNV position. Interestingly, 7.46% of hetSNVs were located in intergenic regions, whereas 68.27% were found within annotated exons (Figure S4). We used Cufflinks to conduct a de novo assembly of transcripts (see Material and Methods) and revealed novel exons specific to single cells and not previously annotated. In total, we retained 9,154 GENCODE genes and 3,875 novel exons specific to single cells for further analysis.
A main challenge was to identify and control for technical noise that could mask genuine biological cell-to-cell allelic expression differences. We carried out a spike-in experiment to assess how our platform performed on synthetic control ERCC RNAs29 to quantify the allelic amount at a single nucleotide position. We concluded that a sensitivity threshold set at 20 RPSM was appropriate for our study objectives (Figure S5). By filtering out sites with fewer than 20 RPSM, we set up the detection threshold at eight molecules per site with a sensitivity of 87%.
One Allele Is Predominantly Detected in Single Cells
We predominantly detected one transcribed allele per interrogated hetSNV of UCF1014 (n = 35,763, RPSM ≥ 20); 76.4% of the hetSNVs displayed an allelic ratio between 0–0.2 and 0.8–1 in 163 single cells (Figure 2). The ratio of single cells expressing one allele to single cells expressing the second allele for all SNVs examined was 49:51. This suggests a stochastic allelic usage, which we confirmed by examining the bulk samples made of 1.5 million cells and samples obtained from an in vitro pool of 78 single cells. Indeed, bulk samples displayed biallelic expression (allelic ratio = 0.2–0.8) for 98.3% of the interrogated hetSNVs (n = 545, RPSM ≥ 20; Figure 2). We obtained the same results from an in vitro single-cell pool sample made of 78 individual cDNAs showing 97.7% of interrogated hetSNVs (n = 480, RPSM ≥ 20) with two transcribed alleles (Figure 2). The skewed monoallelic distribution observed in single cells in a given time point was independent of the number of reads per hetSNV (Figure 3A; Figure S6). Furthermore, our findings were apparently not affected by a bias introduced by the number of PCR cycles given that preparation of single-cell cDNAs with either 12 or 22 PCR cycles revealed similar patterns (Figure 3B). Additionally, the results remained largely unchanged after we removed duplicate reads from the analysis (Figures S7 and S8).
Figure 2.
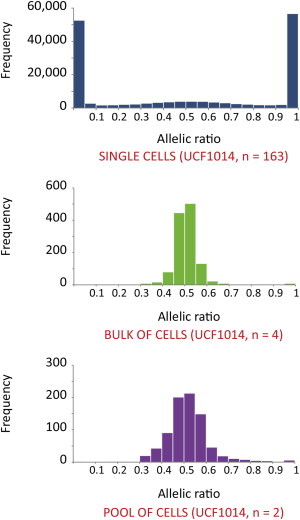
ASE in Single Cells
The histograms show the frequency distribution of the allelic ratio (reference reads per total reads) of 35,763 hetSNVs (≥20 RPSM) in 163 single cells (UCF1014 sample), bulk cell samples, and the pool of single-cell samples.
Figure 3.
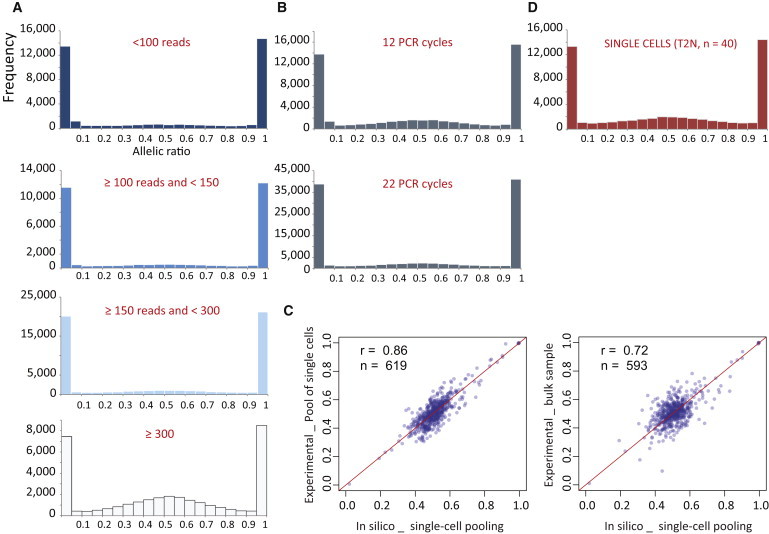
Estimation of the Technical Biases
(A) Frequency-distribution histograms of the allelic ratio (reference reads per total reads) according to the read coverage at hetSNV position (35,763 hetSNVs, 163 UCF1014 single cells, ≥20 RPSM).
