

Abstract
Mitochondria dysfunction was first described in the 1960s. However, the extent and mechanisms of mitochondria dysfunction’s role in cellular physiology and pathology has only recently begun to be appreciated. To adequately evaluate mitochondria-mediated toxicity, it is not only necessary to understand mitochondria biology, but discerning mitochondrial redox biology is also essential. The latter is intricately tied to mitochondrial bioenergetics. Mitochondrial free radicals, antioxidants, and antioxidant enzymes are players in mitochondrial redox biology. This review will provide an across-the-board, albeit not in-depth, overview of mitochondria biology and mitochondrial redox biology. With accumulating knowledge on mitochondria biology and mitochondrial redox biology, we may devise experimental methods with adequate sensitivity and specificity to evaluate mitochondrial toxicity, especially in vivo in living organisms, in the near future.
Keywords: Mitochondria, Redox, Free radical, Antioxidant, Antioxidant enzyme, Toxicology, Bioenergetics
INTRODUCTION
Mitochondria are known as cellular powerhouses. Since more than 60% of cellular ATP is produced through mitochondrial oxidative phosphorylation, calling mitochondria the cellular powerhouse seems appropriate. However, due to an overemphasis on the ATP production of mitochondria, we might have blindly credited the majority of the mitochondria’s pathophysiological involvement to their function as the cellular powerhouse. Such negligence became obvious during the last decade when the interlaced nature of biological events in the nucleus, cytosol, and mitochondria started being uncovered. Among the diverse aspects of mitochondria, mitochondrial redox biology seems to be emerging as the master modulator of mitochondrial, cytosolic, and nuclear biological events.
By no means, it is plausible to provide in-depth insight into specific subjects regarding mitochondria, unless the aforementioned subject is sufficiently narrow, considering the amount of knowledge we have gained during the last two decades. Readers will easily find many excellent recent reviews regarding various aspects of mitochondria. In this regard, this review aims to provide a broad historical and conceptual review of mitochondria and mitochondrial redox biology. In consequence, readers will befittingly find an introductory overview, albeit a bird’s eye view, on the subject of mitochondria, mitochondrial free radical biology, and mitochondrial antioxidants and antioxidant enzymes.
MITOCHONDRIA BIOLOGY
The mitochondrion is an organelle found both in mammalian cells and in lower forms of eukaryotes, such as yeast. The endosymbiosis theory postulates that mitochondria originated from aerobic bacteria (1). Mitochondria are structurally composed of two membranes: an outer membrane (OM) and an inner membrane (IM). The inner membrane has infoldings called cristae, whose structure results in increased surface area for major mitochondrial biochemical reactions. Despite its small volume, the intermembrane space between the two membranes is known to house close to 50 proteins (2). It is also involved in transporting macromolecules in and out of the mitochondria, regulating mitochondrial respiration and apoptosis (3). The inner membrane encloses the mitochondrial matrix where most of the soluble proteins of mitochondria, mitochondrial DNA, and transfer RNA are found (4) (Fig. 1).
Fig. 1. Mitochondrial biology. For detailed information, see the text.
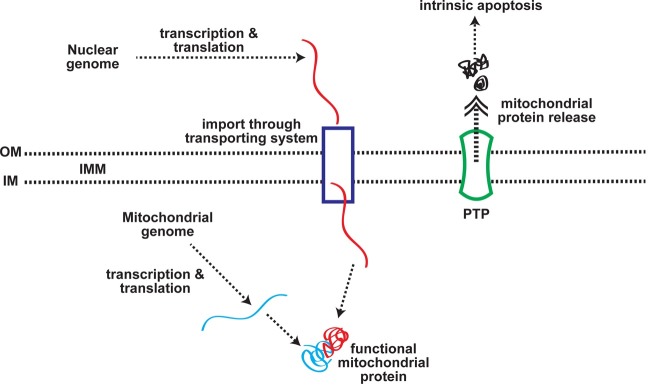
Mitochondrial DNA & RNA. The mitochondrial genome of the eukaryotic phyla frequently studied was shown to exist in the form of super-coiled double stranded DNA, with a size ranging from 15~60 kbp (5). However, as more mitochondrial DNA (mtDNA) is sequenced, a large diversity in the structure and size has been recognized. Much, if not most, of the mtDNA studied bore a linear, multimeric concatenated architecture (6,7). Often, variations in the length and number of introns are important determinants in the size of the mtDNA. Interestingly, the size of the mtDNA did not correlate with the coding capacity of a specific mitochondrial genome. Furthermore, the diversity of mtDNA is demonstrated to be at least partially due to the unusual gene organization.
