

Abstract
Comet assay has been used to estimate cancer risk by quantification of DNA damage and repair in response to mutagen challenge. Our goal was to adopt best practices for the alkaline comet assay to measure DNA repair capacity of white blood cells in whole blood of patients with squamous cell carcinoma of the head and neck (HNSCC). The results show that initial damage by 10 Gy of gamma radiation expressed as percent DNA in comet tail was higher in stimulated lymphocytes (61.1±11.8) compared to whole blood (43.0±12.1) but subsequent repair was similar with comet tail of approximately 20% at 15 minutes and 13% at 45 minutes after exposure. Exposure of whole blood embedded in agarose from 5 to 10 Gy gamma radiation was followed by an approximately 70% repair of the DNA damage within 45 minutes with a faster repair phase in the first 15 minutes. Variability of the measurement was lower within repeated measurements of the same person compared to measurement of different healthy individuals. The repair during first 15 minutes was slower (p=0.01) in ex-/non-smokers (41.0+2.1%) compared to smokers (50.3±2.7%). This phase of repair was also slower (p=0.02) in HNSCC patients (36.8+2.1%) compared to controls matched on age and smoking (46.4+3.0%). The results of this pilot study suggest that quantification of repair in whole blood following a gamma radiation challenge is feasible. Additional method optimization would be helpful to improve the assay for a large population screening.
Introduction
Genome integrity is maintained by an intricate network of DNA repair proteins [1;2]. Defects in this complex machinery are associated with familial predispositions to cancer and other diseases [3]. Increasing evidence links environmental exposures, subtle modification in DNA repair efficiency, and cancer risk [4]. Establishing this connection, however, has been a challenge due to the complexity of interactions that affect the repair pathways [5;6]. Given the differences in DNA repair of lower eukaryotes, model organisms, and humans, molecular epidemiology is expected to provide important supportive evidence that is difficult to establish in targeted mechanistic studies [5;7]. Phenotypic cell-based assays of repair capacity provide a quantitative view of the complex pathways involved in repair [7;8]. Comet assay is one of the screens of DNA integrity and repair capacity that is increasingly used in epidemiological studies exploring the connection of repair deficiency with cancer risk [9;10]. Comet assay quantifies single strand breaks and other damage in terms of migration of the DNA out of the nucleus under alkaline electrophoretic conditions [11]. Specific guidelines were developed to standardize the experimental conditions between laboratories [12]. This allows comparable inter-laboratory testing but further methodological development is needed to facilitate application of this assay to large population studies. In this paper, we describe adoption of best practices for the alkaline comet assay in an initial attempt to screen DNA repair of white blood cells in whole blood. An application of the assay to a population of controls and a small set of patients with squamous cell carcinoma of the head and neck (HNSCC) is discussed.
Materials and Methods
Sample Collection and Processing
Blood samples were collected at the Georgetown University Hospital, Department of Otolaryngology Head and Neck Surgery and at the Lombardi Comprehensive Cancer Center. Age and smoking matched controls were recruited at Georgetown University in collaboration with the National Lung Screening Trial/Lung Screening Study (NLST/LSS) associated study “Recruitment of Control Participants for a Study of Head and Neck Cancer”. All participants signed informed consent and samples were collected in accordance with guidelines of the Georgetown University Institutional Review Board. Patients with newly diagnosed squamous cell carcinoma of the head and neck (HNSCC) were recruited into the study prior to receiving treatment. Participants were interviewed about lifestyle, medical and diet history. Blood samples for reproducibility studies were obtained from healthy volunteers. Blood samples were drawn in green-topped Vacutainer tubes (BD BioSciences, Franklin Lakes, NJ) containing sodium heparin (10-25 IU/mL). Blood samples for the study were processed according to two protocols: 1) Whole blood processing; and 2) Short term culture of isolated lymphocytes (see below). Whole blood used for the comet assay was stored overnight at 4°C. Prior to embedding in agarose, the blood samples were diluted 1:10 in RPMI 1640.
Study Participants
To study the effects of smoking, we analyzed blood samples of a total of 40 non-cancer controls consisting of 17 smokers (mean age 61.8±4.7 years) and 23 non- and ex-smokers (mean age 63.7±5.9). To study the effects of Head and Neck cancer, we analyzed 12 cases (mean age 55.3±12.4; 3 smokers (25%), 9 non- and ex-smokers) and 15 controls (mean age 57.9±4.9; 4 smokers (26%), 11 non- and ex-smoker). Age of the two groups is not statistically different (p=0.46).
