

Abstract
Myotonic dystrophy type 1 (DM1) and type 2 (DM2) are multisystemic disorders linked to two different genetic loci and characterized by several features including myotonia, muscle weakness and atrophy, cardiac dysfunctions, cataracts and insulin-resistance. In both forms, expanded nucleotide sequences cause the accumulation of mutant transcripts in the nucleus deregulating the activity of some RNA-binding proteins and providing an explanation for the multisystemic phenotype of DM patients. However this pathogenetic mechanism does not explain some histopathological features of DM skeletal muscle like muscle atrophy. It has been observed that DM muscle shares similarities with the ageing muscle, where the progressive muscle weakness and atrophy is accompanied by a lower regenerative capacity possibly due to the failure in satellite cells activation. The aim of our study is to investigate if DM2 satellite cell derived myoblasts exhibit a premature senescence as reported for DM1 and if alterations in their proliferation potential and differentiation capabilities might contribute to some of the histopathological features observed in DM2 muscles. Our results indicate that DM myoblasts have lower proliferative capability than control myoblasts and reach in vitro senescence earlier than controls. Differentely from DM1, the p16 pathway is not responsible for the premature growth arrest observed in DM2 myoblasts which stop dividing with telomeres shorter than controls. During in vitro senescence, a progressive decrease in fusion index is observable in both DM and control myotubes with no significant differences between groups. Moreover, myotubes obtained from senescent myoblasts appear to be smaller than those from young myoblasts. Taken together, our data indicate a possible role of DM2 premature myoblast senescence in skeletal muscle histopathological alterations i.e., dystrophic changes and type 2 fibre atrophy.
Key words: Myotonic dystrophy, aging, myoblasts, p16, telomeres
Introduction
Myotonic dystrophies (DM) are autosomal dominant multisystemic disorders characterized by a variety of multisystemic features including myotonia, muscular dystrophy, cardiac dysfunctions, cataracts and insulin-resistance.1 Myotonic dystrophy type 1 (DM1) is the most prevalent form of adult neuromuscular disorder and is caused by an expanded (CTG)n in the 3’ untranslated region of the Dystrophia Myotonic Protein Kinase (DMPK) gene.2-4 The second form of DM, myotonic dystrophy type 2 (DM2), is caused by the expansion of a tetranucleotidic repeat (CCTG)n in the intron 1 of the CCHC-type zinc finger, nucleic acid-binding protein (CNBP) gene.5,6 In both forms, the mutant transcripts accumulate in nuclear foci altering the function of several alternative splicing regulators, which are necessary for the physiological processing of mRNAs.7-10 These alterations lead to a spliceopathy i.e., an aberrant alternative splicing of different genes that explain different features of the DM multisystemic phenotype.11,12 However, at the skeletal muscle level, still there is no mechanistic explanation for the muscle weakness and atrophy observed in DM patients or for the muscle histopathological features characteristic of this disease which includes fibre atrophy-hypertrophy, increased number of central nuclei, and presence of fibres with nuclear clumps.13-15
It has been obseved that in DM muscle tissue there is decreased muscle regeneration in response to the ongoing muscle loss and dystrophy1 thus it is possible that the blunted repair response may result from impaired myogenesis in adult DM muscle. Interestingly, DM muscle shares apparent similarities with the ageing muscle where the progressive muscle weakness and atrophy is accompanied by a slower regenerative capacity.16-20 Satellite cells, the muscle precursor cells, provide the potential for both pre and post-natal growth of skeletal muscles, and for its regeneration following injury.21,22 In normal muscles, satellite cells are quiescently located between the sarcolemma and the basal lamina of mature myofibres; following injury they become activated and then proliferate and fuse into myotubes to regenerate or repair muscle fibres.23,24 It is known that the regenerative capacity of skeletal muscle depends on the number of progenitor cells which declines with age in humans and on their activation, proliferation and differentiation potential.25-27 The proliferative potential of human satellite cells decreases during postnatal muscle growth due to the replicative senescence.25,26 Replicative senescence may be caused by progressive telomere shortening at each cellular division or by additional pathways such as the p16 stress pathway, which could also interfere with the proliferation capacity of human myoblasts by inducing premature senescence.28,29 It is therefore possible that an early senescence of satellite cells could affect both the regeneration process and the maintenance of muscle mass, since the differentiation program of senescent myogenic precursor cells would become defective due to impaired myogenesis and/or the down-regulation of the myogenic regulatory factors. Indeed, Bigot et al. observed that senescent myoblasts are still able to fuse and form myotubes, yet significantly smaller and with a significant reduction in the number of nuclei per myotube and in the fusion index, compared with those obtained from young myoblasts.29 Such defects have been observed in diseases involving repeated cycles of degeneration-regeneration, such as the Duchenne muscular dystrophy (DMD), where satellite cells display a defective phenotype related to the exhaustion of proliferative capacity.30 Defective myoblasts have also been observed from biopsies of patients suffering from other neuromuscular disorders such as oculo-pharyngeal muscular dystrophy, sporadic inclusion body myositis and DM1.31-33 In a recent report, it has been demonstrated that satellite cells isolated from DM1 patients reached the proliferative senescence when their mean telomere lengths were longer than those observed in passage-matched control cells indicating that DM1 cells reach proliferative arrest prematurely, independently of telomere shortening.34 A premature growth arrest of satellite cells from congenital DM1 patients carrying large CTG expansions has also been observed, and the p16 pathway appears to be responsible for this phenomenon.35 As reported for adult DM1 myoblasts, these foetal myoblasts stop dividing when their telomeres are significantly longer than those of control cells.35 The early occurrence of senescence-related features in satellite cell-derived myoblasts suggests that satellite cells from DM1 patients have a reduced regeneration capability, which would contribute to the muscular dystrophic phenotype.
Recently, our group has shown that DM2 myoblasts at early culture passages, when they proliferate at a similar rate as the controls, already show senescence related features mainly consisting in the early appearance of cytological alterations and impairment of the pre-mRNA maturation pathways.36 The aim of our study is to elucidate if satellite cell derived myoblasts obtained from skeletal muscle of DM2 adult patients also exhibit a premature senescence as reported for DM1 and if alterations in their proliferation potential and differentiation capabilities might contribute to some of the clinical and histopathological features observed in DM2 muscles.