In addition to genes split by introns, genes encoding subunits of a protein may be fused together. Genes expressing portions of a single polypeptide are scattered in both mtDNA and nuclear DNA. This structural diversity in mitochondrial DNA may arise from its postulated ‘parasitic lifestyle’ (5)
In humans, the size of the mitochondrial genome is 16,298 bp. Human mitochondrial DNA is present in a double stranded closed circular form (8). Most mammalian cells contain hundreds of mitochondria and, in turn, each mitochondrion contains several (2-10) copies of mtDNA. Despite the fact that most cells in the body have been shown to contain more than 1000 copies of mtDNA, and that mtDNA accumulates mutations more rapidly than does nuclear DNA, it is quite striking that mtDNA has only rarely been observed undergoing recombination. Further, heteroplasmy - the state where more than one mtDNA genotype occurs in an individual - has been observed frequently, only in association with diseases (9).
Three nucleus-encoded transcription factors are necessary for mitochondrial biogenesis: mitochondrial transcription factor A (mtTFA), mitochondrial transcription factors B1 or B2 (TFB1M or TFB2M), and mitochondrial termination factor (mTERF). Also, nuclear-encoded mitochondrial RNA polymerase (mtRNApol) is required for the transcription of mtDNA (10). Since both nuclear and mitochondrialencoded proteins are required for complete mitochondrial biogenesis, the coordination of nuclear and mitochondrial transcriptional activities is necessary. The mechanism(s) for such coordination is gradually elucidated (11). Nuclear respiratory factors 1 and 2 (Nrf-1 and -2) have been linked to mitochondrial biogenesis via the transcriptional activation of mtTFA (12); PPARγ co-activator (PGC)-1α (PGC-1α) has been implicated as an integrative coactivator (13).
Mitochondrial proteins. As yet, the total protein composition of mitochondria is not fully defined. More than 800 proteins of the human mitochondrion have been identified and characterized (14); however, based upon predictions from analyses of the human genome, estimates suggest that the organelle may include over 1000 different polypeptides (15). The human mitochondrial genome contains genes encoding 13 components of the electron transfer chain (ETC), 22 transfer RNA (tRNA), and two ribosomal RNA (rRNA). All other genes encoding the mitochondrial proteins are located in the nuclear genome (10).
During evolution, most of the mitochondrial genome inherited from the proposed endosymbiot is either lost or transferred to the nuclear genome. This must have necessitated the development of systematic protein sorting pathways to correctly target nuclear-encoded mitochondrial proteins. Such a targeting strategy is largely accomplished by stretching 20 to 60 amino acid residues (mitochondrial leading sequence: MLS) in the N-terminus of the mitochondrial precursor proteins. The MLS is often highly positively charged and proteolytically cleaved following import into the mitochondrial matrix. It has been suggested that the positive charges of MLS enhance the accumulation of the mitochondrial proteins to the negatively charged matrix side of the inner membrane (16).
In both yeast and human mitochondria, the translocations of mitochondrial precursor proteins across two mitochondrial membranes are mediated, mainly through multisubunit translocation complexes, called TOM complexes (for the translocation of outer membrane) and TIM complexes (for the translocation of inner membrane), respectively. The channel formed by the Tom40 subunit in the outer membrane serves as an outer membrane protein-conducting channel for mitochondrial proteins with a MLS or an internal targeting signal (16-19). Matrix proteins have been shown to further translocate across the inner membrane by the TIM23 complex in an ATP- and membrane potentialdependent manner (20).
In yeast and plant mitochondria, mitochondrial proteins destined for the inner membrane of mitochondria often utilize one of three sorting pathways: 1) upon interaction with the TIM23 complex, a precursor protein can be inserted laterally into the inner membrane (21), 2) a group of mitochondrial proteins lacking a typical MLS but containing internal targeting signals can employ a specialized inner membrane translocase, the TIM22 complex (22,23), and 3) another group of mitochondrial inner membrane proteins can be completely imported into the matrix before redirection and insertion into the inner membrane of mitochondria. Interestingly, this is the general mechanism used by polytopic membrane proteins of bacterial origin (24).
Mitochondrial membranes. In addition to the trafficking of mitochondrial proteins utilizing translocase to achieve precise and specific targeting, many other molecules move in and out of the mitochondria in order to maintain optimal functioning in the cell. The most important physiological roles of mitochondria in cells are ATP production, heme synthesis, and mixed phospholipid synthesis (25). Historically, the outer membrane of the mitochondria was not considered as a barrier to transport because the voltage-dependent anion channel (VDAC or porin) was assumed to be freely permeable to ions and uncharged molecules because of its high permeability. It is estimated that the pore diameter of VADC is 1.8 nm in the closed state and 3 nm in the main conductance state (26,27). In recent years, however, it was strongly suggested that the pore formed by VDAC can serve as a permeability barrier against small molecules. It can also be regulated by metabolites, substrates, nucleotides, and through interactions with other proteins (28).