(B) Frequency-distribution histograms of the allelic ratio of single cells (35,763 hetSNVs, UCF1014 single cells, ≥20 RPSM), for which the cDNA pre-amplification was performed with 22 or 12 PCR cycles.
(C) Pairwise scatter plots for comparison of the allelic ratio between in silico pooling of single cells and experimental pooling (left panel) or bulk samples (right panel). Listed in the panel are the Pearson correlation coefficients (r) and the numbers of comparisons (n). The diagonal is plotted in red.
(D) Independent experimental replication. Frequency-distribution histograms of the allelic ratio of single cells from T2N single-cell samples (40 single cells, 16,075 hetSNVs, ≥20 RPSM).
We further compared our experimental data to an in-silico-pooled data set. For that, we created an in silico pooling that we derived by averaging the allelic ratios for hetSNVs detected in more than 82 cells (50%). This in silico pooling recapitulated the signal from the in vitro single-cell pool sample (Pearson correlation of 0.86, Figure 3C). Similar results were obtained when we compared the in silico pooling with the bulk sample (Pearson correlation of 0.72; Figure 3C).
In an attempt to validate our findings in a second individual, we used the same method to RNA sequence 40 single cells from another human primary fibroblast female cell line (T2N; Figure 3D). The allele-specific analysis revealed comparable results such that 65.8% of hetSNVs (n = 16,075) expressed only one detectable allele (allelic ratio = 0–0.2 and 0.8–1) and 34.2% of hetSNVs expressed two detectable alleles (allelic ratio = 0.2–0.8; Figure 3D). Our results, in agreement with others,18,19 reveal that snapshot detection of the majority of the protein-coding actively transcribed genes is skewed toward one allele per individual cell. We also describe a random (i.e., non-parental-origin-specific) allelic usage, supporting the stochastic nature of gene transcription.
Stochastic Allelic Expression of Adjacent hetSNV Pairs Is Highly Correlated
We investigated whether two adjacent hetSNVs (RPSM ≥ 20) located in the same gene are likely to be transcribed from the same allele. To do so, we estimated the haplotypes of our sample with SHAPEIT30 by using the phase information contained in sequencing reads and the 1000 Genomes phase 1 haplotypes as a reference.31 Then, we binned every pair of consecutive hetSNVs according to their physical distance (bp) and evaluated whether the allelic ratios agreed with their underlying haplotypes. The relationship between distance and allele expression is reported in Figure 4. We provide evidence that allele expression is highly correlated over the closest adjacent hetSNVs and that this correlation begins to drop from 1 kb to reach 0 at 100 kb. The 1 kb distance corresponds to the average size of cDNA products and might explain this sudden decline (data not shown). It follows that hetSNVs belonging to the same gene are transcribed from the same allele in a single cell. Inversely, it is likely that hetSNVs located on two consecutive genes are not transcribed from the same haplotype in a single cell. Remarkably, compared to hetSNVs in autosomal sites, hetSNVs located on the X chromosome exhibited a higher correlation over longer distances (Figure 4) and a random monoallelic pattern (Figure S10). These results are consistent with the process by which X chromosome inactivation leads to one transcriptionally silenced X chromosome in each 46,XX somatic cell and would thus result in higher allelic correlation across genes on the X chromosome.