Materials and Methods
This study was authorized by the Institutional Ethics Committee of the Local Health Unit (ASL MI2, Melegnano, Milan, Italy) and was conducted according to the principles expressed in the Declaration of Helsinki, the institutional regulation and Italian laws and guidelines. Written informed consent were obtained from the patients for all blood samples and muscle biopsies used in this study.
Patients
Human muscle biopsies from biceps brachii muscle were taken under sterile conditions from DM1 (n=3) and DM2 (n=4) patients enrolled in Ilalian DM Registry and from age matched subjects with no sign of neuromuscular disease (n=4) used as controls. The diagnosis of DM was based upon the clinical diagnostic criteria set by the International Consortium for Myotonic Dystrophy.37 DM2 diagnosis was performed by fluorescence in situ hybridization on muscle frozen sections using a (CAGG)5 probe as previously reported by Cardani et al.38 to verify the presence of ribonuclear inclusions. DM1 and DM2 genotyping were performed on genomic DNA extracted from peripheral blood leukocytes as previously described.39-41
Muscle histopathology and immunohistochemistry
Biceps brachii biopsies were fresh-frozen in isopentane cooled in liquid nitrogen. Histopathological analysis was performed on serial sections (8 µm) processed for routine histological and histochemical stainings. A standard myofibrillar ATPase staining protocol was used after preincubation at pH 4.3, 4.6, and 10.4.42 Immunohistochemical staining was performed on serial sections (6 µm) air-dried and rehydrated in phosphate buffer pH 7.4 (PBS). Non-specific binding sites were blocked with normal goat serum (NGS; Dako, Glostrup, Denmark) at a dilution 1:20 in PBS containing 2% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO, USA) for 20 min at room temperature. Sections were then incubated for 1 h at room temperature with mouse monoclonal primary antibodies against two different myosin heavy chain (MHC): MHCfast, diluited 1:400 in PBS+2%BSA (Sigma-Aldrich); MHCslow, diluited 1:400 in PBS+2%BSA (Sigma-Aldrich). After washing in PBS 3 times for 5 min (3x5 min), sections were incubated for 1 h at room temperature with goat antimouse biotinylated secondary antibody diluted 1:300 in PBS+2%BSA. After washing in PBS 3x5 min, sections were incubated for 30 min with Vectastain ABC complex (Vector Laboratories, Burlingame, CA, USA) and then with 3,3’-Diaminobenzidine (DAB) and hydrogen peroxide for 20 min. Nuclei were counter-stained with Mayer’s hematoxylin. Quantitative evaluation of fiber diameter was made as described previously by Vihola et al.13 with Scion Image (Scion Corporation, Frederick, MD, USA) on images taken with a light microscope (1600x, original magnification). The size of muscle fibers was assessed by measuring the smallest fiber diameter and all data were elaborated using Microcal Origin (Microcal Software Inc., Northampton, MA, USA). The metahistograms were normalized to normal mean diameter for men and women. Relative atrophy and hypertrophy factors were also calculated.
Primary skeletal muscle cell cultures
The human satellite cells were isolated from muscle biopsies as reported by Cardani et al.43 Myoblasts were grown in HAM’s F10 medium (Sigma-Aldrich) supplemented with 15% FBS (Euroclone), 0.5 mg/mL albumin from bovine serum (BSA, Sigma-Aldrich), 0.5 mg/mL fetuin (Sigma-Aldrich), 0.39 µg/mL dexamethasone, 10 ng/mL epidermal growth factor, 0.05 mg/mL insulin, 3 mg/mL glucose, 100 U/mL penicillin and 100 µg/mL streptomycin (proliferative medium). For this study, cells from DM and control patients were plated at a density of 85,000 cells per 60 mm dishes or 50,000 cells per 35 mm dishes. Myogenic purity was evaluated by immunofluorescence using desmin as marker (see below). The percentage of desmin positive myoblasts was calculated as the number of positive cells vs the total number of cells observed. All starting cell populations used in this study had a myogenic purity higher than 80%. Moreover, the myogenic purity was monitored every 5 passages (7 days per passage for a total of 35 days) until senescence was reached. At least 100 cells were counted in at least 10 different optical fields randomly chosen.
Proliferative capacity and senescence
Cells from DM patients and controls were plated in triplicate at the density 50,000 per 35 mm dishes and, after 7 days, were trypsinized, counted with trypan blue and then replated at the same density (I passage; early stage). At each passage, every 7 days, the proliferative capacity was evaluated as mean population doubling (MPD) using the formula log (N/n)/log2 where N is the number of cells counted and n is the number of cells initially plated. Cultures were considered to be in senescence when cells have arrested their growth for 3 consecutive passages (late stage). As biomarker for replicative senescence, SA-β-gal, a β-galactosidase activity detectable in cultured cells undergoing replicative senescence but absent from proliferating cells, was evaluated according to Dimri et al.44 on myoblast at early and late stages of proliferation. Quantification of SA-βGal positive cells was done by counting blue stained cells in 5 randomly selected fields and expressing the percentage of positive cells respective to the total number of cells visualized in each field.
Differentiative capacity
Myoblasts at early and late stages were plated at a high density in 35 mm dishes and, when they reached 80% of confluence, proliferative medium was replaced by differentiative medium (DMEM/High Glucose supplemented with 7% FBS, in presence of 100 U/mL penicillin, 100 µg/mL streptomycin and 0.01 mg/mL insulin) to allow myoblasts fusion. This condition of plating at high density prior to inducing differentiation was chosen in order to eliminate any bias in the quantification of fusion, which could be decreased by the poor migration of senescent cells. After 5 days in differentiative medium (T5), myotubes were analyzed in term of protein expression (MHC-fast myosin and myogenin, a myogenic factor involved in myoblast differentiation; see below) and fusion index to evaluate myoblasts differentiative capacity. Fusion index was determined as number of nuclei in multinucleated myotubes expressed as a percentage of the total number of nuclei. At least 100 nuclei were counted in at least 10 different optical fields randomly chosen.