The mitochondrial inner membrane was originally believed to be impermeable to ions, ensuring efficient chemiosmotic coupling during oxidative phosphorylation. The movements of cations through antiporters, such as K+/H+, Na+/H+, Na+/Ca+2, are considered to be sufficient to maintain mitochondrial volume and Ca+2 homeostasis (29). However, it has become clear that not only are various ion channels present in the inner membrane, but their gating is regulated under specific circumstances (30). The mitochondrial ATP-sensitive K+ channel is of particular interest since it was demonstrated to affect mitochondrial volume, VDAC permeability, matrix pH, and mitochondrial reactive oxygen species (ROS) production (30-32).
Unlike the movements of ions, the shuffling of the substrates necessary for mitochondrial metabolic pathways, such as fatty acid β-oxidation, the Kreb’s cycle, and heme synthesis, are facilitated by mitochondrial carriers. Many mitochondrial carriers belong to a structurally well-defined family. The human genome encodes approximately 48 different mitochondrial carriers (33). Mitochondrial metabolic flux is modeled to involve as many as 89 transport steps across the inner membrane (34). Therefore, many carriers for mitochondrial metabolic flux have yet to be identified. Recently, a mitochondrial pyruvate carrier was finally identified (35).
Cellular respiration through which ATP is produced can be divided into three major pathways: 1) glycolysis, 2) the mitochondrial tricarboxylic acid (TCA) cycle, and 3) mitochondrial ETC. During glycolysis in the cytoplasm, the oxidation of glucose to pyruvate occurs. Subsequent oxidation of pyruvate to CO2 through the TCA cycle liberates electrons to NADH and FADH2. The stepwise transfer of electrons from NADH (via complex I) and/or FADH2 (via complex II) through complex III and IV occurs with concomitant translocation of protons to the mitochondrial intermembrane space. The resulting proton gradient is then utilized by ATP synthase to produce ATP. The glycolytic, TCA, and ETC pathway are tightly regulated and intimately interrelated (36).
Mitochondrial intrinsic apoptosis. Understanding of mitochondrial energy metabolism has become a focus of interest because there are many known examples of malfunctions in mitochondrial energy metabolism, resulting in rare childhood diseases, heart diseases, diabetes, Parkinson’s disease, and cancer (25,37). In addition, mitochondria serve as the site of regulation of programmed cell death (apoptosis). Mitochondrial membrane permeabilization (MMP) is a key event leading to physiological or extrinsic stimulus-induced apoptosis. The mitochondrial megachannel, often called the permeability transition pore (PTP), has been known to cause MMP (38).
Both direct channel formation and the regulation of the PTP by the B-cell lymphocytic leukemia proto-oncogene 2 (Bcl-2) family members are implicated in the mitochondrial apoptosis pathways (28). The majority of BH-3-only proteins of the Bcl-2 family serve as death signal sensors and translocate to the mitochondria, resulting in increased permeability of the outer mitochondrial membrane (39). On the other hand, multi-domain pro-apoptotic proteins either translocate from the cytoplasm to the mitochondria upon stimulation, or they localize in the mitochondria of living cells; Bax is an example for the former and Bak, the latter (39,40).
In receptor-mediated apoptosis, initiator caspase 8 is recruited via its death effecter domain (DED) and is bound to activated death receptors to form a death-inducing signaling complex (DISC). While in the mitochondrial pathway, caspase 9 is recruited to the apoptosome via its caspase recruitment domain (CARD) (41-43). The release of cytochrome C and Smac/Diablo from the mitochondria (second mitochondria-derived activator of the caspase/direct inhibitor of the apoptosis protein (IAP)-binding protein with low pI) is a crucial step in the mitochondrial apoptosis pathway (40,44).
Contrary to caspase-dependent intrinsic and extrinsic apoptosis pathways, three mitochondrial intermembrane proteins are crucial in the caspase-independent cell death: apoptosis inducing factor (AIF) (45), endonuclease G (endoG) (46), and mitochondrial serine protease Omi/HtrA2 (47). Mitochondria are, therefore, reservoirs of caspase-dependent, as well as caspase-independent, effectors of apoptosis.