Figure 4.
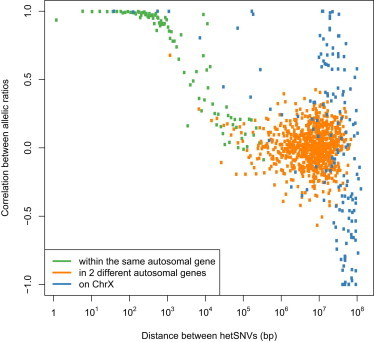
Relationship between hetSNVs Located within Genes or in Two Different Genes
Scatter plots of allelic-ratio correlation versus genomic distance between two adjacent hetSNVs. hetSNVs located in the same autosomal genes are in green, hetSNVs located in different autosomal genes are in orange, and hetSNVs located on the X chromosome are in blue.
Distinct Single-Cell Allelic Pattern
To illustrate the diversity of allelic expression variation across single cells, we selected all genes (n = 568, detected in more than 40 cells) with at least one hetSNV located in coding regions and/or UTRs (RPSM ≥ 20) (Figure 5; Figure S11). As anticipated, genes on the X chromosome (C1GALT1C1 [MIM 300611], ACOT9 [MIM 300862], ZFX [MIM 314980], LAMP2 [MIM 309060], RP11-622K12.1, and TSPAN6 [MIM 300191]) exhibited monoallelic expression in single cells. As a result of lyonization of gene expression,32 cells randomly express only one allele because the second allele is silenced. Consequently, a subset of the cells expressed one allele, and the other subset expressed the second allele. We detected only one or very few cells expressing both alleles. A similar feature was observed for three autosomal genes (RAD52 [MIM 600392], BCLAF1 [MIM 612588], TRBC2 [MIM 615445]), for which fewer than 5% of cells displayed biallelic expression (allelic ratio = 0.2–0.8). Interestingly, we observed 32 autosomal genes with a stochastic single-cell skewed allelic expression, i.e., for which fewer than 10% of cells expressed one type of allele and the remaining cells expressed either the second allele or both alleles (<80% of cells with allelic ratio = 0.2–0.8). As an example, Figure 5 schematically shows the profile of some of those genes (VAMP3 [MIM 603657], CNN3 [MIM 602374], RP11-166D19.1, ATL3 [MIM 609369], RAD52 [MIM 600392], C12orf75, FAM101B [MIM 615928], CCDC80, SPCS3, WDR36 [MIM 609669], BCLAF1 [MIM 612588], and TRBC2 [MIM 615445]). Next, we observed a class of 16 genes (MINOS1, CAP1, ITGB1 [MIM 135630], CD44 [MIM 107269], NACA [MIM 601234], DAD1 [MIM 600243], UQCR11, PPP1CB [MIM 600590], SRSF6 [MIM 601944], RPS21 [MIM 180477], SSR3 [MIM 606213], SPARC [MIM 182120], SRSF3 [MIM 603364], CALU [MIM 603420], TOMM7 [MIM 607980], and COL1A2 [MIM 120160]) for which more than 90% of cells expressed both alleles (allelic ratio = 0.2–0.8). We termed those genes “single-cell biallelic genes” because both alleles were expressed in almost all individual cells.
Figure 5.
