

Abstract
Background
Parasitological methods for the diagnosis of Visceral leishmaniasis (VL) require invasive procedures, so serological and molecular approaches have been developed but are not generally applicable in the field. We evaluated a loop mediated isothermal amplification (LAMP) assay using blood from VL patients and compared it to nested PCR.
Methods
Forty-seven subjects with clinical features (fever, hepatosplenomegaly and anemia) were confirmed positive for VL by the direct agglutination test (DAT) at titers >3200. Forty DAT negative individuals from non-endemic areas with no clinical signs or symptoms of VL served as controls. A LAMP assay was performed using a set of six primers targeting Leishmania infantum kinetoplast DNA (kDNA) minicircle gene under isothermal (64 °C) conditions. For nested PCR we used primers targeting the kDNA minicircle gene.
Results
The LAMP assay provided a detection limit of 1 parasite in 1 ml of peripheral blood and detected L. infantum DNA in 44 of 47 DAT-confirmed VL cases, with diagnostic sensitivity of 93.6% (95% CI). No L. infantum DNA was amplified in controls, indicating a specificity of 100%. The nested PCR yielded sensitivity of 96% (95% CI) and a specificity of 100% (95% CI).
Conclusion
The LAMP assay gave results similar to those of nested PCR but in a shorter time. The LAMP method is simple; requires no sophisticated equipment; has a short reaction time; and results, indicated by turbidity of the reaction mixture, are observable with the naked eye.
Keywords: Visceral leishmaniasis, Human, LAMP, Nested PCR, Peripheral blood, Iran
Introduction
Visceral leishmaniasis (VL) is a major public health problem in Iran, with endemic areas including Ardabil and East Azerbaijan in the northwest and the provinces of Fars and Bushehr in the south (1). The disease is frequently fatal if untreated, with a worldwide incidence of almost 500 000 and 50 000 deaths annually (2). Early definitive diagnosis of VL is of crucial to the initiation of therapy to prevent severe complications. The parasitological techniques conducted on bone marrow aspirates that are generally used for detection and identification of VL lack sensitivity (microscopy) or are time consuming (culture). They require invasive procedures and are difficult to repeat for patient follow-up. Serological methods such as the Direct Agglutination Test (DAT) have been used successfully for the diagnosis of VL (3-4). Direct Agglutination is appropriate for the detection of VL in immunocompetent patients when large numbers of specific antibodies are present but has a low sensitivity in immunocompromized patients, including HIV-infected, and is of limited value in evaluating treatment effectiveness, since the antibodies remain detectable for an extended time after clinical cure (5-6). Antigen detection in urine through the latex agglutination test (KAtex) has been suggested as a tool for diagnosis of VL, but several studies have reported poor to moderate sensitivity (7-10). Molecular methods based on DNA amplification, such as nested PCR and real-time quantitative PCR, have been developed and evaluated as potential tools for rapid and sensitive detection of leishmaniasis (11-13). To avoid invasive procedures, peripheral blood and buffy coat is often used. Reported sensitivity levels of PCR with blood have ranged from 70% to 96% (11, 14-16). However, cost and the necessity for sophisticated laboratory equipment and skilled technicians render this technology inappropriate for routine diagnosis in hospital laboratories and under field conditions in areas where VL is endemic. More recently, a rapid, simple, and sensitive technique called loop-mediated isothermal amplification (LAMP) was developed (17). The assay has been used for detection of bacterial, viral, protozoan, and fungal diseases (18-21), and used successfully for the diagnosis of several parasitic infections (22-24).
The goal of this study was to use the kineto DNA (kDNA) region to design LAMP primers for detection of Leishmania infantum DNA in human peripheral blood samples and compare the results to those of nested PCR. This is the first report of the LAMP assay to detect L. infantum DNA in human peripheral blood samples in Iran.
Materials and Methods
Ethical approval
This study was reviewed and approved by the Tehran University of Medical Sciences Ethics Committee and all subjects provided written informed consent.
Patients and controls
Peripheral blood samples were obtained from 47 patients at the Tehran University of Medical Sciences Hospitals (Children’s Medical Centers, Aliasghar Child Hospital, Bahrami Children’s Hospital) with confirmed VL. The patients presented characteristic clinical symptoms of fever, hepatosplenomegaly, and anemia consistent with infection in bone marrow aspirate detected by microscopy (n=22) and serology (DAT ≥ 3200). Thirty individuals from non-VL endemic areas with no clinical signs or symptoms of VL and negative DAT results, as well as 10 patients exhibiting non-VL infectious diseases (cutaneous leishmaniasis, malaria, tuberculosis, toxoplasmosis, hepatitis, herpes virus) constituted controls.