At least three known regulators of PTP are involved in cellular energy metabolism: 1) hexokinase, 2) glyceraldehyde-3-phosphate dehydrogenase in the cytosol, and 3) creatine kinase in the mitochondrial intermembrane space (38,48,49). In addition, it has been proposed that ANT in the PTP coordinates cellular metabolism with mitochondrial energy metabolism (48). Therefore, cytoplasmic metabolic pathways, mitochondrial energy metabolism, and mitochondrial apoptotic pathways appear to be tightly regulated via the mitochondrial structure and biochemistry to achieve optimal cellular function.
From the point of view of their biogenesis and cellular functions, mitochondria are unique organelles since they require the contribution and coordination of two physically-separated genomes and subcellular compartments. With the discovery of an increasing number of human diseases caused by or associated with abnormal mitochondrial physiology and their functions, there is an increased need to better understand the molecular mechanisms utilized for the coordination of the nucleus, cytoplasm, and mitochondria.
MITOCHONDRIAL FREE RADICAL BIOLOGY
Generation of superoxide anion. According to a recent mitochondrial proteomics study, approximately 36% of the 615 total distinct mitochondrial proteins are involved in the TCA cycle, oxidative phosphorylation (OXPHOS), carbohydrate metabolism, amino acid metabolism, and lipid metabolism (34,50). The maximum ATP generation per glucose, palmitate, or glutamate molecule is calculated as 31.5, 106, and 20.5 ATP molecules, respectively (25). Mitochondria appear to have been structurally and biochemically evolved to maximize ATP production per substrate. Examples of this include the cristae and mitochondrial carriers; the former ensures more metabolic and OXPHOS machineries per mitochondrion, while the latter facilitates an efficient substrate supply from the cytoplasm to the mitochondria.
Although highly efficient in transporting protons across the mitochondrial inner membrane, the nature of coenzyme Q as an electron carrier, which functions by alternating electron oxidation-reduction reactions, results in the mitochondria being a major intracellular source of oxygen radicals under physiological conditions. Superoxide anion (![]() ) is generated in the mitochondria at a rate of approximately 2~5% of the total electrons transported through the electron transport chain (ETC). It is also 1~2% of the total daily oxygen consumption (25,51). The loss of 2% of the electrons due to the electron transfer to oxygen, therefore, would produce 3% fewer ATP molecules from glucose metabolism (25). More importantly, mitochondrial
) is generated in the mitochondria at a rate of approximately 2~5% of the total electrons transported through the electron transport chain (ETC). It is also 1~2% of the total daily oxygen consumption (25,51). The loss of 2% of the electrons due to the electron transfer to oxygen, therefore, would produce 3% fewer ATP molecules from glucose metabolism (25). More importantly, mitochondrial ![]() must have been a challenging obstacle to overcome during the evolution of mitochondria. It has been suggested that the oxygen radical-rich environment of mitochondria may have been one of the driving forces of mitochondrial gene transfer to the nuclear genome (16), and that 6% of the total number of mitochondrial proteins are calculated to be involved in redox modulation (50).
must have been a challenging obstacle to overcome during the evolution of mitochondria. It has been suggested that the oxygen radical-rich environment of mitochondria may have been one of the driving forces of mitochondrial gene transfer to the nuclear genome (16), and that 6% of the total number of mitochondrial proteins are calculated to be involved in redox modulation (50).
In 1954, Gerschman and colleagues proposed that the toxic effects of elevated oxygen levels on aerobes mediated by ionizing radiation was due to free radical formation (52). However, the relevance of their hypothesis to the life sciences was not appreciated until McCord and Fridovich’s discovery of superoxide dismutase (SOD) in 1969 (53). ![]() disproportionates quite rapidly to form hydrogen peroxide (H2O2). This spontaneous reaction alone can maintain the steady state level of
disproportionates quite rapidly to form hydrogen peroxide (H2O2). This spontaneous reaction alone can maintain the steady state level of ![]() in mitochondria at micromolar concentrations (51). However, with 3 μM of manganese superoxide dismutase (MnSOD) in the matrix, mitochondria maintains picomolar and nanomolar concentrations of
in mitochondria at micromolar concentrations (51). However, with 3 μM of manganese superoxide dismutase (MnSOD) in the matrix, mitochondria maintains picomolar and nanomolar concentrations of ![]() and H2O2, respectively (51).
and H2O2, respectively (51).
![]() possesses an unpaired electron, and is, therefore, a free radical. In hydrophobic environments,
possesses an unpaired electron, and is, therefore, a free radical. In hydrophobic environments, ![]() is a strong proton acceptor, nucleophile, and reducing agent. It becomes an oxidant when proton donors, such as catechols or vitamins C and E, are available. In aqueous environments,
is a strong proton acceptor, nucleophile, and reducing agent. It becomes an oxidant when proton donors, such as catechols or vitamins C and E, are available. In aqueous environments, ![]() rapidly loses its basic, nucleophilic, and oxidizing characteristics, ultimately resulting in its functioning as a reducing agent (54).
rapidly loses its basic, nucleophilic, and oxidizing characteristics, ultimately resulting in its functioning as a reducing agent (54).