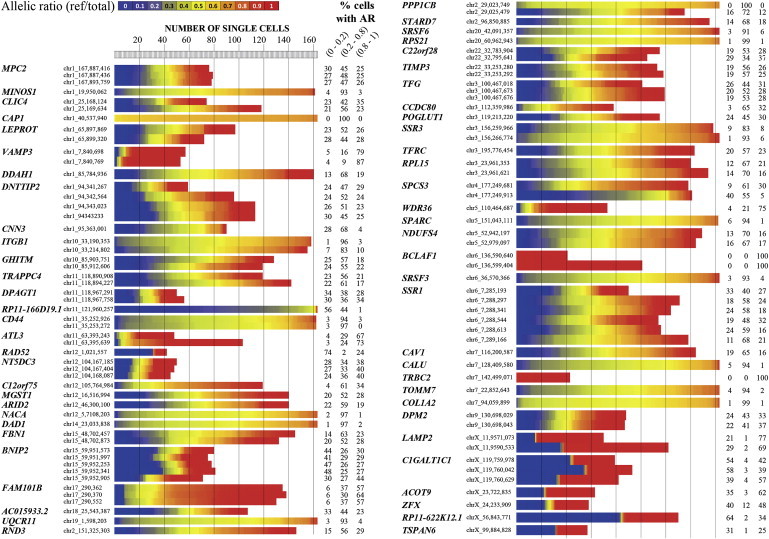
Pattern of Allelic Expression in Single Cells
We selected genes for a representative view of the allelic expression in single cells. A complete overview is given in Figure S11. The index bar indicates the color coding for the allelic-ratio values (reference reads per total reads). Vertical dashed lines delineate a set of 20 single cells.
Transcription Rate and ASE in Single Cells
We asked whether the total transcript level could correlate with ASE in single cells. We quantified the steady-state mRNA level of detectable genes in 163 single cells from UCF1014 by averaging the RPSM values for the hetSNVs located in coding regions and/or UTRs. We plotted the minimum allelic ratio against mRNA level (RPSM average) for all detectable genes (Figure 6A). The results indicated that genes with higher mRNA levels were enriched in the single-cell biallelic genes described above (see Figure 6A). Analysis of functional-pathway enrichment was performed with DAVID (Database for Annotation, Visualization, and Integrated Discovery)33 on genes with high mRNA levels (average RPSM per gene > 90). Genes associated with cellular function and maintenance were enriched with gene-ontology terms related to response to nutrients, protein-complex assembly, the ER, organelle membranes, catalytic activities, and others (modified Fisher exact p value < 0.05; Figure 6A). This is consistent with the fact that constitutively expressed genes, essential for the maintenance of basic cellular functions, generally maintain constantly high mRNA levels across cells.
Figure 6.

Relationship of mRNA Level and Allelic Ratio in Single Cells
(A) A composite figure made of a scatter plot and a table. The scatter plot represents the mRNA level from single-cell RNA-seq data (UCF1014) against the minimum allelic ratio. The minimum allelic ratio is the absolute value of the difference between 0.5 and the allelic ratio (reference reads per total reads). For each gene, the average of RPSM values (coding regions and UTRs) for all single cells was calculated, log transformed (log2), and plotted on the y axis. Each data point represents one gene. Genes located on the X chromosome are included. The table on the right is the list of enriched gene-ontology (GO) terms with their respective p values (DAVID) for genes with RPSM values > 90.
(B) Box plots of RNA half-life (left) or CAGE nuclear RNA (right) in three different groups of sites with variable allelic ratio (AR). tpm stands for the normalized raw CAGE tag count per million.
Because steady-state mRNA abundance is determined by both the rate of transcription and mRNA decay, we used published RNA half-life data from HeLa cells to examine the relationship between the single-cell allelic ratio of gene expression and the half-life of these transcripts.34 Tani et al. developed an inhibitor-free method named BRIC-seq (5′-bromo-uridine-immunoprecipitation chase-deep sequencing) to determine mRNA decay. We selected the genes commonly expressed by the two different data sets and further divided the sites into three groups according to their single-cell allelic ratio. Compared to the single-cell biallelic group (allelic ratio = 0.2–0.8), the single-cell monoallelic group (allelic ratio <0.2 or >0.8) contained sites with significantly shorter half-lives (p value < 2 × 10−16, ANOVA; Figure 6B). This finding suggests that genes detected as biallelic in single cells are more likely to have a longer RNA half-life. We then tested the relationship between transcriptional initiation rates of genes in single cells. Thus, we introduced in our analysis Cap Analysis of Gene Expression (CAGE35) sequencing data from nuclear-enriched RNAs of human dermal fibroblasts of fetal origin (HDF-f).36 CAGE technology on nuclear-enriched RNA allows reliable transcription start site (TSS) identification and transcript quantification of mostly nascent messenger RNAs. We identified 2,572 nuclear CAGE clusters from HDF-f overlapping TSSs of protein-coding genes expressed in our single-cell RNA-seq data. Figure 6B shows a correlation between the initiation rate (determined by nuclear CAGE tags) and ASE. Sites with monoallelic expression show a lower initiation rate than do biallelic sites (p value < 2 × 10−16, ANOVA).