Immunofluorescence
Immunofluorescence has been performed on proliferating myoblasts at early stage, on senescent myoblasts at late stage and on myotubes obtained from myoblasts at early and late stages. Myoblast and myotube (T5) cultures were fixed in 4% paraformaldehyde for 15 min at 4°C. After fixation, cells were washed several times in PBS and then permeabilized in 0.4% Triton X-100 in PBS for 5 min. After washing in PBS, non-specific binding sites were blocked with NGS (Dako-Cytomation) at a dilution 1:20 in PBS+2% BSA for 20 min at room temperature. Myoblast and myotubes were then incubated respectively with a primary antibody mouse monoclonal anti-desmin (CD33, Dako, 1:100 in PBS+2%BSA) and a mouse monoclonal anti MHC-fast (Sigma-Aldrich, 1:600 in PBS+2%BSA) for 1 h at room temperature. After washing in PBS 3x5 min, cells were incubated for 1 h at room temperature with secondary antibodies (goat anti mouse Alexa 488-labeled; Molecular Probes, Eugene, OR, USA; 1:400 in PBS+2%BSA). After washing in PBS 3x5 min, nuclei were stained with 165 nM 4,6-diamidino-2-phenylindole (DAPI). Cells were finally mounted with Mowiol and examined using a fluorescence microscope (LEICA FW4000).
Western blot analysis
During the in vitro ageing, the expression of different proteins involved in proliferation and/or in differentiation was analyzed by western blot (WB) on myoblasts or on myotubes (T5) protein extracts. WB analyses were performed on proliferating (early stage) and non-proliferating (late stage) myoblasts and on myotubes obtained from myoblasts at early and late stage. Whole cell extracts were prepared by the addition to the plates of lysis buffer [50 mM Tris–HCl, pH 7.4 (TBS), 200 mM NaCl, 1% NP-40, 1 mM phenylmethylsulfonyl fluoride, 0.1% SDS, 0.5% sodium deoxycholate, 10 mM b-glycerophosphate, 10 mM sodium fluoride, 1 mM sodium vanadate, Protease inhibitor cocktail (Sigma Aldrich)] and rubbed with a cell scraper. After incubating on ice for 15 min, samples were centrifuged at 5700g for 15 min at 4°C, and supernatant was collected and stored at -80°C. Pellets were suspended in 50 mM TBS with 5% SDS and stored at -80°C. Protein concentration in each sample was determined by using BCA assay kit (Pierce, Rockford, IL, USA). Eight µg of protein for each sample was loaded per lane and electrophoresed on 8% or 15% sodium dodecyl sulfate-polyacrylamide gels, and than transferred to nitrocellulose membranes (Bio-Rad Laboratories). After blocking non specific sites in TBS containing 5% BSA for 30 min at 42°C, membranes were incubated overnight at 4°C with different primary antibodies diluted in TBS+5%BSA+0.1%Tween20 for mouse monoclonal antibodies, and in TBS+5%BSA+ 0.3%Tween20 for rabbit polyclonal antibodies. The expression of desmin (1:500; Clone D33; Dako-Cytomation), MyoD (1 µg/mL; Clone 5.2F; Sigma-Aldrich), p16 (1:200; Clone H-156; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and PCNA (1:5000; Clone PC10; Dako-Cytomation) was analyzed in myoblast cultures, while MHCf (1:5000; Sigma-Aldrich) and myogenin (1 µg/mL; Clone F12B; Sigma-Aldrich) were analyzed in myotubes cultures. After several washes in TBS+0.2%Tween20, membranes were incubated with horseradish peroxidase conjugated goat anti-mouse or anti-rabbit secondary antibodies diluted respectively 1:5000 and 1:10,000 in TBS+5% BSA+0.2%Tween20 (Jackson Immuno Research Laboratories, West Grove, PA, USA). Membranes were washed and immune complexes were detected using the ECL detection system (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Blots were performed in triplicate.
DNA extraction and telomere length analysis
Genomic DNA has been extracted from 1x106 DM1 (n=2), DM2 (n=4) and control (n=3) myoblasts at early and late stages with FlexiGene kit (QIAGEN, Hilden, Germany), following the manufacture’s instructions. The quantification of telomere lengths has been assessed through Southern blot analysis of terminal restriction fragments (TRF) using the TAGGG Telomere Length Assay (Roche). Briefly, 2 µg of genomic DNA from each sample were digested with HinfI and RsaI enzymes, digested fragments were resolved by gel electrophoresis in a 0.8% agarose gel and transferred onto a nylon membrane. The blotted DNA fragments were hybridized to a digoxigenin (DIG)-labeled (TTAGGG)4 probe and incubated with a anti DIG-specific antibody covalently coupled to alkaline-phosphate. Metabolized CDP-Star substrate is then detected by chemiluminescence indicating the location of the TRF on the blot. The average TRF length was determined by comparing the location of the TRF on the blot relative to a molecular weight standard. The TRF signal has been scanned and digitalized by STORM 860 (Molecular Dynamics, Sunnyvale, CA, USA) and ImageQuant software. Mean TRF lengths have been calculated for each sample on three independent gels, as reported previously by Kimura et al.45 The number of cell division being known, the shortening velocity of telomeres has been determined, and it has been calculated whether in DM cells the telomere loss is higher than in controls.
Statistical analysis
To determine significance between two groups, comparisons were made using an unpaired Student’s t-test. The groups were considered statistically different at P<0.05.
Results
Patients and skeletal muscle histopathology
This study was performed on DM1 and DM2 age-matched patients compared to subjects with no sign of neuromuscular disease used as controls. Although the number of patients analyzed in this study was small, DM1 and DM2 groups were homogeneous: the DM1 cohort was represented by 3 classical adult patients mildly affected with a range of CTG repeat expansion of 361.6±102.8 (E2) and the MIRS was 3-4, the DM2 cohort was represented by 4 patients with classical PROMM phenotype and fluctuating myotonia, except for patient #3 (Table 1).
Table 1.