Microscopy
Bone marrow samples were obtained from VL patients and smears were prepared on glass slides, dried, fixed in methanol, stained with Giemsa in PBS, and examined microscopically at 1000 x for Leishmania sp.
Serology
Leishmania antibodies were detected by DAT (1, 3). DAT antigens were prepared from L. infantum promastigotes at the Protozoology Unit of the School of Public Health at Tehran University of Medical Sciences. Sera were diluted with 0.9% saline containing 0.8% mercaptoethanol. Initially, for screening purposes, two dilutions of 1: 800 and 1: 3200 were made and tested. If both of these two dilutions were positive, the samples with titers 1: 800 were diluted further to give end-point titers of 1: 102400. Negative control wells (antigen only) and known negative and positive controls were tested in each plate daily. Fifty μl of antigen was added to wells containing 50 μl of diluted serum and incubated overnight. The cut-off value was defined as the highest dilution at which agglutination was visible as a blue dot compared to negative control wells, which had clear blue dots. Specific Leishmania antibodies ≥ 1:3200 were considered positive.
DNA preparation
The DNA template for the nested PCR and LAMP assay was extracted from the buffy coat layer obtained from 2-5 ml whole peripheral blood collected in EDTA using the QI Aamp DNA Blood mini kit (Qiagen, Cortaboeuf, France) according to manufacturer’s instructions. Following centrifugation and washing, DNA was eluted from the spin column in 100 μl elution buffer and stored at -20 oC before use in the PCR-based assay. The concentration and purity of extracted DNA were assessed by the ratio of the optical density at 260 and 280 nm. Five-μl and 2 μl portions of the extracted DNA were used for nested PCR amplification and LAMP assay, respectively.
Nested PCR-based Assay
For nested PCR, the specific nucleotide sequences of variable segments of kDNA minicircles from L. infantum were amplified as described by Noyes et al. with slight modification (25). Each 25 μl first-round reaction mixture contained 5 μl template DNA, 0.2 mM each dNTPs (Roche, Penzberg, Germany), 1.5 mM MgCl2, 1.0 U Taq polymerase, 50 mMTris-HCl (pH 7.6), and 20 pmol of each of CSB1XR and CSB2XF primers (first-round primer). The DNA amplification was carried out under the following conditions: 95 °C for 5 min; 35 cycles of 30 s at 95 °C, 1 min at 59 °C, and 45 min at 72 °C; and a final extension of 10 min at 72 °C. The first PCR product was diluted 10-fold in ultrapure water. One μl was used as a template for the second amplification, under the same conditions and reaction mixture as the first round, except that LiR and 13Z (second-round primer) were used as the primers. The amplified products of the second round were visualized in 2% agarose gels stained with ethidium bromide. The DNA purified from promastigote cultures of the reference strain of L. infantum (MHOM/TN/80/IPT1) were amplified in each PCR experiment and used as positive control. Negative controls (the products of PCR in which ultrapure water replaced the template DNA) were also run. An internal control (β-glubulin housekeeping gene) was used to ensure that negative results were genuine and did not reflect PCR inhibition, DNA loading, sample degradation, or absence of human cells.
Loop-mediated isothermal amplification (LAMP)
A set of six oligonucleotide primers were used for the LAMP assay, targeting eight re in the sequence of L. infantum kDNA (GenBank accession no. Z35271). The outer forward primer (F3), outer backward primer (B3), forward inner primer (FIP), backward inner primer (BIP), and loop forward (LF) and backward (LB) primers were designed using the online Primer Explorer V4 software (Eiken Chemical Co. Ltd, Tokyo, Japan-http://primerexplorer.jp/elamp4.0.0/index.html). kDNA-specific LAMP primers were designed according to the general criteria described by Notomi et al. (17). The location and nucleotide sequences of all primers are shown in Fig. 1 and Table 1, respectively. The LAMP reaction contained the following components: 40 pmol each of FIP and BIP primers, 5 pmol each of fig3 and B3 primers, 20 mMTris-HCl (pH 8.8), 10mM KCl, 10 mM (NH4)2 SO4, 8 mM MgSO4, 0.8 M betaine (Sigma-Aldrich), 2mM each of deoxynucleo triphosphate, 0.1%TritonX-100 (Roche Applied Science), 8 units Bst DNA Polymerase, Large Fragment (New England Biolabs) and 2 μl extracted nucleic acid in a total volume of 25 μl. The reaction was incubated at 64 °C for 90 min in a heat block for amplification and held at 90 °C for 2 min to inactivate the reaction.