Cell Culture and Cell Exposures
Jurkat T-cells were cultured at 37 °C in 5% CO2 in RPMI 1640 with 10 percent FBS and 1 percent sodium pyruvate. To obtain primary cultures, peripheral blood lymphocytes were isolated by gradient centrifugation using Histopaque-1077 (Sigma-Aldrich, St. Louis, MO) in Accuspin™ tubes (Sigma-Aldrich, St. Louis, MO). Isolated lymphocytes were seeded at a concentration of 5×105 cells per mL in RPMI 1640 (GIBCO-Invitrogen, Gaithersburg, MD) containing the following: 15% (v/v) Fetal Calf Serum, 2mM L-glutamine (GIBCO-Invitrogen, Gaithersburg, MD), 10U/mL Sodium Heparin (Sigma-Aldrich, St. Louis, MO), 16U/mL phytohemaglutinin (GIBCO-Invitrogen, Gaithersburg, MD), 1U/mL Penicillin and 1μg/mL Streptomycin (GIBCO-Invitrogen, Gaithersburg, MD). The cells were incubated at 37 °C in 5% CO2 for 65 hours. The cultured lymphocytes were then resuspended in media without PHA and supplemented with IL2 (20U/ml) for an additional 28 hours of culture.
DNA damage was induced using either the radiomimetic bleomycin or gamma radiation. For the bleomycin experiments, cells embedded in agarose were incubated for 30 minutes at 37 °C in RPMI 1640 containing 3μg/mL bleomycin sulfate (Sigma-Aldrich, St. Louis, MO). For the irradiation experiments, cells were exposed to gamma rays derived from a Cs-137 source in a Research Irradiator. Cells in suspension in PBS were treated in a tissue culture flask (Corning Life Sciences, Corning, NY). Cells embedded in agarose on glass slides prior to irradiation were treated in a glass staining dish (Thermo-Fisher, Waltham, MA) containing PBS (4 °C, pH 7.4; GIBCO-Invitrogen, Gaithersburg, MD).
Alkaline Comet Assay
The alkaline comet assay was conducted in accordance with the guidelines summarized by Tice et al, with some modifications [12]. Our comet slides were made in-house using a three-layer procedure where the cells were embedded in the topmost layer. Frosted slides (Thermo-Fisher, Waltham, MA) were coated in 1% agarose (Sigma-Aldrich, St. Louis, MO) and dried in an oven at 60 °C for 20 minutes. The second layer consisted of 0.75% normal melting agarose (Invitrogen, Gaithersburg, MD). Cells were mixed with 0.75% low-melting agarose (BioWhittaker-Cambrex, East Rutherford, NJ) kept at 37 °C under a yellow-frosted incandescent light (Philips Electronics North America, New York, NY) to prevent DNA damage. Both the second and third layers were flattened using a 22×22mm coverslip (Thermo-Fisher, Waltham, MA) and the slides were placed on ice to facilitate the gelling process. Duplicate slides were made for each dose/repair point. After the embedding procedure, the cells were exposed to a mutagen to generate DNA damage. Following mutagen challenge, cells were either placed in lysis buffer (2.5M NaCl, 100mM EDTA, 10mM Tris, 200mM NaOH, 1 percent (v/v) Triton X-100, 10 percent (v/v) DMSO) adjusted to pH 10 or allowed to repair. Cells allowed to repair the DNA damage were incubated in RPMI 1640 at 37°C for a fixed length of time (typically 15 and 45 minutes) and then transferred to the lysis buffer, pH 10. The slides were kept in the lysis buffer at 4 °C in the dark for 3-24 hours. The slides were transferred to ice-cold electrophoresis buffer (pH 13; 300mM NaOH, 1mM EDTA) for 40 minutes. Electrophoresis was carried out at 1.3 V/cm for 45 minutes at 4°C in the dark. The volume of the buffer was adjusted so that the current at the start was 300 mA. Slides were neutralized using three five minute washes with autoclaved 0.4M Tris buffer (pH 7.4). DNA was fixed with methanol for 10 minutes. Slides were then washed with autoclaved distilled water twice for 5 minutes and allowed to dry overnight. The dried slides kept in a storage box (Thermo-Fisher, Waltham, MA) until scoring.