Clinical data of patients analyzed in this study.
| Patient | Sex | Age at onset | Age at biopsy | CTG expansion | Myotonia | MIRS° | MRC# |
|---|---|---|---|---|---|---|---|
| CTR-1 | F | - | 43 | - | Not present | - | - |
| CTR-2 | M | - | 44 | - | Not present | - | - |
| CTR-3 | M | - | 48 | - | Not present | - | - |
| CTR-4 | F | - | 48 | - | Not present | - | - |
| DM1-1 | M | 38 | 39 | 370 | ± | 3 | 128.35 |
| DM1-2 | F | 35 | 45 | 460 | ++ | 4 | 128.35 |
| DM1-3 | M | 39 | 47 | 255 | + | 4 | 105.36 |
| DM2-1 | F | 35 | 51 | - | + | - | 118.70 |
| DM2-2 | F | 35 | 53 | - | ± | - | 132.99 |
| DM2-3 | F | 19 | 45 | - | ++ | - | 128.68 |
| DM2-4 | F | 54 | 59 | - | + | - | 109.68 |
°Muscle Impairment Rating Scale, stage of the disease for myotonic dystrophy type 1 (DM1) patients.53
#Medical Research Council, scale for muscle strength scale (0-5 grade) on 15 muscles at both sides in the upper and lower limbs for a total of 150 maximum score. CTR, control; DM1, myotonic dystrophy type 1; DM2, myotonic dystrophy type 2; F, female; M, male.
Analysis of muscle sections immunostained for MHC fast or slow myosin allows us to detect and measure fibers smaller than 5 µm, including all nuclear clump fibers which are recognizable by the presence of a thin rim of immunoreaction around the nuclei (Figure 1 A,B). All patients used in this study show an increase of fibre atrophy. In particular, an increase of both type 1 and type 2 fiber atrophy is present in DM1 patients and a prevalent type 2 fibre atrophy is evident in DM2 patients. Hypertrophic type 1 and type 2 fibers were also present in DM patients (Figure 1C). The evaluation of atrophy (AF) and hypertrophy (HF) factors for each patient group are reported in Figure 1D.
Figure 1.
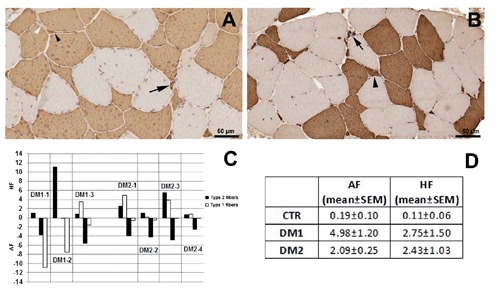
Cross-sections of skeletal muscle from DM1 (A) and DM2 (B) patient after immunostaining for fast myosin. Note the presence of type 1 (white arrowhead) and type 2 (black arrowhead) atrophic fibers in DM1 section (A), while in DM2 section (B) type 2 fibre atrophy (black arrowhead) is evident. Black arrows indicate fast positive nuclear clumps. Relative hypertrophy factor (HF) and atrophy factor (AF) of each DM1 and DM2 patients (C) and mean values ± standard error of the mean (SEM) of HF and AF for each patient group (D). The results are based on the morphometric analysis of sections immunostained for MHC fast or slow myosin.
Myoblast cultures and proliferative capacity
The myogenic purity of cell preparations was evaluated from the percentage of cells expressing desmin. Immunofluorescence assay revealed that the cell preparations used in this study were quite similar in terms of the desmin expression. The myogenic purity was around 75-80% throughout the experimental procedures (data not shown). Also MyoD protein expression indicated that the myogenic purity remained similar in the early and late stages both in DM and control cultures (Figure 2 A,B).
Figure 2.

A) Representative western blot analysis of MyoD expression in myoblasts at early (e) and late (l) passages of proliferative lifespan obtained from biceps brachii muscle samples from healthy (CTR), DM1 and DM2 patients; density of the bands has been normalized to β-tubulin expression used as internal control. B) Histograms representing mean values of MyoD protein expression analysed by densitometry of CTR (n=4), DM1 (n=3) and DM2 (n=4) patients; scale bars are for SEM. MyoD expression is similar in young and senescent cells indicating that the myogenic purity persists to be high throughout the experiment. C) Lifespan plots of control and DM myoblasts. D) Mean of max number of divisions of CTR (n=4), DM1 (n=3) and DM2 (n=4) patients; scale bars represent SEM; the average proliferative lifespan of the DM myoblasts is reduced as compared to that of control cells. E) Representative western blot analyses of PCNA in the early (e) and late (l) stages of myoblast proliferative lifespan; the results have been normalized to the expression of β-tubulin. F) Histograms representing mean values of PCNA protein expression analyzed by densitometry of CTR (n=4), DM1 (n=3) and DM2 (n=4) patients; scale bars represent SEM; levels of PCNA are significantly decreased in both DM and control senescent cells compared to young cells. *P<0.05, **P<0.01.
The in vitro proliferative capacity of myoblasts obtained from skeletal muscle biopsies of 3 DM1 and 4 DM2 patients was compared to that of myoblasts obtained from 4 age-matched unaffected individuals used as controls. As shown in Figure 2C, an interindividual variability was evident in both DM1 and DM2 patient group, however the average proliferative lifespan of the DM1 and DM2 myoblasts was reduced by 50% and 36% respectively as compared to that observed in control cells. Moreover, DM1 (9 passages) and DM2 (10 passages) myoblasts stopped dividing earlier compared with control cultures (15 passages) (Figure 2C). Myoblasts from DM1 and DM2 patients showed a clear reduction of the maximum number of divisions compared to those observed in controls cells (Figure 2D). Analysis of PCNA expression showed a significant reduction of protein expression in the late passages compared to the early stages both in DM and control cells (Figure 2E,F). The expression of PCNA in the late passages indicated that the majority of DM and control cells were arrested in G1 phase of the cell cycle.
Senescence analysis
To evaluate if a mechanism similar to senescence was responsible for the early proliferative growth arrest of DM myoblasts, we analyzed biomarkers usually observed in senescent cells. At early passages DM and control cells were actively dividing with rare or no senescent cells being detected but as the cells reached their late passage, the number of senescent cells positive for SA-β gal increased dramatically in both DM and control cells (Figure 3). Moreover, DM and control myoblasts at the late stages of proliferation showed a flattened morphology with an enlarged cytoplasm and extended cytosolic processes, a morphology reminiscent of that observed when myoblasts reach a senescent state (Figure 4).
Figure 3.