Fig 1.

Location and sequence of LAMP targets and priming sites in the L. infantum reference sequence (GenBank accession no. Z35271). Forward outer primer (FOP) and backward outer primer (BOP) are respectively homologous to the sequences of F3 and B3 regions of the target. Forward inner primer (FIP) contains the sense sequence of F2 region at the 3′ end directly linked to the F1c (complementary sequence to F1 region) at the 5′ end, and backward inner primer (BIP) contains the sense sequence of B2 region at the 3′ end directly linked to the B1c (complementary sequence to B1 region) at the 5′ end. Forward loop primer (FLP) and backward loop primer (BLP), respectively, contain the sequence complementary to a part of sequence between F1 and F2 and between B1 and B2 regions. The red box indicates the Hha I cutting site
Table 1.
Primer sets used for amplification of kDNA genes in LAMP assay for L. infantum
| Primer | Type | Length mer | Sequence (5 ′-3′) |
|---|---|---|---|
| FIP(F1C+F2) | Forward Inner primer | 38 | GTGGTCTGGGTAGTGGCTT-GGTT-GGATTGGCCTGAAAC |
| BIP(B1C+B2) | Backward Inner primer | 36 | TGCGCCTTGGACTTTCGG-TAATGTTGGGCACATGCT |
| F3 (FOP) | Forward outer primer | 19 | TGGTTGGAAATTGGCTCTC |
| B3 (BOP) | Backward outer primer | 22 | GATTGAGTTGGATTCATTGACG |
| FLP | Forward Loop primer | 20 | CTGGGTCTGGAGGCTTTATT |
| BLP | Backward Loop primer | 22 | GGCTTTTGTTGGGATTTTGTTG |
Determination of analytical sensitivity and specificity of LAMP assay
The sensitivity of the LAMP assay was determined by spiking a defined number of cultured promastigotes of L. infantum (MHOM/TN/80/IPT1) into a negative Leishmania blood sample. Ten-fold serial dilutions ranging from 100 000 to 1 parasites per ml blood were used. After extraction of nucleic acid, the dilutions were amplified by both LAMP and nested PCR, and products were visualized by 2% agarose gel electrophoresis. The species specificity of the LAMP primers was determined by testing against DNA of L. tropica and L. major and non-leishmanial infections of malaria, toxoplasmosis, tuberculosis, and HBV, HCV, HSV, and CMV.
Clinical sensitivity and specificity
The clinical sensitivity and specificity of the LAMP assay were calculated using 47 whole-blood samples and DAT as the reference standard. Due to the agreement between the DAT and BM results, DAT was considered the gold standard. Sensitivity was calculated as the (number of true positives)/(number of true positives+number of false negatives), and specificity was calculated as (number of true negatives)/(number of true negatives+number of false positives). Sensitivity of LAMP and nested PCR were compared with McNemar’s test using SPSS16 software.
Results
Detection and confirmation of LAMP products
The LAMP reaction causes turbidity, observable with the naked eye, in the reaction tube proportional to the amount of amplified DNA, which is linked to the production of insoluble salt of magnesium pyrophosphate (Fig. 2A). Positive reactions turned green upon addition of SYBR Green I, while negative reactions remained orange (Fig. 2B). For further confirmation, the LAMP products were subjected to electrophoresis on 2% agarose gel containing 0.5 μg/ml ethidium bromide and visualized under UV light. A positive result was defined as the appearance of a typical ladder-like pattern on the gel (Fig. 3).
Fig 2.

Visual endpoint detection method of LAMP reaction. (A) Assessment of LAMP reaction by turbidity. 1A, with template DNA (positive); 2A, without template (negative). (B) Detection of LAMP reaction by fluorescence using SYBR Green I. 1B, with template (positive, color of reaction mixture changes from original orange to bright green); 2B, without template (negative, color of reaction mixture remains unchanged)
Fig 3.