Discussion
The transcriptional activity of alleles is of interest because it determines the steady-state level of mRNA of the cell and the nature of transcripts available for translation. By analyzing single-cell transcriptomes, we and others have confirmed that gene transcription is stochastic and extremely variable among cells.18,19,37,38 Our main observation suggests that, at any point in time, a cell contains mostly transcripts from one allele (76.4% of hetSNVs with >20 RPSM). This stochastic monoallelic expression at the single-cell level is independent of the parent of origin of the allele, given that we randomly detected one or the other allele in each single cell. Our results are in agreement with recent studies in human lymphoblastoid lines and mouse embryonic progenitors and cultured fibroblasts,18,19 confirming the existence of an essential process of eukaryotic cells.
Does this necessarily mean that the cellular machinery transcribes one allele at a time? Transcription occurs as either a constitutive or an episodic bursty mode.39,40 For most eukaryotic genes, transcription in mammals is discontinuous and occurs in transcriptional bursts interspersed by refractory periods of gene inactivity.41–43 It has been demonstrated that transcriptional bursting is gene specific, and thus the frequency and amplitude of the bursts and intervals of gene inactivity modulate the extent of temporal variations in mRNA in a cell.44 Two main alternative scenarios could explain the observation of stochastic monoallelic expression in single cells. In the first scenario, a single cell transcribes one allele at a time. The transcription machinery could switch from one allele to the second. This requires the assembly of the pre-initiation complex on one allele and the subsequent dissociation of the complex and clearance of the promoter region before the next round of transcription initiation.45 This model of transcription was previously referred to as the dynamic flip-flop transcription cycle model with a long period of gene inactivity and suggests a cross-talk between the two alleles.11,46–49 This model could predict heterogeneity within a population of cells expressing one allele at a time. In the second scenario, we hypothesized that the cellular machinery simultaneously transcribes both alleles of all autosomal genes, with the exception of the imprinted genes. The first alternative explanation of our results is that transcription is indeed biallelic but asynchronous, i.e., the transcription machinery is associated with both alleles, but the bursts of transcription of each allele are not synchronous. In such a scenario, in a single cell at a particular point in time, the allelic transcription appears biallelic if the mRNA half-life is very long, i.e., longer than the refractory period of gene inactivity. Conversely, the allelic transcription appears random and monoallelic for a gene that is poorly transcribed (below the threshold of detection) with a long refractory period of gene inactivity and/or for genes with very short half-lives. Our findings are compatible with this scenario because we revealed that monoallelic transcripts in single cells are short lived and less actively transcribed and that biallelic transcripts are long lived and more actively transcribed.
The stochastic monoallelic expression in single cells could be theoretically linked to phenotypic variability in humans; examples of this include (1) penetrance of a dominant developmental disorder, (2) expressivity of a dominant disorder, (3) cellular or tissue phenotype in carriers of recessive disorders, (4) cellular heterogeneity in cancers, (5) differential cellular response to environmental agents, (6) predisposition to a complex phenotype, and (7) phenotypic variability in monozygotic twins.
In conclusion, the allelic expression of single cells might be an important determinant of the developmental fate and specific function of each cell and might contribute to the phenotypic variability of the organism.