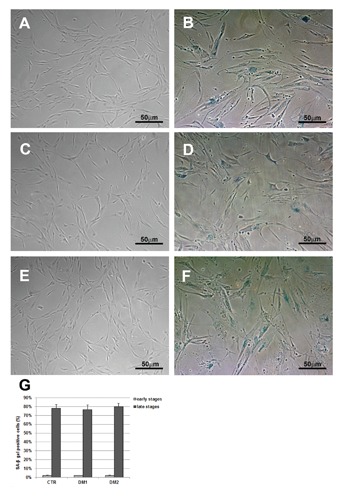
Representative images of CTR (A,B), DM1 (C,D) and DM2 (E,F) myoblasts stained for the senescence-associated β-galactosidase (SA-βGal) activity. At the proliferative (early) stages, very few cells were positive (A,C,E). At the high cell passage (late stages), myoblasts appear large, with the characteristic cytoplasmic blue staining typical of senescence (B,D,F). G) Quantification of the amount of SA-βGal positive cells shows a dramatic increase in the percentage of positive cells at the late stages; scale bars represent SEM.
Figure 4.

Representative images of CTR (A,B), DM1 (C,D) and DM2 (E,F) myoblasts immunostained for the myogenic marker, desmin. Both DM and control myoblasts at early stages of proliferation were relatively small and elongated (A,C,E), while at the late stages of proliferation they showed a flattened morphology with enlarged cytoplasm and extended cytosolic processes (B,D,F). Nuclei have been visualized with DAPI (blue).
We examined the expression of p16 protein at the beginning and at the end of DM lifespan to verify if this mechanism was also involved in the premature proliferative arrest observed in DM2 myoblasts in vitro. Our analysis indicate that p16 was overexpressed in DM1 cells both at the early and late stages compared to controls, while p16 protein expression appeared to be similar in DM2 and control myoblasts at both stages analyzed (Figure 5 A,B). Critically short telomeres trigger loss of cell viability in tissues, which has been related to alteration of tissue function and loss of regeneration tissue capabilities. Since it has been observed that DM1 myoblasts stopped dividing prematurely independently of telomere shortening,34,35 we have analyzed telomere loss induced by in vitro aging in satellite-cell-derived myoblasts, isolated from muscles of DM1 (n=2), DM2 (n=4) patients and from age-matched control subjects (n=3). As shown in Figure 5C, satellite cells isolated from DM1 patients stopped growing prematurely with telomeres longer than controls (respectively 11.4±1.1 Kb and 9.5±0.3 Kb), differently from what observed in DM2 myoblasts which stop dividing with a median telomere length of 8.3±1.7 Kb even if a wide variability in mean TRF length can be observed in DM samples. It should be noted that the two DM2 cultures (#2 and #3, Figure 2C) showing the most premature growth arrest also showed the shortest telomere length at the early stages probably indicating an excessive proliferation in vivo before their isolation (data not shown). Moreover these two DM2 patients also show the highest values of atrophy factor in vivo (Figure 1C). As shown in Figure 5D, DM2 satellite cells has a clear two-fold decrease in the amount of telomeric DNA at every cell passage compared to controls whereas in DM1 cells the mean bp loss per division is 30% lower than controls.
Figure 5.
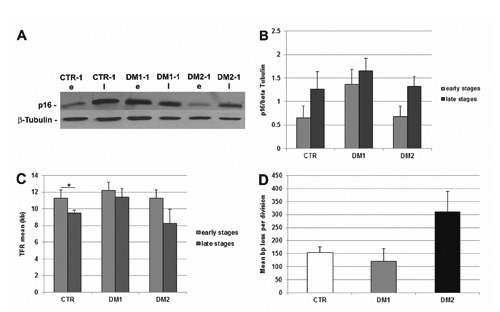
DM premature senescence pathway. A) Representative western blot analyses of p16 in the early (e) and late (l) stages of myoblast proliferative lifespan; the results have been normalized to the expression of β-tubulin. B) Histograms representing mean values of p16 protein expression analysed by densitometry in healthy (CTR; n=4), DM1 (n=3) and DM2 (n=4) patients; at both stages analyzed, p16 was more expressed in DM1 cells as compared to the controls while the expression was similar in DM2 and control myoblasts; scale bars represent SEM. C) Mean length of telomeric restriction fragments (TRF) measured on DM2 (n=4) and control (n=4) myoblasts at proliferative and senescent stage; senescent DM2 cells had shorter telomeres than the proliferating cells (*P<0.05); scale bars represent SEM. D) Mean length (in bps) of telomeric DNA lost per division in control and DM2 myoblasts; DM2 cells lost more bps per division than control cells; scale bars represent SEM.
Differentiative capacity
Myoblasts at early and late stages of proliferation were induced to fuse into myotubes during 5 days of differentiation (T5) in order to evaluate their differentiative capacity. Although the senescent cells were still able to form myotubes, they appeared to be significantly smaller than those formed by the young cells, which also formed branched myotubes not observable in senescent cultures (Figure 6 A-F). As observed in Figure 6G, a significant reduction in fusion index is observable both in DM and control myoblasts which has reached a proliferative arrest compared to proliferating myoblasts. This reduction appears to be more evident in senescent myoblasts from DM1 patients who also show higher value of atrophy factor compared to DM2. Moreover, the expression of myogenin appeared to be decreased both in DM and control myotubes obtained from non-proliferating myoblasts as compared to those obtained from proliferating myoblasts (Figure 6 H,I).
Figure 6.
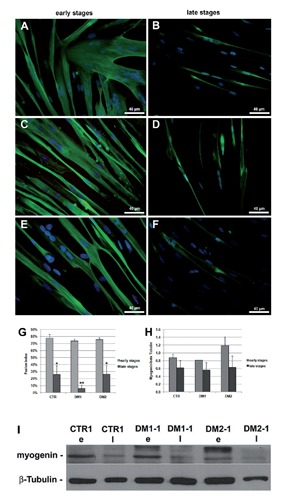
Differentiation capability of young and senescent myoblasts. A-F) Fast myosin immunofluorescence (green) of control (A,B), DM1 (C,D) and DM2 (E,F) myotubes from myoblasts at early and late stages of their proliferative lifespan, after 5 days of differentiation (T5). Nuclei have been visualized with DAPI (blue). Young myoblasts generate well-differentiated and cross-striated (C,E) myotubes larger than those formed by the senescent cells. G) Fusion index, i.e. the number of nuclei incorporated into myotubes as a percentage of the total number of nuclei, has been calculated in young and senescent myoblasts after 5 days of differentiation; a significant reduction of fusion index is observable in both DM and control myotubes obtained from senescent cells (*P<0.05, **P<0.01). H) Histograms represents mean values of myogenin protein expression analysed by densitometry of control (n=4), DM1 (n=3) and DM2 (n=4) patients; a reduced myogenin expression is observed in myotubes from senescent cells as compared to those obtained from young cells; scale bars represent SEM. I) Representative western blot analyses of myogenin in myotubes obtained from myoblasts at the early (e) and late (l) stages of their proliferative lifespan; the results have been normalized to the expression of β-tubulin.