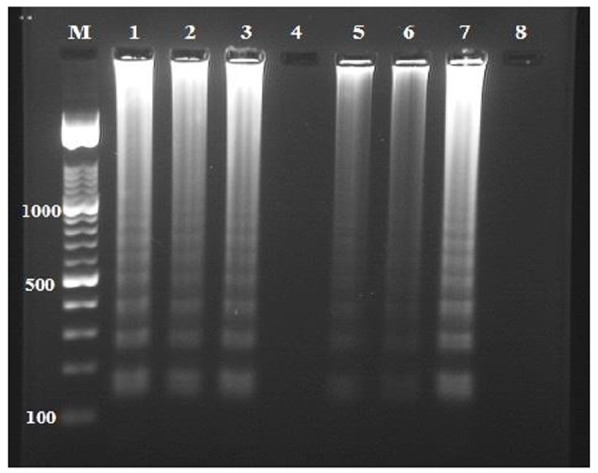
Detection of L. infantum DNA by loop-mediated isothermal amplification (LAMP). Lane M, 100 bp DNA ladder marker (Roche Applied Science); lanes 1–3, 5 and 6, DNA sample from each patient; lane 7, L. infantum positive control, lanes 4 and 8, DNA sample from a healthy human and PCR grade water (negative control), respectively
This pattern is due to the formation of an assembly of stem-loop DNAs with cauliflower-like structures of various stem lengths. The accuracy of the LAMP reaction was confirmed by digestion with restriction enzyme Hha I to ensure that the LAMP products exhibited the corresponding sequence of the kDNA gene of L. infantum. The size of fragments produced by digestion was in agreement with that predicted from the expected DNA structures: 76 and 121 bp (Fig. 4).
Fig 4.
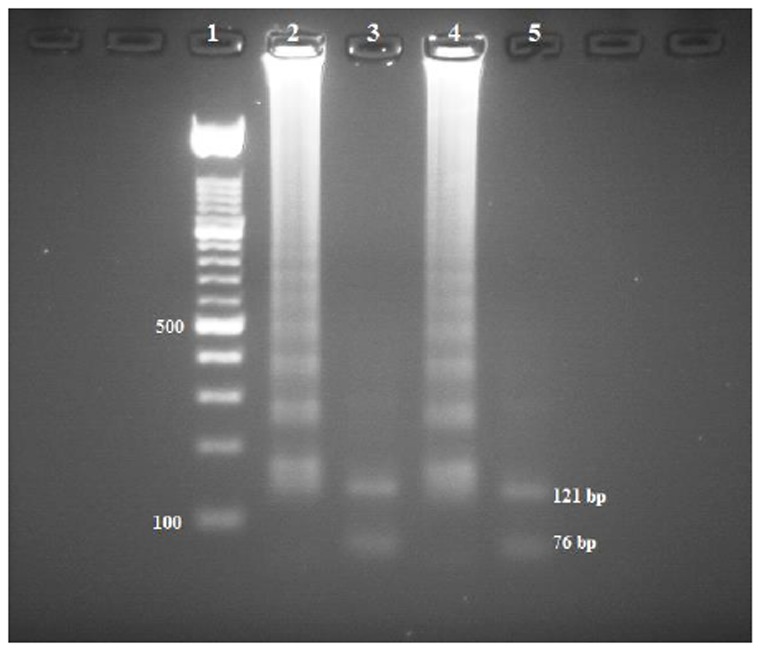
Restriction enzyme analysis of the LAMP products amplified from the kDNA gene. The digestion products were run on a 3% agarose gel. Lane 1, 100 bp DNA ladder marker (Roche Applied Science); lanes 2 and 4 LAMP reactions performed on the DNA extracted from the clinical sample and L. infantum (MHOM/TN/80/IPT1), respectively; lanes 3 and 5, LAMP product of lanes 2 and 4, respectively, after digestion with Hha I (121- and 76-bp bands were expected)
Analytical sensitivity and specificity of LAMP assay
To estimate the sensitivity, serially diluted blood samples containing 1 to 100 000 L. infantum promastigotes per ml blood were examined. The detection limit of both LAMP and nested PCR was 1 parasite in a 1 ml blood (Fig. 5). To evaluate the specificity of the LAMP assay, reactions were also conducted for amplification of other Leishmania parasites as well as non-leishmanial infections. Non-target DNA was not amplified, showing the LAMP assay to be specific for detection of L. infantum. Results were similar to those obtained with nested PCR (data not shown).
Fig 5.