Comet Scoring and Data Collection
All samples were coded to blind the experimenter to case/control status. Additionally, the dose and repair information on the slides was masked prior to scoring to prevent bias in the selection of comets for imaging. Slides were rehydrated for 45-60 minutes in autoclaved distilled water and stained for 10 minutes using a 1μg/mL ethidium bromide solution (Invitrogen, Gaithersburg, MD). Excess stain was washed away using three 5 minute washes with autoclaved distilled water. Slides were scored wet with a cover glass over the gel. Comet images were obtained using an Olympus BX-51 microscope (Opelco-Olympus, Center Valley, PA) equipped with a 100W mercury burner and a wide green fluorescent mirror (U-MWG2) and a cooled 5 megapixel digital CCD camera (QImaging Micropublisher 5.0 RTV, QImaging, Surrey, BC, Canada). Two neutral density filters (U-25ND25 and U-25ND50) were inserted into the light path to reduce photobleaching of the fluorophore. Images were scored using a semi-automated Comet Analysis System (Loats Associates, Westminster, MD). A minimum of fifty images were recorded at 400× total magnification for each slide with two slides per dose-repair point. In accordance with the idea that “hedgehogs” were dead cells that do not offer information regarding DNA repair [13], we tracked them as a categorical variable but did not include them in the calculation of the summary statistics. We observed 2.1 “hedgehogs” per 100 cells on average. The Loats software calculated a number of parameters but we focused primarily on percent DNA in tail, the most reliable parameter for inter laboratory comparisons [14]. For completeness, data on tail length and Olive tail moment is provided in supplemental materials.
Data Analysis
One hundred comet images were recorded for each dose-repair point (2 slides 50 images each). These images were analyzed by a semi automated scoring system (Loats Associates, Westminster, MD). The percent DNA in tail for each image was used as the variable of interest. Experiments involving cell lines were typically repeated a minimum of three times and the summary statistics (mean, standard deviation, median, range, and variance) were calculated according to standard formulas. Experiments using whole blood samples from the population were done once per sample. The sampling distribution of the mean and standard error for this population of samples was computed for each dose-repair point. Percent repaired was calculated as follows: [(Initial damage - Damage at time T)/Initial damage]×100. ANOVA was used to check for differences between cell types (Jurkat T-cells, primary lymphocytes, and whole blood) and for differences between bleomycin induced damage and gamma radiation induced damage.
To determine the variability “within” a sample, the assay was conducted nine times on blood samples obtained from one control. To determine the variability between samples, forty healthy controls were selected for analysis. The effects of smoking were examined by sorting the forty controls by smoking status (current smoker vs ex-smoker/non-smoker). Ex-smokers were defined as subjects that quit smoking for at least 6 months; non-smokers smoked less than 100 cigarettes during their life time. Data from a small pilot study of 27 men and women recruited for a study of HNSCC (12 HNSCC cases and 15 cancer free controls) was examined for differences in DNA repair capacity. A two-sided T-test was used to compare the mean “percent DNA in tail” measurements for each dose-repair point as well as the percent repaired at 15 and 45 minutes. The p-values reported in the text are based on two sided t-test unless otherwise indicated.
Results
Jurkat T-cell line was used to establish the comet assay for comparison of lymphocyte and whole blood assays in our laboratory. We focused on DNA repair measured at several time points following a mutagen challenge. Comparison of repair in Jurkat T-cells, stimulated primary lymphocytes, and whole blood is presented in Figure 1. Jurkat T-cells showed the highest initial damage following exposure to 10 Gy gamma radiation with progressively less damage observed in primary lymphocytes and whole blood. Jurkat T-cells also showed fastest DNA repair with approximately 90% of the DNA damage repaired within 15 minutes. The repair in primary lymphocytes and whole blood was similar (no significant difference at 8 to 45 minutes after exposure) with both repairing approximately 70% damage within 45 minutes. As reported previously, the repair kinetic appears biphasic with a faster repair within first 15 minutes and a slower repair from 15 to 45 minutes [9;15]. Except for the Jurkat T, the cells did not completely repair the damage even at 45 minutes. To minimize variability of the repair measurement, we evaluated the possibility to embed the cells into agarose prior to exposure [16;17]. The results for lymphocytes suspended in PBS in tissue culture flasks or embedded in agarose prior to irradiation were nearly identical (Table 1). A uniform quick start and end of the repair process was achieved by transfer of the slides with embedded cells from PBS to RPMI at the start and from RPMI to lysis buffer at the end of the assay. We found similar results for Jurkat t-cells and whole blood (data not shown). These results suggest that the cells embedded in agarose can be used for quantification of repair. Because of similar repair kinetic in whole blood and cultured lymphocytes and because of ease of handling of whole blood, we chose to continue our experiments with embedded whole blood. For all experiments, whole blood was analyzed after an overnight storage at 4 °C [18;19] which was previously reported to minimize variability of the measurement [20].
Figure 1.