Discussion
Many symptoms of adult form of DM1 and DM2, such as muscle weakness and wasting, cataracts, and cardiac arrhythmias, are reminiscent to normal aging. Recent data on dystrophic skeletal muscle myonuclei have demonstrated alterations of mRNA pathways similar to those observed during aging.19,46-48 Increasing evidence has demonstrated that in age-related myopathies, such as sarcopenia and myotonic dystrophy, the progressive muscle weakness and atrophy are characterized by impaired muscle regeneration due to satellite cells premature senescence that limits their proliferative potential.1,35,49,50 In DM1 muscles the number of satellite cells is increased compared to muscles from non-affected individuals; however, DM1 cells do not seem to be able to counteract the progressive muscle atrophy due to a reduced proliferative capacity triggered by a mechanism of premature growth arrest.34
It is known that primary DM myoblasts do not show evident morphological abnormalities and are capable of normally differentiating;43,51,52 however, we have recently observed that DM2 myoblasts are characterized by senescence related features mainly consisting in the early appearance of cytological alterations and impairment of the pre-mRNA maturation pathways.36 To further investigate DM2 myoblasts aging, we have analyzed if cultured myoblasts from DM2 patients differ from myoblasts of age-matched DM1 and normal individuals in terms of cell proliferation, morphology, differentiation and senescence during in vitro aging and if alterations in their proliferation potential and differentiation capabilities might contribute to some of the clinical and histopathological features observed in DM2 muscles.
Our results seem to indicate that DM1 and DM2 myoblasts are characterized by a premature proliferative growth arrest compared to healthy myoblasts. Moreover, a mechanism similar to senescence appear is to be responsible for the early proliferative growth arrest because DM cells expressed biomarkers usually observed in senescent cells much earlier in their replicative lifespan as compared with age-matched control cultures. These data suggest that the in vivo regenerative capacity of DM satellite cells might be constitutively impaired. Furthermore, while the p16 pathway appear to be responsible for the premature growth arrest in DM1 probably in response to a CTG-related stress,35 our results suggest that this mechanism is not responsible for the proliferative arrest observed in DM2 myoblasts. On the contrary, differently from what observed in DM1 myoblasts and reported in literature,33-35 DM2 myoblasts stop dividing with telomeres shorter than controls suggesting that the signaling involved in premature senescence depend on a telomere-driven pathway. CCTG expansion might interfere with the telomere homeostasis in DM2 cells, yet the mechanism causing an accelerated telomere shortening has to be clarified. Critically short telomeres trigger loss of cell viability in tissues, which has been related to alteration of tissue function and loss of regeneration tissue capabilities. Each time a satellite cell divides, its DNA loses a small portion of this telomeric sequence;53 once a critical length is reached, the cell becomes senescent and can no longer play a role in regeneration. Telomere length is an important predictive biomarker indicative for the regenerative capacity of human satellite cells as shown in Duchenne muscular dystrophy in which a telomere shortening after continuous cycles of degeneration and regeneration has been reported.30 Hence, it appears that CTG and CCTG expansions trigger in vitro a mechanism of myoblast premature senescence through two different pathways, which could explain the different histological alterations observed between DM1 and DM2 skeletal muscle as for example the selective type 2 fibre atrophy present in DM2 muscle. However, further studies will be necessary to better understand how different senescence mechanisms involved in DM1 and DM2 myoblasts could lead to the selective involvement of type 1 or type 2 fibers in DM1 and DM2 respectively. As previously reported,29,54 our results indicate that replicative senescence deregulates the myogenic programme resulting in impaired myogenesis. A decrease of the expression of myogenin, a myogenic factor involved in myoblast differentiation, is observable in myotubes obtained from senescent myoblasts as compared to those observed from proliferating myoblasts. Moreover, even though the senescent myoblasts are still able to fuse, a significant reduction in the fusion index has been observed when compared with young cells. It should be noted that the reduction in fusion index appear to be more evident in senescent myoblasts obtained from DM1 patients indicating that myoblasts deficiency could be responsible of the more severe muscle histopathology observed in DM1 compared to DM2. The reduced differentiating capacity is due to defect in differentiation and/or fusion properties of the senescent myoblasts and not to a decrease in the motility of senescent cells, since differentiation in senescent cultures was induced in myoblasts seeded at very high density. Thus, the histopathological defects observed in DM muscle such as fibre atrophy and nuclear clumps, could be due at least in part to the inability of premature aged myoblasts to generate myotubes able to produce mature skeletal muscle fibers or to fuse with existing fibers and prevent them from atrophy. Although this study has been carried out on a limited number of cases, it seems indicate that, as in DM1 patients, myoblast premature senescence could explain the skeletal muscle wasting observed in patients affected by DM2.