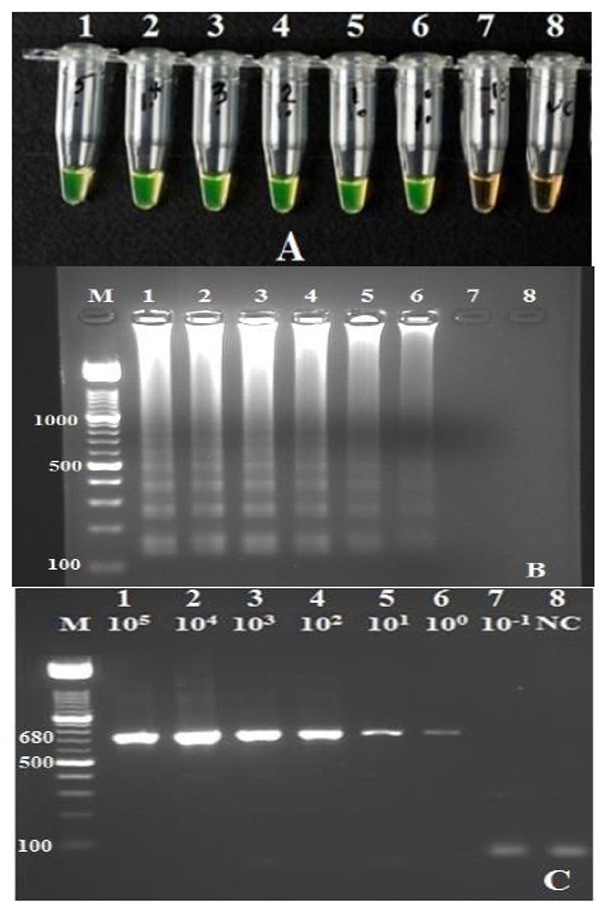
Comparison of sensitivity of loop-mediated isothermal amplification (LAMP) to nested PCR. Lane M, 100 bp DNA ladder marker (Roche Applied Science); tubes and lanes 1-7, reactions contained 105, 104, 103, 102, 101, 10, and 10-1 cultured promastigote of L. infantum per 1 ml blood, respectively; lane 8, negative control (PCR grade water). (A) Detection limit of the LAMP reaction based on color change in SYBR Green I by the naked eye under visible light: The color of positive reactions (tubes1–6) became bright green while the color of the negative reactions (tubes7–8) remained unchanged (orange). (B) Detection limit of the LAMP reaction using electrophoresis of 2 μl of each reaction mixture in 2% agarose gel followed by ethidium bromide staining: lanes 1–6 showed the typical ladder-shaped pattern of a positive reaction. (C) Sensitivity of the nested PCR and producing a 680-basepair fragment
Detection of L. infantum DNA from blood samples from patients with VL
The LAMP products were examined in light and electrophoresed in agarose gels, and similar positive results were observed. Among 47 confirmed VL cases, 45 were positive with nested PCR and all controls were negative, resulting in a sensitivity of 96% (95% CI) and specificity of 100% (95% CI). Forty-four of 47 VL cases were positive with LAMP, and all controls gave negative results, yielding a sensitivity of 93.6% (95% CI) and a specificity of 100% (95% CI). All samples positive by LAMP were also positive by nested PCR (Table 2). We compared the sensitivity and specificity of the LAMP assay with that of nested PCR using the McNemar test and found no significant difference (P=0.31). As all the controls were negative by both methods, the McNemar test was not performed for specificity.
Table 2.