Generation of hydroxyl radical. It is generally accepted that the generation of hydroxyl radical (HO•) through ‘Fenton chemistry’ is crucial for reported ![]() -mediated deleterious effects on biological systems (55). The first step in ‘superoxide-driven Fenton chemistry’ is the reduction of Fe(III) to Fe(II) (55). It is argued that the direct reduction of ferric ion to ferrous ion by
-mediated deleterious effects on biological systems (55). The first step in ‘superoxide-driven Fenton chemistry’ is the reduction of Fe(III) to Fe(II) (55). It is argued that the direct reduction of ferric ion to ferrous ion by ![]() may not occur (56). However, evidence for this reaction has been documented: 1) despite the strictly regulated availability of iron in vivo, a labile pool of iron has been found in the cytoplasm (57), 2) independent of heme and iron-sulfur clusters, mitochondria maintains a compartmentalized ‘non-heme non-iron-sulfur iron’ pool in the matrix and inner membrane (58), and 3)
may not occur (56). However, evidence for this reaction has been documented: 1) despite the strictly regulated availability of iron in vivo, a labile pool of iron has been found in the cytoplasm (57), 2) independent of heme and iron-sulfur clusters, mitochondria maintains a compartmentalized ‘non-heme non-iron-sulfur iron’ pool in the matrix and inner membrane (58), and 3) ![]() enhances the release of iron from storage proteins and iron-sulfur clusters (59).
enhances the release of iron from storage proteins and iron-sulfur clusters (59).
Once reduction of ferric iron to ferrous iron occurs, hemolytic fission of the oxygen-oxygen bond in H2O2 by Fe(II) produces two HO• radicals (54,55). The biological significance of HO• generation via ‘superoxide-driven Fenton chemistry’ has been attributed to the conversion of the less reactive ![]() and H2O2 to the highly reactive HO•, the latter being capable of directly oxidizing proteins to produce a long-lived protein radical (60). It has also been postulated that iron released from iron-sulfur clusters by
and H2O2 to the highly reactive HO•, the latter being capable of directly oxidizing proteins to produce a long-lived protein radical (60). It has also been postulated that iron released from iron-sulfur clusters by ![]() may interact with DNA, suggesting the possibility that DNA is a prime target for HO•
(61). Direct reduction of iron by
may interact with DNA, suggesting the possibility that DNA is a prime target for HO•
(61). Direct reduction of iron by ![]() with a resulting HO• generation has been documented, despite the presence of other biological reducing agents, such as NADPH, NADH, GSH, and cysteine (62,63). In contrast, MnSOD decreases HO• generation (51). Therefore, MnSOD in the mitochondrial matrix may serve as a guardian of mitochondrial DNA against
with a resulting HO• generation has been documented, despite the presence of other biological reducing agents, such as NADPH, NADH, GSH, and cysteine (62,63). In contrast, MnSOD decreases HO• generation (51). Therefore, MnSOD in the mitochondrial matrix may serve as a guardian of mitochondrial DNA against ![]() and HO•. It is controversial whether HO• is capable of initiating lipid peroxidation; it has been proposed that ferryl radicals are more likely to initiate this reaction (54,55).
and HO•. It is controversial whether HO• is capable of initiating lipid peroxidation; it has been proposed that ferryl radicals are more likely to initiate this reaction (54,55).
Generation of peroxynitrite anion. Peroxynitrite anion (ONOO−) may be formed either from the interaction between ![]() and nitric oxide (NO•) at the diffusion-limited rate of 1.6 × 1010 M−1 S−1
(64) or through the reaction of nitroxyl anion (NO−) with molecular oxygen (65); the importance of the latter reaction in vivo is yet to be determined. In general, the formation of ONOO− from
and nitric oxide (NO•) at the diffusion-limited rate of 1.6 × 1010 M−1 S−1
(64) or through the reaction of nitroxyl anion (NO−) with molecular oxygen (65); the importance of the latter reaction in vivo is yet to be determined. In general, the formation of ONOO− from ![]() and NO• has an unfavorable reaction (66), although it has been proposed that
and NO• has an unfavorable reaction (66), although it has been proposed that ![]() may serve as a sink for NO•, thereby regulating the availability of NO• as a vasodilator (67).
may serve as a sink for NO•, thereby regulating the availability of NO• as a vasodilator (67).