Funding Statement
Funding: this research was funded by the FMM - Fondazione Malattie Miotoniche (www.fondazionemalattiemiotoniche.org) and AFM-Association Française contre les Myopathies. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Harper PS. Myotonic dystrophy, 3th ed.Sanders, London, UK, 2001. [Google Scholar]
- 2.Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcipt encoding a protein kinase family member. Cell 1992;68:799-808. [DOI] [PubMed] [Google Scholar]
- 3.Fu YH, Pizzuti A, Fenwick RG, Jr, King J, Rajnarayan S, Dunne PW, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992;255:1256-8. [DOI] [PubMed] [Google Scholar]
- 4.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3’ untranslated region of the gene. Science 1992;255:1253-5. [DOI] [PubMed] [Google Scholar]
- 5.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001;293:864-7. [DOI] [PubMed] [Google Scholar]
- 6.Ranum LP, Day JW. Myotonic dystrophy: RNA pathogenesis comes into focus. Am J Hum Genet 2004;74:793-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller JW, Urbinati CR, Teng-umnuay P, Stenberg MG, Byrne BJ, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 2000;19:4439-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, et al. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet 2001;10:2165-70. [DOI] [PubMed] [Google Scholar]
- 9.Timchenko NA, Cai ZJ, Welm AL, Reddy S, Ashizawa T, Timchenko LT. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem 2001;276:7820-6. [DOI] [PubMed] [Google Scholar]
- 10.Perdoni F, Malatesta M, Cardani R, Giagnacovo M, Mancinelli E, Meola G, et al. RNA/MBNL1-containing foci in myoblast nuclei from patients affected by myotonic dystrophy type 2: an immunocytochemical study. Eur J Histochem 2009; 53:151-58. [DOI] [PubMed] [Google Scholar]
- 11.Osborne RJ, Thornton CA. RNA-dominant diseases. Hum Mol Genet 2006;15:R162-9. [DOI] [PubMed] [Google Scholar]
- 12.Meola G, Cardani R. RNA binding proteins in myotonic dystrophies, pp. 153-66. In: Denman RB. (ed.) RNA binding proteins in development and disease. Research Signpost, Kerala, 2009. [Google Scholar]
- 13.Vihola A, Bassez G, Meola G, Zhang S, Haapasalo H, Paetau A, et al. Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology 2003;60:1854-7. [DOI] [PubMed] [Google Scholar]
- 14.Bassez G, Chapoy E, Bastuji-Garin S, Radvanyi-Hoffman H, Authier FJ, Pellisser JF, et al. Type 2 myotonic dystrophy can be predicted by the combination of type 2 muscle fibre central nucleation and scattered atrophy. J Neuropathol Exp Neurol 2008;67:319-25. [DOI] [PubMed] [Google Scholar]
- 15.Pisani V, Panico MB, Terracciano C, Bonifazi E, Meola G, Novelli G, et al. Preferential central nucleation of type 2 myofibers is an invariable feature of myotonic dystrophy type 2. Muscle Nerve 2008;38:1405-11. [DOI] [PubMed] [Google Scholar]
- 16.Shavlakadze T, Grounds M. Of bears, frogs, meat, mice and men: complexity of factors affecting skeletal muscle mass and fat. Bioessays 2006;28:994-1009. [DOI] [PubMed] [Google Scholar]
- 17.Mouly V, Aamiri A, Bigot A, Cooper RN, Di Donna S, Furling D, et al. The mitotic clock in skeletal muscle regeneration, disease and cell mediated gene therapy. Acta Physiol Scand 2005;184:3-15. [DOI] [PubMed] [Google Scholar]
- 18.Schiaffino S, Dyar KA, Ciciliot S, Blaauw B, Sandri M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J 2013;280:4294-314. [DOI] [PubMed] [Google Scholar]
- 19.Moss FP, Leblond CP. Satellite cells as the source of nuclei in muscles of growing rats. Anat Rec 1971;170:421-35. [DOI] [PubMed] [Google Scholar]
- 20.Malatesta M. Skeletal muscle features in myotonic dystrophy and sarcopenia: do similar nuclear mechanisms lead to skeletal muscle wasting? Eur J Histochem 2012;56:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Relaix F, Zammit PS. Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development 2012;139:2845-56. [DOI] [PubMed] [Google Scholar]
- 22.Bischoff R, Heintz C. Enhancement of skeletal muscle rigeneration. Dev Dyn 1994;201:41-54. [DOI] [PubMed] [Google Scholar]
- 23.Cooper RN, Tajbakhsh S, Mouly V, Cossu G, Buckingham M, et al. In vivo satellite cell activation via Myf5 and MyoD in rigenerating mouse skeletal muscle. J Cell Sci 1999; 112:2895-901. [DOI] [PubMed] [Google Scholar]
- 24.Renault V, Thornell LE, Butler-Browne G, Mouly V.Human skeletal muscle satellite cells: aging, oxidative stress and the mitotic clock. Exp Gerontol 2002;37:1229-36. [DOI] [PubMed] [Google Scholar]
- 25.Renault V, Thornell LE, Eriksson PO, Butler-Browne G, Mouly V. Regenerative potential of human skeletal muscle during aging. Aging Cell 2002;1:132-9. [DOI] [PubMed] [Google Scholar]
- 26.Wright WE, Shay JW. Historical claims and current interpretations of replicative aging. Nat Biotechnol 2002;20:682-8. [DOI] [PubMed] [Google Scholar]
- 27.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science 1998;279:349-52. [DOI] [PubMed] [Google Scholar]
- 28.Zhu CH, Mouly V, Cooper RN, Mamchaoui K, Bigot A, Shay JW, et al. Cellular senescence in human myoblasts is overcome by human telomerase reverse transcriptase and cyclin dependent kinase 4: consequences in aging muscle and therapeutic strategies for muscular dystrophies. Aging Cell 2007;6:515-23. [DOI] [PubMed] [Google Scholar]
- 29.Bigot A, Jacquemin V, Debacq-Chainiaux F, Butler-Browne G, Toussaint O, Furling D, et al. Replicative aging down-regulates the myogenic regulatory factors in human myoblasts. Biol. Cell 2008;100:189-99. [DOI] [PubMed] [Google Scholar]
- 30.Decary S, Hamida CB, Mouly V, Barbet JP, Hentati F, Butler-Browne GS. Shorter telomeres in dystrophic muscle consistent with extensive regeneration in young children. Neuromuscul Disord 2000;10:113-20. [DOI] [PubMed] [Google Scholar]