Acknowledgments
We thank Michel Strubin and David Suter for critical comments on the manuscript, Mark Lynch for helpful technical suggestions, Audrey Letourneau and Cedric Howald for processing raw data, and the Institute of Genetics and Genomics of Geneva genomics platform at the University of Geneva. This work was supported by Swiss National Science Foundation (SNF-144082) and European Research Council (ERC-249968) grants to S.E.A. We thank the ChildCare foundations for supporting the S.E.A. laboratory. This work was also supported by Louis-Jeantet Foundation, Swiss National Science Foundation, and European Research Council grants to E.T.D. The computations were performed at the Vital-IT High Performance Computing Center of the Swiss Institute of Bioinformatics.
Contributor Information
Emmanouil T. Dermitzakis, Email: emmanouil.dermitzakis@unige.ch.
Stylianos E. Antonarakis, Email: stylianos.antonarakis@unige.ch.
Accession Numbers
RNA and DNA sequencing data have been deposited in the European Genome-phenome Archive for controlled accesses under accession number EGAS00001001009.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
European Genome-phenome Archive, https://www.ebi.ac.uk/ega/
Gemtools, http://github.com/gemtools
Online Mendelian Inheritance in Man (OMIM), http://omim.org/
SAMtools, http://samtools.sourceforge.net/
UCSC Genome Browser, http://genome.ucsc.edu/
Vital-IT, http://www.vital-it.ch
References
- 1.Ferguson-Smith A.C. Genomic imprinting: the emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011;12:565–575. doi: 10.1038/nrg3032. [DOI] [PubMed] [Google Scholar]
- 2.Reik W., Walter J. Genomic imprinting: parental influence on the genome. Nat. Rev. Genet. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- 3.Augui S., Nora E.P., Heard E. Regulation of X-chromosome inactivation by the X-inactivation centre. Nat. Rev. Genet. 2011;12:429–442. doi: 10.1038/nrg2987. [DOI] [PubMed] [Google Scholar]
- 4.Chow J.C., Yen Z., Ziesche S.M., Brown C.J. Silencing of the mammalian X chromosome. Annu. Rev. Genomics Hum. Genet. 2005;6:69–92. doi: 10.1146/annurev.genom.6.080604.162350. [DOI] [PubMed] [Google Scholar]
- 5.Chess A., Simon I., Cedar H., Axel R. Allelic inactivation regulates olfactory receptor gene expression. Cell. 1994;78:823–834. doi: 10.1016/s0092-8674(94)90562-2. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez I., Feinstein P., Mombaerts P. Variable patterns of axonal projections of sensory neurons in the mouse vomeronasal system. Cell. 1999;97:199–208. doi: 10.1016/s0092-8674(00)80730-8. [DOI] [PubMed] [Google Scholar]
- 7.Rimm I.J., Bloch D.B., Seidman J.G. Allelic exclusion and lymphocyte development. Lessons from transgenic mice. Mol. Biol. Med. 1989;6:355–364. [PubMed] [Google Scholar]
- 8.Gimelbrant A.A., Ensminger A.W., Qi P., Zucker J., Chess A. Monoallelic expression and asynchronous replication of p120 catenin in mouse and human cells. J. Biol. Chem. 2005;280:1354–1359. doi: 10.1074/jbc.M411283200. [DOI] [PubMed] [Google Scholar]
- 9.Gimelbrant A., Hutchinson J.N., Thompson B.R., Chess A. Widespread monoallelic expression on human autosomes. Science. 2007;318:1136–1140. doi: 10.1126/science.1148910. [DOI] [PubMed] [Google Scholar]
- 10.Wang J., Valo Z., Smith D., Singer-Sam J. Monoallelic expression of multiple genes in the CNS. PLoS ONE. 2007;2:e1293. doi: 10.1371/journal.pone.0001293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wijgerde M., Grosveld F., Fraser P. Transcription complex stability and chromatin dynamics in vivo. Nature. 1995;377:209–213. doi: 10.1038/377209a0. [DOI] [PubMed] [Google Scholar]
- 12.Levsky J.M., Shenoy S.M., Pezo R.C., Singer R.H. Single-cell gene expression profiling. Science. 2002;297:836–840. doi: 10.1126/science.1072241. [DOI] [PubMed] [Google Scholar]