Evidence suggests that both ONOO− and peroxynitrous acid (ONOOH) can diffuse across biomembranes (68). NO• is neutral and therefore should be able to diffuse across mitochondrial membranes (66). Cytosolic NO• is mainly produced from L-arginine through a catalytic reaction by the enzyme ‘nitric oxide synthase (NOS)’ (69). While the three isoforms of NOS have been relatively well characterized, the existence of a mitochondrial NOS isoform is currently controversial (70-73); nevertheless, evidence for the physiological function of mitochondrial NOS in the heart is accumulating (74).
The maximal production of ONOO− can be achieved at a NO•/![]() flux ratio of two (75). Although this reaction occurs at the diffusion-limited rate (64), NO• must compete with mitochondrial SODs for
flux ratio of two (75). Although this reaction occurs at the diffusion-limited rate (64), NO• must compete with mitochondrial SODs for ![]() to produce ONOO−, e.g. with copper and zinc superoxide dismutase (CuZnSOD) in the intermembrane space and/or with MnSOD in the matrix. With concentrations of SODs frequently found at μM levels, NO• cannot outcompete SODs in obtaining
to produce ONOO−, e.g. with copper and zinc superoxide dismutase (CuZnSOD) in the intermembrane space and/or with MnSOD in the matrix. With concentrations of SODs frequently found at μM levels, NO• cannot outcompete SODs in obtaining ![]() to form ONOO− under physiological conditions (60,66,76). However, under pathological conditions where local NO• concentrations reach μM levels, NO• may be able to compete with SODs for
to form ONOO− under physiological conditions (60,66,76). However, under pathological conditions where local NO• concentrations reach μM levels, NO• may be able to compete with SODs for ![]() , and thus generate ONOO−
(66).
, and thus generate ONOO−
(66).
Alternatively, NO• may react with thiyl radical (GS•) and form S-nitrosoglutathione (GSNO). Nitrosothiols, including GSNO, are found at μM concentrations in human plasma (77). With a half-life of longer than 40 min in plasma, nitrosothiols are storage/carrier forms of NO• and decomposed to release NO•, a reaction mediated by catalytic Cu+1 (66). CuZnSOD has been implicated as a source of the protein-bound catalytic copper that is necessary to decompose nitrosothiols, though this reaction is GSH concentration-dependent (78). Therefore, nitrosothiols in the mitochondrial intermembrane space can serve as a source of NO•. In addition, the availability of NO• to the mitochondrial matrix may be determined by CuZnSOD and GSH contents in the mitochondrial intermembrane space.
The generation of ONOO− from ![]() and NO• also has biological significance because of the subsequent generation of three additional biologically-reactive radicals, HO•, nitrogen dioxide (•NO2), and carbonate radical anion (
and NO• also has biological significance because of the subsequent generation of three additional biologically-reactive radicals, HO•, nitrogen dioxide (•NO2), and carbonate radical anion (![]() ). Upon protonation of ONOO−, ONOOH can either isomerize to nitrate (
). Upon protonation of ONOO−, ONOOH can either isomerize to nitrate (![]() ) or decompose to •NO2 and HO•. Homolytic cleavage of ONOOH to •NO2 and HO• occurs at a constant rate of 0.38 s−1 (Fig. 2) (79,80).
) or decompose to •NO2 and HO•. Homolytic cleavage of ONOOH to •NO2 and HO• occurs at a constant rate of 0.38 s−1 (Fig. 2) (79,80).
Fig. 2. Mitochondrial 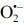 conversion to HO•, •NO2, and
conversion to HO•, •NO2, and 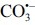 . For detailed information, see the text. Adapted from (51) with modifications. , superoxide anion; H2O2, hydrogen peroxide; HO•, hydroxyl radical; NO•, nitric oxide; ONOO−, peroxynitrite anion; ONOOH, peroxynitrous acid; •NO2, nitrogen dioxide;
. For detailed information, see the text. Adapted from (51) with modifications. , superoxide anion; H2O2, hydrogen peroxide; HO•, hydroxyl radical; NO•, nitric oxide; ONOO−, peroxynitrite anion; ONOOH, peroxynitrous acid; •NO2, nitrogen dioxide; 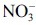 , nitrate; CO2, carbon dioxide;
, nitrate; CO2, carbon dioxide; 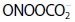 , nitrosoperoxocarboxylate anion; , carbonate radical anion.
, nitrosoperoxocarboxylate anion; , carbonate radical anion.