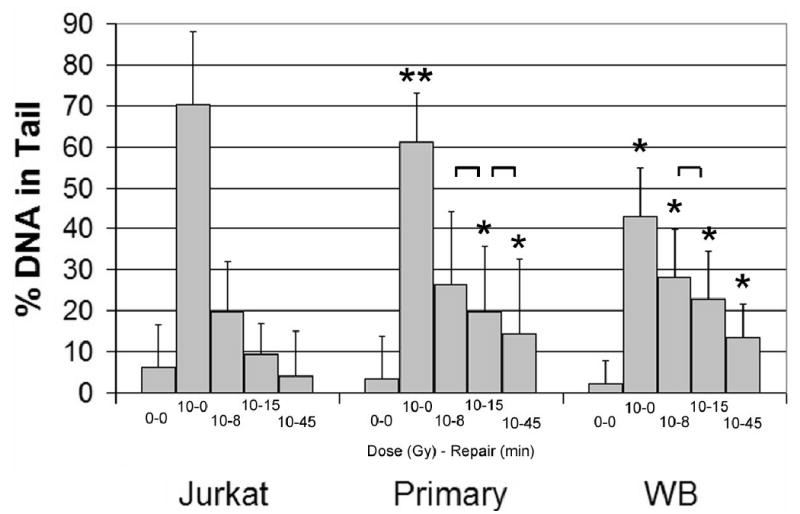

Kinetic of repair over 45 minutes at 37 °C in RPMI media in three types of cell preparations (n=3 for each) exposed to 10 Gy of gamma radiation. Whole blood was obtained from a healthy (non-cancer) volunteer.
A. *Significantly different from the corresponding dose-time point in Jurkat T-cells (p<0.05). **Significantly different from from the corresponding dose-time point in Jurkat T-cells and whole blood (p<0.05). Brackets indicate the only differences within each series that were not significantly different.
B. Percent of repaired DNA in tail compared to 10 Gy 0 minutes. Triangles, Jurkat T-cells; Squares, primary cultured lymphocytes; Circles, whole blood.
Table 1.
Comparison of repair following exposure of primary cultured lymphocytes to 10 Gy of gamma radiation. Lymphocytes were either embedded in agarose or in suspension during irradiation. Cells treated in suspension were embedded in agarose prior to repair. Values shown are the Mean ± S.D. for three replicates of each condition.
| Dose (Gy) - Repair (min) | Embedded | Suspension |
|---|---|---|
| Mean ± SD | Mean ± SD | |
| 0-0 | 3.40 ± 10.51 | 4.71 ± 12.54 |
| 0-8 | 1.97 ± 5.30 | 4.11 ± 12.81 |
| 0-15 | 2.00 ± 7.96 | 5.10 ± 14.59 |
| 0-45 | 2.95 ± 10.18 | 5.38 ± 13.12 |
| 10-0 | 61.16 ± 11.79 | 60.23 ± 14.07 |
| 10-8 | 26.32 ± 17.84 | 31.34 ± 21.64 |
| 10-15 | 19.9 ± 15.85 | 20.38 ± 17.62 |
| 10-45 | 14.28 ± 18.35 | 13.52 ± 15.69 |
Figure 2 shows a dose response for whole blood embedded in agarose prior to treatment with gamma radiation. The initial damage increased linearly with dose (r2 = 0.92). Our criteria for an “optimum dose” included initial damage close to 50% and a high ratio of initial damage-to-damage at 45 minutes. Both 9 and 10 Gy of gamma irradiation induced about 50% DNA in tail with 9 Gy showing the greatest differential at 45 minutes. The cells repaired about 70% of the initial damage at 45 minutes for almost every dose which suggests that majority of the cells are able to repair the damage induced by increasing doses of radiation. This supports the notion that the embedded cells are intact and functioning during the repair period.
Figure 2.

Dose response of whole blood from a healthy (non-cancer) volunteer to gamma radiation exposure. Each bar represents average percent DNA in tail at a given dose (0 to 10 Gy) at two time points (0 or 45 minutes of repair).
In addition to ionizing radiation, the radiomimetic bleomycin is often used in studies of DNA damage and repair [9;21]. We compared treatment of whole blood with bleomycin, 3μg/ml, to treatment with gamma radiation. Bleomycin, induced about 90% DNA in tail, compared to 40% DNA in Tail after 10 Gy of gamma radiation (Figure 3). The repair of the bleomycin-induced damage was fast, with about 70% of the damage repaired at 15 minutes. Bleomycin exposure would be therefore a good model for evaluation of the repair kinetic but we observed a large day-to-day variability in the bleomycin experiments (data not shown). The dosing with gamma radiation was therefore selected for further experiments. The damage within each series (bleomycin and gamma radiation) was significantly different at all time points except the 8 and 15 minute time points in the gamma radiation series. The 8 minute time point was not included in further experiments. The day-to-day variability of the assay following gamma radiation was evaluated on whole blood from a single donor measured nine separate times (Table 2). The subject-to-subject variability was measured by performing the assay once for each of 40 non-cancer controls. The means and medians for each dose-repair point were similar between the two sets of data but the variance and range (maximum minus minimum) were smaller for each dose-repair point in the day-to-day measurements compared to measurements between subjects. The higher inter individual variability suggests that the assay could provide a useful comparison of repair in a population.