Comparison of loop-mediated isothermal amplification (LAMP) to nested PCR for diagnosis of L. infantum
| Diagnostic assay | n (%) with Nested PCR | |||
|---|---|---|---|---|
| Positive | Negative | Total n. (%) | ||
| n (%) with LAMP PCR | Positive | 42(89.3) | 2(4.3) | 44(93.6) |
| Negative | 3(6.4) | 0(0) | 3(6.4) | |
| Total n (%) | 45(95.7) | 2(4.3) | 47(100) | |
Discussion
Accurate and sensitive procedures for diagnosis of VL are required for development of an effective program of control and treatment to reduce the morbidity and mortality rate. The LAMP assay reported by Notomi et al. (16) is a low cost, simple, and rapid DNA amplification technique based on a unique primer design. This technique has been used to detect DNA of protozoan’s such as several African trypanosome species (18, 26) as well as Malaria (27), Giardia (28), Cryptosporidium (29), and tuberculosis (30). In this study, we developed a LAMP assay capable of detecting a single parasite in 1 ml blood, which corresponds to 0.025 parasites per reaction. This high sensitivity was achieved through the presence of 10 000 copies per cell (31-32). The specificity of LAMP is generally high, as it uses six primers for the recognition of eight distinct regions on the target DNA sequences (17). However, the LAMP assay will need further investigation with more samples from other disease-endemic areas. In this study, peripheral blood samples collected from patients with confirmed VL (n=47) and from healthy individuals (n=30) were analyzed using LAMP and nested PCR assays simultaneously. Of 47 patients, 44 (93.6%) were positive by LAMP and 45 (95.7%) were positive by nested PCR. These results showed efficacy of the LAMP assay to be comparable to nested PCR, which is widely used for the diagnosis of leishmaniasis. Takagi et al. (22) reported 8 of 10 (80%) confirmed VL patients to be positive for parasite DNA with LAMP. Adam et al. (21) in a study of 30 confirmed VL patients, found diagnostic sensitivity of 83% and specificity of 98%. A recent LAMP-based assay for the detection of Leishmania DNA in buffy coat of blood from 75 confirmed VL patients reported sensitivity of 90.7% and specificity of 83% (33). In our study, we found a higher sensitivity of 93.6% and specificity of 100%. The LAMP developed in the present study demonstrated sensitivity and specificity similar to that of nested PCR for the detection of L. infantum parasites. Although the sensitivity level of 93.6% falls slightly below the WHO requirement of >95% for an acceptable test, there are several advantages of the LAMP assay. Diagnosis by LAMP assay does not require a complicated thermal cycler, a turbidimeter, or skilled technicians. The assay is performed under isothermal conditions using a simple incubator such as a heat block or water bath to provide a constant temperature of 60-65 °C, making it more practical and economical than nested PCR. The time requirement for the procedure is also an advantage. Only 1.5 h was needed to perform the LAMP assay compared to 5 h for nested PCR. In addition, the LAMP assay is interpreted by observing turbidity due to magnesium pyrophosphate accumulation as a by-product of DNA amplification, which reduces time and cost compared with conventional PCR analysis. In all reactions, positive samples from negative samples simply, by the turbidity of the reaction mixtures. Alternatively, inspection for amplification can be performed with the naked eye by using SYBR green I, which turns green in the presence of amplified DNA. A major drawback of this assay is a high risk of production of secondary LAMP products. This is probably caused by the extremely high efficacy of the reaction. The risk can be reduced by changing gloves between assays and conducting preparation of the reagents and the detection of the products in separate areas.
Conclusion
LAMP is an assay that, for the first time, enables a molecular diagnosis of L. infantum where there are minimal facilities or under field conditions. The technique presents sensitivity and specificity similar to that of nested PCR, does not require sophisticated equipment, and is simpler and more cost-effective than nested PCR. To assess the on-site performance of LAMP for rapid diagnosis of VL in the field further evaluation of this technique is necessary.
Acknowledgments
This research was financially supported by Tehran University of Medical Sciences (Project No: p-993). We express our thanks to Z. Zarei and S. Charedar for laboratory help and valuable assistance with collection of blood samples. The authors declare that there is no conflict of interests.