- 31.Périé S, Mamchaoui K, Mouly V, Blot S, Bouazza B, Thornell LE, et al. Premature proliferative arrest of cricopharyngeal myoblasts in oculo-pharyngeal muscular dystrophy: therapeutic perspectives of autologous myoblast transplantation. Neuromuscul Disord 2006;16:770–81. [DOI] [PubMed] [Google Scholar]
- 32.Morosetti R, Broccolini A, Sancricca C, Gliubizzi C, Gidaro T, Tonali PA, et al. Increased aging in primary muscle cultures of sporadic inclusion-body myositis. Neurobiol Aging 2010;31:1205-14. [DOI] [PubMed] [Google Scholar]
- 33.Furling D, Coiffier L, Mouly V, Barbet JP, Lacau St Guily J, Taneja K, et al. Defective satellite cells in congenital myotonic dystrophy. Hum Mol Genet 2001;10:2079-87. [DOI] [PubMed] [Google Scholar]
- 34.Thornell LE, Lindstöm M, Renault V, Klein A, Mouly V, Ansved T, et al. Satellite cell dysfunction contributes to the progressive muscle atrophy in myotonic dystrophy type 1. Neuropathol Appl Neurobiol 2009;35: 603-13. [DOI] [PubMed] [Google Scholar]
- 35.Bigot A, Klein AF, Gasnier E, Jacquemin V, Ravassard P, Butler-Brown G, et al. Large CTG repeats trigger p16-dependent premature senescence in myotonic dystrophy type 1 muscle precursor cells. Am J Pathol 2009;174:1435-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malatesta M, Giagnacovo M, Renna LV, Cardani R, Meola G, Pellicciari C. Cultured myoblasts from patients affected by myotonic dystrophy type 2 exhibit senescence-related features: ultrastructural evidence. Eur J Histochem 2011;55:e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moxley RT, 3rd, Meola G, Udd B, Ricker K. Report of the 84th ENMC workshop: PROMM (proximal myotonic myopathy) and other myotonic dystrophy-like syndromes: 2nd workshop. 13-15th October. 2000, Loosdrecht: The Netherlands: Neuromuscul Disord 2002;12:306-17. [DOI] [PubMed] [Google Scholar]
- 38.Cardani R, Mancinelli E, Sansone V, Rotondo G, Meola G. Biomolecular identification of (CCTG)n mutation in myotonic dystrophy type 2 (DM2) by FISH on muscle biopsy. Eur J Histochem 2004;48:437-42. [DOI] [PubMed] [Google Scholar]
- 39.Botta A, Bonifazi E, Vallo L, Gennarelli M, Garrè C, Salesi L, et al. Italian Guidelines for molecular analysis in myotonic dystrophy. Acta Myol 2006;25:23-33. [PubMed] [Google Scholar]
- 40.Bonifazi E, Vallo L, Giardina E, Botta A, Novelli G. A long PCR-based molecular protocol for detecting normal and expanded ZNF9 alleles in myotonic dystrophy type 2. Diagn Mol Pathol 2004;13:164-6. [PubMed] [Google Scholar]
- 41.Valaperta R, Sansone V, Lombardi F, Verdelli C, Colombo A, Valisi M, et al. Identification and characterization of DM1 patients by a new diagnostic certified assay: neuromuscular and cardiac assessments. Biomed Res Int 2013;2013:958510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dubowitz V. Muscle biopsy, pp. 19-40. In: Dubowitz V. (ed.) A practical approach. Bailliere Tindall, London, 1985. [Google Scholar]
- 43.Cardani R, Baldassa S, Botta A, Rinaldi F, Novelli G, Mancinelli E, et al. Ribonuclear inclusions and MBNL1 nuclear sequestration do not affect myoblast differentiation but alter gene splicing in myotonic dystrophy type 2. Neuromuscul Disor 2009;19:335-43. [DOI] [PubMed] [Google Scholar]
- 44.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995;92:9363-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kimura M, Stone RC, Hunt SC, Skurnick J, Lu X, Cao X, et al. Measurement of telomere length by the Southern blot analysis of terminal restriction fragment lengths. Nat Protoc 2010;5:1596-607. [DOI] [PubMed] [Google Scholar]
- 46.Malatesta M, Giagnacovo M, Cardani R, Meola G, Pellicciari C. RNA processing is altered in skeletal muscle nuclei of patients affected by myotonic dystrophy. Histochem Cell Biol 2011;135:419-25. [DOI] [PubMed] [Google Scholar]
- 47.Malatesta M, Perdoni F, Muller S, Zancanaro C, Pellicciari C. Nuclei of aged myofibres undergo structural and functional changes suggesting impairment in RNA processing. Eur J Histochem 2009; 53:97-106. [DOI] [PubMed] [Google Scholar]
- 48.Malatesta M, Giagnacovo M, Costanzo M, Cisterna B, Cardani R, Meola G. Muscleblind-like1 undergoes ectopic relocation in the nuclei of skeletal muscles in myotonic dystrophy and sarcopenia. Eur J Histochem 2013;57:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Machida S, Narusawa M. The roles of satellite cells and hematopoietic stem cells in impaired regeneration of skeletal muscle in old rats. Ann N Y Acad Sci 2006;1067: 349-53. [DOI] [PubMed] [Google Scholar]
- 50.Verdijk LB, Koopman R, Schaart G, Meijer K, Savelberg HH, van Loon LJ. Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. Am J Physiol Endocrinol Metab 2007;292:E151-7. [DOI] [PubMed] [Google Scholar]
- 51.Pelletier R, Hamel F, Beaulieu D, Patry L, Haineault C, Tarnopolsky M, et al. Absence of a differentiation defect in muscle satellite cells from DM2 patients. Neurobiol Dis 2009;36:181-90. [DOI] [PubMed] [Google Scholar]
- 52.Loro E, Rinaldi F, Malena A, Masiero E, Novelli G, Angelini C, et al. Normal myogenesis and increased apoptosis in myotonic dystrophy type-1 muscle cells. Cell Death Differ 2010;17:1315-24. [DOI] [PubMed] [Google Scholar]
- 53.Decary S, Mouly V, Hamida CB, Sautet A, Barbet JP, Butler-Browne GS. Replicative potential and telomere length in human skeletal muscle: implications for satellite cell-mediated gene therapy. Hum Gene Ther 1997;8:1429-38. [DOI] [PubMed] [Google Scholar]
- 54.Lorenzon P, Bandi E, de Guarrini F, Pietrangelo T, Schäfer R, Zweyer M, et al. Ageing affects the differentiation potential of human myoblasts. Exp Gerontol 2004; 39:1545-54. [DOI] [PubMed] [Google Scholar]
- 55.Mathieu J, Boivin H, Meunier D, Gaudreault M, Bégin P. Assessment of a disease-specific muscular impairment rating scale in myotonic dystrophy. Neurology 2001;56:336-40. [DOI] [PubMed] [Google Scholar]