Figure 3.

Exposure of whole blood from a healthy (non-cancer) volunteer to bleomycin and gamma radiation. Black bars, cells exposed to bleomycin, 3μg/ml; Dotted bars, cells exposed to gamma irradiation, 10 Gy.
Table 2.
Variability of the measurement determined by the following: A. “within 9 replicates”, day-to-day variability of the whole blood assay measured nine times on a single healthy volunteer; and B. “between 40 non-cancer controls”, subject-to-subject variability of the whole blood assay measured once for each of 40 population controls. SD, standard deviation; SEM, standard error of the mean; Range, maximum minus minimum.
| Dose (Gy)-Repair (min) | 9-0 | 9-15 | 9-45 |
|---|---|---|---|
| A. Within 9 Replicates | |||
| Mean ± SD | 41.95 ± 7.17 | 22.74 ± 4.59 | 10.53 ± 4.77 |
| Variance | 51.42 | 21.04 | 22.79 |
| Median | 40.53 | 23.11 | 8.78 |
| Range | 27.22 | 15.33 | 14.30 |
| B. Between 40 Non-Cancer Controls | |||
| Mean ± SEM | 44.17 ± 1.45 | 24.84 ± 1.14 | 15.76 ± 0.93 |
| Variance | 83.56 | 51.74 | 34.66 |
| Median | 44.06 | 25.32 | 16.02 |
| Range | 39.99 | 26.57 | 22.00 |
To evaluate the effect of smoking on DNA repair, forty non-cancer controls used in the measurement of variability were sorted according to smoking status. Non-smokers were grouped with ex-smokers and compared to current smokers. The values for percent DNA in tail for each dose-repair point were similar between the two groups (Table 3). The percent repair of DNA in tail between 9-0 and 9-15 minutes, i.e. the fast phase of the repair kinetic, was higher for smokers than non-/ex-smokers (p=0.01). This observation suggests that single strand break repair might be induced in white blood cells by the exposure to cigarette smoke.
Table 3.
Effect of smoking on the repair kinetic in forty non-cancer controls. Whole blood of 17 smokers and 23 non-/ex-smokers embedded in agarose was exposed to 9 Gy gamma radiation following overnight storage at 4 °C. 9-0, 9 Gy 0 minutes of repair; 9-15, 9 Gy 15 minutes of repair; 9-45, 9 Gy 45 minutes of repair; % Repair for 0 to 15 and 15 to 45 minutes was calculated as described in the methods section. P-values are based on a two sided t-test.
| Dose (Gy) - Repair (min) | 9-0 | 9-15 | 9-45 | % Repair 0 to 15 | % Repair 15 to 45 |
|---|---|---|---|---|---|
| Non-Smokers (4) and Ex-Smokers (19) | |||||
| Mean | 41.83 | 24.69 | 15.84 | 40.98 | 21.26 |
| SEM | 1.94 | 1.37 | 1.14 | 2.13 | 1.82 |
| Smokers (17) | |||||
| Mean | 40.90 | 20.76 | 12.31 | 50.30 | 21.09 |
| SEM | 3.11 | 1.98 | 1.34 | 2.74 | 2.18 |
| T-Test | |||||
| p-value | 0.27 | 0.40 | 0.18 | 0.01 | 0.95 |
The optimized assay conditions were evaluated in a pilot study of HNSCC. We compared HNSCC cases (n=12) with controls frequency matched on age and smoking (n=15). HNSCC cases consisted of 3 smokers (25%) and 9 non-/ex-smokers with mean age 55.3 years; controls consisted of 4 current smokers (26%) and 11 non-/ex-smokers with mean age 57.9. Age of the two groups is not statistically different (p=0.46). Two participants (1 case and 1 control) did not provide exact quit date to determine current or ex-smoker categories. The percent repaired at 15 minutes was significantly lower in HNSCC cases (p=0.02) and percent DNA in the comet tail at 15 and 45 minutes of repair was marginally higher in HNSCC patients (Table 4). This small pilot supports the previously observed association of lower DNA repair capacity with increased risk of HNSCC [21-23]. The study would have to be expanded to confirm this pilot observation.
Table 4.