References
- 1.Mohebali M, Edrissian GhH, Shirzadi MR, Akhoundi B, Hajjaran H, Zarei Z, et al. An observational study on the current distribution of visceral leishmaniasis in different geographical zones of Iran and implication to health policy. Travel Med Infect Dis. 2011 Mar;9(2):67–74. doi: 10.1016/j.tmaid.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis. 2004 Sep;27(5):305–18. doi: 10.1016/j.cimid.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Mohebali M, Edrissian GH, Nadim A, Hajjaran H, Akhoundi B, Hooshmand B. Application of direct agglutination test (DAT) for the diagnosis and seroepidemiological studies of visceral leishmaniasis in Iran. Iranian J Parasitol. 2006;1(1):15–25. [Google Scholar]
- 4.Mohebali M, Hamzavi Y, Edrissian GH, Forouzani A. Seroepidemiological study of visceral leishmaniasis among humans and animal reservoirs in Bushehr province, Islamic Republic of Iran. East Mediterr Health J. 2001 Nov;7(6):912–7. [PubMed] [Google Scholar]
- 5.Alvar J, Canavate C, Gutierrez-Solar B, Jimenez M, Laguna F, Lopez-Velez R, et al. Leishmania and human immunodeficiency virus coinfection: the first 10 years. Clin Microbiol Rev. 1997 Apr;10(2):298–319. doi: 10.1128/cmr.10.2.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desjeux P, Alvar J. Leishmania/HIV co-infections: epidemiology in Europe. Ann Trop Med Parasitol. 2003 Oct;97(Suppl 1):3–15. doi: 10.1179/000349803225002499. [DOI] [PubMed] [Google Scholar]
- 7.Rijal S, Boelaert M, Regmi S, Karki BM, Jacquet D, Singh R, et al. Evaluation of a urinary antigen-based latex agglutination test in the diagnosis of kala-azar in eastern Nepal. Trop Med Int Health. 2004 Jun;9(6):724–9. doi: 10.1111/j.1365-3156.2004.01251.x. [DOI] [PubMed] [Google Scholar]
- 8.Salam MA, Khan MG, Mondal D. Urine antigen detection by latex agglutination test for diagnosis and assessment of initial cure of visceral leishmaniasis. Trans R Soc Trop Med Hyg. 2011 May;105(5):269–72. doi: 10.1016/j.trstmh.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 9.Singh DP, Goyal RK, Singh RK, Sundar S, Mohapatra TM. In search of an ideal test for diagnosis and prognosis of kala-azar. J Health Popul Nutr. 2010 Jun;28(3):281–5. doi: 10.3329/jhpn.v28i3.5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghatei MA, Hatam GR, Hossini MH, Sarkari B. Performance of latex agglutination test (KAtex) in diagnosis of visceral leishmaniasis in Iran. Iranian Journal of Immunology. 2009;6(4):202–7. doi: 10.22034/iji.2009.17093. [DOI] [PubMed] [Google Scholar]
- 11.Antinori S, Calattini S, Longhi E, Bestetti G, Piolini R, Magni C. Clinical use of polymerase chain reaction performed on peripheral blood and bone marrow samples for the diagnosis and monitoring of visceral leishmaniasis in HIV-infected and HIV-uninfected patients: a single-center 8-year experience in Italy and review of the literature. Clin Infect Dis. 2007 Jun 15;44(12):1602–10. doi: 10.1086/518167. [DOI] [PubMed] [Google Scholar]
- 12.Cruz I, Canavate C, Rubio JM, Morales MA, Chicharro C, Laguna F, et al. A nested polymerase chain reaction (Ln-PCR) for diagnosing and monitoring Leishmania infantum infection in patients co-infected with human immunodeficiency virus. Trans R Soc Trop Med Hyg. 2002 Apr;96(Suppl 1):S185–9. doi: 10.1016/s0035-9203(02)90074-x. [DOI] [PubMed] [Google Scholar]
- 13.Lachaud L, Chabbert E, Dubessay P, Reynes J, Lamothe J, Bastien P. Comparison of various sample preparation methods for PCR diagnosis of visceral leishmaniasis using peripheral blood. J Clin Microbiol. 2001 Feb;39(2):613–7. doi: 10.1128/JCM.39.2.613-617.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reithinger R, Dujardin JC. Molecular diagnosis of leishmaniasis: current status and future applications. J Clin Microbiol. 2007 Jan;45(1):21–5. doi: 10.1128/JCM.02029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh RK, Pandey HP, Sundar S. Visceral leishmaniasis (kala-azar): challenges ahead. Indian J Med Res. 2006 Mar;123(3):331–44. [PubMed] [Google Scholar]