At a physiological pH, spontaneous decomposition of ONOO− is too slow to be biologically important. ONOO− reacts with biological targets at rate constants ranging from 102~106 M−1 S−1
(60,81); among possible biological targets are reactions with carbon dioxide (CO2) or bicarbonate (HCO3−) at a pH-independent rate constant of 3 × 104 M−1 S−1
(60,82). Through the formation of a very unstable intermediate nitrosoperoxocarboxylate anion (![]() ), ONOO− rapidly decomposes to either
), ONOO− rapidly decomposes to either ![]() and CO2 at about 65% or •NO2 and
and CO2 at about 65% or •NO2 and ![]() at approximately 35% of the total intermediates (83). With
at approximately 35% of the total intermediates (83). With ![]() at 5~10 mM concentrations (84), mitochondria most likely favor the decomposition of ONOO− through
at 5~10 mM concentrations (84), mitochondria most likely favor the decomposition of ONOO− through ![]() , thereby generating •NO2 and
, thereby generating •NO2 and ![]() .
.
Biological significance of mitochondrial reactive oxygen species. Biological consequences of ONOO− formation are determined, to a large extent, by radicals generated during the decomposition processes of ONOO−. HO• formation, which is likely favored at low pH levels and in the absence of CO2, results in one-electron oxidation of most DNA bases and amino acids, with rate constants near the diffusion limit (109~1010 M−1 S−1). These targets are indiscriminate and often present at the site of HO• production (60,81). It should be noted that HO• is one of very few agents that can directly oxidize CO2/![]() to produce
to produce ![]() , although its rate constant is low (106~108 M−1 S−1) (85). Simultaneous generation of •NO2 and
, although its rate constant is low (106~108 M−1 S−1) (85). Simultaneous generation of •NO2 and ![]() through CO2- mediated ONOO− decomposition efficiently nitrates protein-tyrosine residues and nucleic acid-guanine residues. Formation of 3-nitrotyrosine or 8-nitroguanine is promoted mainly by a combination of target-derived radical formation and a diffusion-controlled termination reaction with •NO2, where
through CO2- mediated ONOO− decomposition efficiently nitrates protein-tyrosine residues and nucleic acid-guanine residues. Formation of 3-nitrotyrosine or 8-nitroguanine is promoted mainly by a combination of target-derived radical formation and a diffusion-controlled termination reaction with •NO2, where ![]() preferentially oxidizes tyrosine and guanine via one-electron abstraction and •NO2 promoted nitration on tyrosyl and guanine radicals (86-88). In addition, one-electron oxidations by •NO2 and
preferentially oxidizes tyrosine and guanine via one-electron abstraction and •NO2 promoted nitration on tyrosyl and guanine radicals (86-88). In addition, one-electron oxidations by •NO2 and ![]() and two-electron oxidation reactions by ONOO− contributed to oxidative and nitrative stress/damage mediated by
and two-electron oxidation reactions by ONOO− contributed to oxidative and nitrative stress/damage mediated by ![]() and •NO (76,81).
and •NO (76,81).
Under physiological conditions, mitochondrial ![]() is the most important reactant of the mitochondrial free radical pool. Superoxide anions serve as a continuous source of mobile electrons whose movement cannot be completely controlled by cellular biochemical mechanisms, thereby disturbing the normal cellular reactions mediated by reduction-oxidation (redox). As its formation is unavoidable, mitochondrial
is the most important reactant of the mitochondrial free radical pool. Superoxide anions serve as a continuous source of mobile electrons whose movement cannot be completely controlled by cellular biochemical mechanisms, thereby disturbing the normal cellular reactions mediated by reduction-oxidation (redox). As its formation is unavoidable, mitochondrial ![]() must efficiently be eliminated once it is produced, thereby preventing the generation of other free radical species, such as HO•, •NO2 and
must efficiently be eliminated once it is produced, thereby preventing the generation of other free radical species, such as HO•, •NO2 and ![]() . Mitochondrial antioxidants and antioxidant enzymes must have been evolved with this in mind; their complexity and their intricate involvement in cellular functions suggest this to be true.
. Mitochondrial antioxidants and antioxidant enzymes must have been evolved with this in mind; their complexity and their intricate involvement in cellular functions suggest this to be true.
MITOCHONDRIAL ANTIOXIDANTS AND ANTIOXIDANT ENZYMES
Mitochondria are a source of ![]() production. For example, for a 60 kg woman, 160~320 mmol, and for a 80 kg man, 215~430 mmol of
production. For example, for a 60 kg woman, 160~320 mmol, and for a 80 kg man, 215~430 mmol of ![]() are produced each day (51). Mitochondria must have evolved to sufficiently protect themselves from
are produced each day (51). Mitochondria must have evolved to sufficiently protect themselves from ![]() -mediated damage. To suppress radical formation as well as oxidant-induced reactions, antioxidants and antioxidant enzymes whose primary functions are designed to counteract reactive radicals and oxidants must have also evolved (Fig. 3).