Effect of HNSCC on the repair kinetic. Whole blood of 12 HNSCC patients and 15 cancer free controls embedded in agarose was exposed to 9 Gy gamma radiation following overnight storage at 4 °C. 9-0, 9 Gy 0 minutes of repair; 9-15, 9 Gy 15 minutes of repair; 9-45, 9 Gy 45 minutes of repair; % Repair for 0 to 15 and 15 to 45 minutes was calculated as described in the methods section. P-values are based on a two sided t-test.
| Dose (Gy) - Repair (min) | 9-0 | 9-15 | 9-45 | % Repair 0 to 15 | % Repair 15 to 45 |
|---|---|---|---|---|---|
| Cases (n = 12) | |||||
| Mean ± SEM | 46.88 ± 3.58 | 29.98 ± 2.71 | 19.04 ± 1.85 | 36.77 ± 2.14 | 22.26 ± 3.34 |
| Median | 51.13 | 31.38 | 18.52 | 35.46 | 27.32 |
| Range | 34.20 | 31.57 | 23.14 | 24.79 | 37.55 |
| Controls (n = 15) | |||||
| Mean ± SEM | 43.27 ± 2.70 | 23.02 ± 1.80 | 13.93 ± 1.71 | 46.37 ± 2.99 | 21.84 ± 2.84 |
| Median | 44.87 | 21.70 | 11.95 | 41.48 | 20.53 |
| Range | 39.37 | 26.06 | 21.77 | 46.54 | 48.61 |
| T-Test | |||||
| p-values | 0.43 | 0.04 | 0.06 | 0.02 | 0.93 |
Discussion
Measurement of DNA strand breaks by comet assay has been used to evaluate genotoxicity, to monitor exposures, or to quantify DNA damage and repair in molecular epidemiology [24-26]. In particular, comet assay has been used to examine the association of DNA repair capacity in peripheral white blood cells with risk of various cancers [9;10;17] including HNSCC [23;27]. Palyvoda et al measured DNA damage at six time points between 0-180 minutes after gamma radiation exposure in 48 HNSCC patients and 38 healthy controls [27]. The study showed increased baseline damage and decreased repair in HNSCC patients. The authors also reported high variability in background DNA damage which suggests that standardization of the assay conditions is important. Iwakawa et al compared residual DNA damage in 10 healthy controls and 87 HNSCC patients [23]. The mean residual damage after 15 minutes of repair was significantly higher (p<0.01) in HNSCC cases (56.8+24.4) compared to healthy controls (42.9+19.6). This study also showed that such differences were not observed in EBV-transformed cell lines. In this study, we present initial results of our attempt to adopt standard assay condition for quantification of DNA repair in HNSCC patients and controls.
The monitoring of repair in the population would be ideally done on target tissue. This is not practical and peripheral white blood cells are typically used as a surrogate. The assumption is that the DNA repair capacity of an individual is a genetic predisposition measurable in various cell types. This notion is supported by the results of studies of relatives and twins showing that the repair phenotypes are heritable [8;21]. Besides the use of isolated lymphocytes, several studies reported use of whole blood for the population studies [19;23]. Additional purification and culturing of the lymphocytes does not provide a clear advantage; it is not clear at present which subtype would be a closer approximation of the target tissue or why. We therefore adopted analysis of whole blood in our study. The performance of the assay on freshly isolated blood samples is complicated by the timing of patient sample collection and inability to store/repeat analyses. It would be advantageous to use cryo-preserved or EBV-transformed cells. Our experience and publications of others show, however, that the measurement of DNA repair in cryo-preserved and EBV-transformed cells is not feasible at present [23;28].
Bleomycin is a radiomimetic commonly used in DNA repair studies [9;21]. In our hands, the initial damage in bleomycin treated cells is high and repaired fast (Figure 3). However, the day to day variability is higher than in the gamma radiation experiments which prompted us to select the exposure to radiation for the population study.
In our study, the highest initial damage and the fastest repair was observed in Jurkat T-cells. It appears that these actively replicating cells are most sensitive to gamma radiation, followed by the lymphocytes stimulated with PHA and IL-2. The whole blood is less sensitive possibly due to higher fraction of the cells in G0 [29]. The repair in primary cultured lymphocytes and fresh whole blood is comparable as demonstrated in Figure 1. The repair was about 70% complete at 45 minutes at all doses examined (Figure 2). We chose a dose of 9 Gy for the study of DNA repair capacity; this dose is comparable with other studies of DNA repair [14;17]. Initially, we tested multiple time points along the repair kinetic (data not shown) but for analysis of patient samples we chose two time points (15 minutes and 45 minutes) to approximate the fast and slow portion of the repair [9;15]. Analysis of additional time points in a large population study would not, in our experience, be feasible. With two repair time points, we are able to process slides for a maximum of 4 patients at a time with a minimum of two days needed to complete the sample processing.