- 16.Sundar S, Rai M. Laboratory diagnosis of visceral leishmaniasis. Clin Diagn Lab Immunol. 2002 Sep;9(5):951–8. doi: 10.1128/CDLI.9.5.951-958.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000 Jun 15;(12):E63. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuboki N, Inoue N, Sakurai T, Di Cello F, Grab DJ, Suzuki H, et al. Loop-mediated isothermal amplification for detection of African trypanosomes. J Clin Microbiol. 2003 Dec;41(12):5517–24. doi: 10.1128/JCM.41.12.5517-5524.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maruyama F, Kenzaka T, Yamaguchi N, Tani K, Nasu M. Detection of bacteria carrying the stx2 gene by in situ loop-mediated isothermal amplification. Appl Environ Microbiol. 2003 Aug;69(8):5023–8. doi: 10.1128/AEM.69.8.5023-5028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Endo S, Komori T, Ricci G, Sano A, Yokoyama K, Ohori A, et al. Detection of gp43 of Paracoccidioides brasiliensis by the loop-mediated isothermal amplification (LA-MP) method. FEMS Microbiol Lett. 2004 May;234(1):93–7. doi: 10.1016/j.femsle.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 21.Okafuji T, Yoshida N, Fujino M, Motegi Y, Ihara T, Ota Y, et al. Rapid diagnostic method for detection of mumps virus genome by loop-mediated isothermal amplification. J Clin Microbiol. 2005 Apr;43(4):1625–31. doi: 10.1128/JCM.43.4.1625-1631.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams ER, Schoone GJ, Ageed AF, Safi SE, Schallig HD. Development of a reverse transcriptase loop-mediated isothermal amplification (LAMP) assay for the sensitive detection of Leishmania parasites in clinical samples. Am J Trop Med Hyg. 2010 Apr;82(4):591–6. doi: 10.4269/ajtmh.2010.09-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takagi H, Itoh M, Islam MZ, Razzaque A, Ekram AR, Hashighuchi Y, et al. Sensitive, specific, and rapid detection of Leishmania donovani DNA by loop-mediated isothermal amplification. Am J Trop Med Hyg. 2009 Oct;81(4):578–82. doi: 10.4269/ajtmh.2009.09-0145. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Thekisoe OM, Aboge GO, Kyan H, Yamagishi J, Inoue N, et al. Toxoplasma gondii: sensitive and rapid detection of infection by loop-mediated isothermal amplification (LA-MP) method. Exp Parasitol. 2009 May 12;2(1):47–50. doi: 10.1016/j.exppara.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 25.Noyes HA, Reyburn H, Bailey JW, Smith D. A nested-PCR-based schizodeme method for identifying Leishmania kinetoplast minicircle classes directly from clinical samples and its application to the study of the epidemiology of Leishmania tropica in Pakistan. J Clin Microbiol. 1998 Oct;36(10):2877–81. doi: 10.1128/jcm.36.10.2877-2881.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Njiru ZK, Mikosza AS, Matovu E, Enyaru JC, Ouma JO, Kibona SN, et al. African trypanosomiasis: sensitive and rapid detection of the sub-genus Trypanozoon by loop-mediated isothermal amplification (LAMP) of parasite DNA. Int J Parasitol. 2008 Apr;38(5):589–99. doi: 10.1016/j.ijpara.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poon LL, Wong BW, Ma EH, Chan KH, Chow LM, Abeyewickreme W, et al. Sensitive and inexpensive molecular test for falciparum malaria: detecting Plasmodium falciparum DNA directly from heat-treated blood by loop-mediated isothermal amplification. Clin Chem. 2006 Feb;52(2):303–6. doi: 10.1373/clinchem.2005.057901. [DOI] [PubMed] [Google Scholar]
- 28.Plutzer J, Karanis P. Rapid identification of Giardia duodenalis by loop-mediated isothermal amplification (LAMP) from faecal and environmental samples and comparative findings by PCR and real-time PCR methods. Parasitol Res. 2009 Jun;104(6):1527–33. doi: 10.1007/s00436-009-1391-3. [DOI] [PubMed] [Google Scholar]
- 29.Bakheit MA, Torra D, Palomino LA, Thekisoe OM, Mbati PA, Ongerth J. Sensitive and specific detection of Cryptosporidium species in PCR-negative samples by loop-mediated isothermal DNA amplification and confirm-ation of generated LAMP products by sequencing. Vet Parasitol. 2008 Nov 25;158(1-2):11–22. doi: 10.1016/j.vetpar.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 30.Aryan E, Makvandi M, Farajzadeh A, Huygen K, Bifani P, Mousavi SL, et al. A novel and more sensitive loop-mediated isothermal amplification assay targeting IS6110 for detection of Mycobacterium tuberculosis complex. Microbiol Res. 2010 Mar 31;165(3):211–20. doi: 10.1016/j.micres.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 31.Brewster S, Aslett M, Barker DC. Kinetoplast DNA minicircle database. Parasitol Today. 1998 Nov;14(11):437–8. doi: 10.1016/s0169-4758(98)01330-1. [DOI] [PubMed] [Google Scholar]
- 32.Rogers WO, Wirth DF. Kinetoplast DNA minicircles: regions of extensive sequence divergence. Proc Natl Acad Sci U S A. 1987 Jan;84(2):565–9. doi: 10.1073/pnas.84.2.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khan MG, Bhaskar KR, Salam MA, Akther T, Pluschke G, Mondal D. Diagnostic accuracy of loop-mediated isothermal amplification (LA-MP) for detection of Leishmania DNA in buffy coat from visceral leishmaniasis patients. Parasit Vectors. 2012;5:280. doi: 10.1186/1756-3305-5-280. [DOI] [PMC free article] [PubMed] [Google Scholar]