- 13.Osborne C.S., Chakalova L., Brown K.E., Carter D., Horton A., Debrand E., Goyenechea B., Mitchell J.A., Lopes S., Reik W., Fraser P. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat. Genet. 2004;36:1065–1071. doi: 10.1038/ng1423. [DOI] [PubMed] [Google Scholar]
- 14.Osborne C.S., Chakalova L., Mitchell J.A., Horton A., Wood A.L., Bolland D.J., Corcoran A.E., Fraser P. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 2007;5:e192. doi: 10.1371/journal.pbio.0050192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fraser P. Transcriptional control thrown for a loop. Curr. Opin. Genet. Dev. 2006;16:490–495. doi: 10.1016/j.gde.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 16.Chakalova L., Fraser P. Organization of transcription. Cold Spring Harb. Perspect. Biol. 2010;2:a000729. doi: 10.1101/cshperspect.a000729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levsky J.M., Shenoy S.M., Chubb J.R., Hall C.B., Capodieci P., Singer R.H. The spatial order of transcription in mammalian cells. J. Cell. Biochem. 2007;102:609–617. doi: 10.1002/jcb.21495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng Q., Ramsköld D., Reinius B., Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343:193–196. doi: 10.1126/science.1245316. [DOI] [PubMed] [Google Scholar]
- 19.Marinov G.K., Williams B.A., McCue K., Schroth G.P., Gertz J., Myers R.M., Wold B.J. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res. 2014;24:496–510. doi: 10.1101/gr.161034.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dimas A.S., Deutsch S., Stranger B.E., Montgomery S.B., Borel C., Attar-Cohen H., Ingle C., Beazley C., Gutierrez Arcelus M., Sekowska M. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dahoun S., Gagos S., Gagnebin M., Gehrig C., Burgi C., Simon F., Vieux C., Extermann P., Lyle R., Morris M.A. Monozygotic twins discordant for trisomy 21 and maternal 21q inheritance: a complex series of events. Am. J. Med. Genet. A. 2008;146A:2086–2093. doi: 10.1002/ajmg.a.32431. [DOI] [PubMed] [Google Scholar]
- 22.Marco-Sola S., Sammeth M., Guigó R., Ribeca P. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods. 2012;9:1185–1188. doi: 10.1038/nmeth.2221. [DOI] [PubMed] [Google Scholar]
- 23.Lappalainen T., Sammeth M., Friedländer M.R., ’t Hoen P.A., Monlong J., Rivas M.A., Gonzàlez-Porta M., Kurbatova N., Griebel T., Ferreira P.G., Geuvadis Consortium Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baker S.C., Bauer S.R., Beyer R.P., Brenton J.D., Bromley B., Burrill J., Causton H., Conley M.P., Elespuru R., Fero M., External RNA Controls Consortium The External RNA Controls Consortium: a progress report. Nat. Methods. 2005;2:731–734. doi: 10.1038/nmeth1005-731. [DOI] [PubMed] [Google Scholar]
- 30.Delaneau O., Marchini J., Zagury J.F. A linear complexity phasing method for thousands of genomes. Nat. Methods. 2012;9:179–181. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 31.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lyon M.F. Gene action in the X-chromosome of the mouse (Mus musculus L.) Nature. 1961;190:372–373. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- 33.Sherman B.T., Huang W., Tan Q., Guo Y., Bour S., Liu D., Stephens R., Baseler M.W., Lane H.C., Lempicki R.A. DAVID Knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinformatics. 2007;8:426. doi: 10.1186/1471-2105-8-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tani H., Mizutani R., Salam K.A., Tano K., Ijiri K., Wakamatsu A., Isogai T., Suzuki Y., Akimitsu N. Genome-wide determination of RNA stability reveals hundreds of short-lived noncoding transcripts in mammals. Genome Res. 2012;22:947–956. doi: 10.1101/gr.130559.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi H., Lassmann T., Murata M., Carninci P. 5′ end-centered expression profiling using cap-analysis gene expression and next-generation sequencing. Nat. Protoc. 2012;7:542–561. doi: 10.1038/nprot.2012.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fort A., Hashimoto K., Yamada D., Salimullah M., Keya C.A., Saxena A., Bonetti A., Voineagu I., Bertin N., Kratz A., FANTOM Consortium Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat. Genet. 2014;46:558–566. doi: 10.1038/ng.2965. [DOI] [PubMed] [Google Scholar]