The workflow selected based on the above results includes sampling of whole blood with an overnight storage at 4 °C, exposure of cells embedded in agarose to 9 Gy of gamma radiation, and measurement of repair at 15 and 45 minutes after exposure. Under these conditions, the variability of the measurement between people is higher than the day-to-day variability for the same person sampled repeatedly (Table 2). The entire workflow focuses on quantification of repair as opposed to quantification of initial damage. The disappearance of the comet tail is only a rough approximation of the repair process and does not evaluate in detail any of the relevant enzymatic processes. In addition, the phenotype may be further influenced by factors only indirectly connected with DNA repair including chromatin structure [30]. However, such a global view may generate hypotheses stimulating further research of the repair pathways [7]. We used these conditions to compare DNA repair in smokers and in HNSCC patients. The repair was slightly higher during the first 15 minutes in smokers compared to ex-/non-smokers (Table 3). This is possibly due to induction of repair by exposure to cigarette smoke. The repair was also slightly higher in controls compared to HNSCC patients matched on age and smoking status (Table 4). This could reflect a genetic predisposition similar to the heritable phenotype of mutagen sensitivity previously associated with increased risk of HNSCC [22].
In conclusion, our results show that the described workflow is appropriate for quantification of DNA repair on blood samples in a population study. We were able to detect a slower repair in HNSCC patients in a small pilot study of DNA repair. However, the throughput of the assay is limited and the application to a large population would benefit from further methodological improvements including an efficient cell preservation protocol and automated image processing.
Supplementary Material
Acknowledgments
We want to thank Dr. Bozena Novotna for an expert introduction to comet assay analyses. The project was supported in part by Grant Number M01RR-023942-01 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH. Additional support was provided by U.S. Army Medical Research and Material Command, Prostate Cancer Research Program award W81XWH-04-1-0294, American Cancer Society award CRTG-02-245-01-CCE, and Flight Attendant Medical Research Institute award 052444 to RG. Most importantly, we acknowledge the study participants for their contributions to making this study possible.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 2.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 3.Bohr VA. DNA damage and its processing: relation to human disease. J Inherit Metab Dis. 2002;25:215–222. doi: 10.1023/a:1015681929316. [DOI] [PubMed] [Google Scholar]
- 4.Mohrenweiser HW, Wilson DM, III, Jones IM. Challenges and complexities in estimating both the functional impact and the disease risk associated with the extensive genetic variation in human DNA repair genes. Mutat Res. 2003;526:93–125. doi: 10.1016/s0027-5107(03)00049-6. [DOI] [PubMed] [Google Scholar]
- 5.Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 6.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 7.Jones IM, Thomas CB, Xi T, Mohrenweiser HW, Nelson DO. Exploration of methods to identify polymorphisms associated with variation in DNA repair capacity phenotypes. Mutat Res. 2007;616:213–220. doi: 10.1016/j.mrfmmm.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Berwick M, Vineis P. Markers of DNA repair and susceptibility to cancer in humans: an epidemiologic review. J Natl Cancer Inst. 2000;92:874–897. doi: 10.1093/jnci/92.11.874. [DOI] [PubMed] [Google Scholar]
- 9.Schmezer P, Rajaee-Behbahani N, Risch A, Thiel S, Rittgen W, Drings P, Dienemann H, Kayser KW, Schulz V, Bartsch H. Rapid screening assay for mutagen sensitivity and DNA repair capacity in human peripheral blood lymphocytes. Mutagenesis. 2001;16:25–30. doi: 10.1093/mutage/16.1.25. [DOI] [PubMed] [Google Scholar]
- 10.Schabath MB, Spitz MR, Grossman HB, Zhang K, Dinney CP, Zheng PJ, Wu X. Genetic instability in bladder cancer assessed by the comet assay. J Natl Cancer Inst. 2003;95:540–547. doi: 10.1093/jnci/95.7.540. [DOI] [PubMed] [Google Scholar]
- 11.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 12.Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu JC, Sasaki YF. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen. 2000;35:206–221. doi: 10.1002/(sici)1098-2280(2000)35:3<206::aid-em8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 13.Burlinson B, Tice RR, Speit G, Agurell E, Brendler-Schwaab SY, Collins AR, Escobar P, Honma M, Kumaravel TS, Nakajima M, Sasaki YF, Thybaud V, Uno Y, Vasquez M, Hartmann A. Fourth International Workgroup on Genotoxicity testing: Results of the in vivo Comet assay workgroup, Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 2007;627:31–35. doi: 10.1016/j.mrgentox.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 14.Kumaravel TS, Jha AN. Reliable Comet assay measurements for detecting DNA damage induced by ionising radiation and chemicals. Mutat Res. 2006;605:7–16. doi: 10.1016/j.mrgentox.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Frankenberg-Schwager M. Review of repair kinetics for DNA damage induced in eukaryotic cells in vitro by ionizing radiation. Radiother Oncol. 1989;14:307–320. doi: 10.1016/0167-8140(89)90143-6. [DOI] [PubMed] [Google Scholar]
- 16.McNamee JP, McLean JR, Ferrarotto CL, Bellier PV. Comet assay: rapid processing of multiple samples. Mutat Res. 2000;466:63–69. doi: 10.1016/s1383-5718(00)00004-8. [DOI] [PubMed] [Google Scholar]
- 17.Smith TR, Miller MS, Lohman KK, Case LD, Hu JJ. DNA damage and breast cancer risk. Carcinogenesis. 2003;24:883–889. doi: 10.1093/carcin/bgg037. [DOI] [PubMed] [Google Scholar]
- 18.Anderson D, Yu TW, Dobrzynska MM, Ribas G, Marcos R. Effects in the Comet assay of storage conditions on human blood. Teratog Carcinog Mutagen. 1997;17:115–125. [PubMed] [Google Scholar]
- 19.Chuang CH, Hu ML. Use of whole blood directly for single-cell gel electrophoresis (comet) assay in vivo and white blood cells for in vitro assay. Mutat Res. 2004;564:75–82. doi: 10.1016/j.mrgentox.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 20.Novotna B. DNA Repair Workshop. Smolenice, Slovakia: 2000. Increased DNA fragmentation detected by comet assay in peripehral blood of heterozygotes for a frameshift mutation in the DNA repair protein nibrin; p. 33. Abstract. [Google Scholar]
- 21.Wu X, Gu J, Spitz MR. Mutagen sensitivity: a genetic predisposition factor for cancer. Cancer Res. 2007;67:3493–3495. doi: 10.1158/0008-5472.CAN-06-4137. [DOI] [PubMed] [Google Scholar]
- 22.Cloos J, Spitz MR, Schantz SP, Hsu TC, Zhang ZF, Tobi H, Braakhuis BJ, Snow GB. Genetic susceptibility to head and neck squamous cell carcinoma. J Natl Cancer Inst. 1996;88:530–535. doi: 10.1093/jnci/88.8.530. [DOI] [PubMed] [Google Scholar]
- 23.Iwakawa M, Goto M, Noda S, Sagara M, Yamada S, Yamamoto N, Kawakami Y, Matsui Y, Miyazawa Y, Yamazaki H, Tsuji H, Ohno T, Mizoe J, Tsujii H, Imai T. DNA repair capacity measured by high throughput alkaline comet assays in EBV-transformed cell lines and peripheral blood cells from cancer patients and healthy volunteers. Mutat Res. 2005;588:1–6. doi: 10.1016/j.mrgentox.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 24.Collins AR, Horvathova E. Oxidative DNA damage, antioxidants and DNA repair: applications of the comet assay. Biochem Soc Trans. 2001;29:337–341. doi: 10.1042/0300-5127:0290337. [DOI] [PubMed] [Google Scholar]
- 25.Rojas E, Lopez MC, Valverde M. Single cell gel electrophoresis assay: methodology and applications. J Chromatogr B Biomed Sci Appl. 1999;722:225–254. doi: 10.1016/s0378-4347(98)00313-2. [DOI] [PubMed] [Google Scholar]
- 26.Olive PL, Banath JP. The comet assay: a method to measure DNA damage in individual cells. Nat Protocols. 2006;1:23–29. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 27.Palyvoda O, Mukalov I, Polanska J, Wygoda A, Drobot L, Widel M, Rzeszowska-Wolny J. Radiation-induced DNA damage and its repair in lymphocytes of patients with head and neck cancer and healthy donors. Anticancer Res. 2002;22:1721–1725. [PubMed] [Google Scholar]
- 28.Duthie SJ, Pirie L, Jenkinson AM, Narayanan S. Cryopreserved versus freshly isolated lymphocytes in human biomonitoring: endogenous and induced DNA damage, antioxidant status and repair capability. Mutagenesis. 2002;17:211–214. doi: 10.1093/mutage/17.3.211. [DOI] [PubMed] [Google Scholar]
- 29.Maluf SW. Monitoring DNA damage following radiation exposure using cytokinesis-block micronucleus method and alkaline single-cell gel electrophoresis. Clin Chim Acta. 2004;347:15–24. doi: 10.1016/j.cccn.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 30.Morrison AJ, Shen X. DNA repair in the context of chromatin. Cell Cycle. 2005;4:568–571. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.