- 37.Shalek A.K., Satija R., Adiconis X., Gertner R.S., Gaublomme J.T., Raychowdhury R., Schwartz S., Yosef N., Malboeuf C., Lu D. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–240. doi: 10.1038/nature12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu A.R., Neff N.F., Kalisky T., Dalerba P., Treutlein B., Rothenberg M.E., Mburu F.M., Mantalas G.L., Sim S., Clarke M.F., Quake S.R. Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods. 2014;11:41–46. doi: 10.1038/nmeth.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suter D.M., Molina N., Naef F., Schibler U. Origins and consequences of transcriptional discontinuity. Curr. Opin. Cell Biol. 2011;23:657–662. doi: 10.1016/j.ceb.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 40.Levine J.H., Lin Y., Elowitz M.B. Functional roles of pulsing in genetic circuits. Science. 2013;342:1193–1200. doi: 10.1126/science.1239999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suter D.M., Molina N., Gatfield D., Schneider K., Schibler U., Naef F. Mammalian genes are transcribed with widely different bursting kinetics. Science. 2011;332:472–474. doi: 10.1126/science.1198817. [DOI] [PubMed] [Google Scholar]
- 42.Harper C.V., Finkenstädt B., Woodcock D.J., Friedrichsen S., Semprini S., Ashall L., Spiller D.G., Mullins J.J., Rand D.A., Davis J.R., White M.R. Dynamic analysis of stochastic transcription cycles. PLoS Biol. 2011;9:e1000607. doi: 10.1371/journal.pbio.1000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muramoto T., Cannon D., Gierlinski M., Corrigan A., Barton G.J., Chubb J.R. Live imaging of nascent RNA dynamics reveals distinct types of transcriptional pulse regulation. Proc. Natl. Acad. Sci. USA. 2012;109:7350–7355. doi: 10.1073/pnas.1117603109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Molina N., Suter D.M., Cannavo R., Zoller B., Gotic I., Naef F. Stimulus-induced modulation of transcriptional bursting in a single mammalian gene. Proc. Natl. Acad. Sci. USA. 2013;110:20563–20568. doi: 10.1073/pnas.1312310110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grimaldi Y., Ferrari P., Strubin M. Independent RNA polymerase II preinitiation complex dynamics and nucleosome turnover at promoter sites in vivo. Genome Res. 2014;24:117–124. doi: 10.1101/gr.157792.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trimborn T., Gribnau J., Grosveld F., Fraser P. Mechanisms of developmental control of transcription in the murine alpha- and beta-globin loci. Genes Dev. 1999;13:112–124. doi: 10.1101/gad.13.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gribnau J., de Boer E., Trimborn T., Wijgerde M., Milot E., Grosveld F., Fraser P. Chromatin interaction mechanism of transcriptional control in vivo. EMBO J. 1998;17:6020–6027. doi: 10.1093/emboj/17.20.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fraser P., Grosveld F. Locus control regions, chromatin activation and transcription. Curr. Opin. Cell Biol. 1998;10:361–365. doi: 10.1016/s0955-0674(98)80012-4. [DOI] [PubMed] [Google Scholar]
- 49.Jackson D.A., Pombo A., Iborra F. The balance sheet for transcription: an analysis of nuclear RNA metabolism in mammalian cells. FASEB J. 2000;14:242–254. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.