-mediated damage. To suppress radical formation as well as oxidant-induced reactions, antioxidants and antioxidant enzymes whose primary functions are designed to counteract reactive radicals and oxidants must have also evolved (Fig. 3).
Fig. 3. Mitochondrial reactive oxygen species and antioxidant enzymes. For detailed information, see the text.

Cellular antioxidant systems can be divided into two major groups, enzymatic and non-enzymatic; the former is often called an ‘antioxidant enzyme (AOE) system’, whereas low-molecular weight organic, as well as inorganic, antioxidants belong to the latter and are collectively called the ‘antioxidants’ (89). They also can be categorized according to their main modes of action. ‘Directly operating antioxidants’ efficiently scavenge oxidants/radicals (scavenging function) and/or regenerate oxidized biomolecules (repair function), whereas ‘indirectly working antioxidants’ are often cellular redox couples whose redox states significantly contribute to the cellular reduction potential, thereby promoting the regeneration of oxidized antioxidants (90,91).
Dismutation of superoxide anion. The movement of electrons liberated from highly-reduced organic molecules to molecular oxygen provides cellular energy to maintain a low entropy state necessary for initiating biochemical reactions. Most biochemical reactions where electrons move are two-electron processes to avoid the formation of products with unpaired electrons (90). Therefore, there seems to be at least two choices to detoxify mitochondrial ![]() : First, the unpaired electron of
: First, the unpaired electron of ![]() can be removed to generate ground state oxygen (3O2). Second, one additional electron can be added to pair with the unpaired electron on
can be removed to generate ground state oxygen (3O2). Second, one additional electron can be added to pair with the unpaired electron on ![]() , thereby generating peroxide ion (
, thereby generating peroxide ion (![]() ). The former strategy utilizes the removal of an electron from
). The former strategy utilizes the removal of an electron from ![]() by vitamin C or E (92,93), whereas superoxide dismutases (SODs) pair up the unpaired electron on
by vitamin C or E (92,93), whereas superoxide dismutases (SODs) pair up the unpaired electron on ![]() with the unpaired electron from another
with the unpaired electron from another ![]() , thus utilizing both strategies.
, thus utilizing both strategies.
Two forms of SOD with redox-active metal cofactor(s) are present in mitochondria: MnSOD in the matrix, and CuZnSOD in the intermembrane space (94,95). Mitochondrial ![]() is produced mainly at Complex I and III of the ETC with different topological preferences. Complex I releases
is produced mainly at Complex I and III of the ETC with different topological preferences. Complex I releases ![]() exclusively to the matrix side, whereas complex III-driven
exclusively to the matrix side, whereas complex III-driven ![]() is released on both sides of the inner membrane of mitochondria with equal probability (96). Anionic
is released on both sides of the inner membrane of mitochondria with equal probability (96). Anionic ![]() is highly membrane impermeable, and therefore, compartmentalized (96), which may explain the presence of two SODs in each compartment of mitochondria, that is, the matrix and the intermembrane space.
is highly membrane impermeable, and therefore, compartmentalized (96), which may explain the presence of two SODs in each compartment of mitochondria, that is, the matrix and the intermembrane space.
SOD catalyzes the dismutation of two ![]() into O2 and H2O2, during which SOD serves both as an oxidant and as a reductant. The midpoint reduction potential of MnSOD is measured as 393 ± 29 mV, which is approximately the median value between the oxidation of O2 (−160 mV) and the reduction of
into O2 and H2O2, during which SOD serves both as an oxidant and as a reductant. The midpoint reduction potential of MnSOD is measured as 393 ± 29 mV, which is approximately the median value between the oxidation of O2 (−160 mV) and the reduction of ![]() (+850 mV), resulting in favorable thermodynamics for the catalytic activity (97).
(+850 mV), resulting in favorable thermodynamics for the catalytic activity (97).
Scavenging of free radicals. To be an effective ‘directly operating antioxidant’, the following must be true: (1) both antioxidants and antioxidant radicals must be relatively non-reactive, (2) the antioxidant radical should decay to harmless products, (3) there should be no O2 addition to the antioxidant radical, and (4) a renewal mechanism must exist to result in molecular regeneration (98).
Vitamins C and E satisfy all of the above requirements for functioning as effective antioxidants. It is necessary to maintain sufficient amounts of co-antioxidants to regenerate antioxidants from antioxidant radicals generated in redox reactions in order to avoid the pro-oxidant properties of the antioxidant radical (99,100).
Vitamin E concentrations in mitochondria are estimated to be 0.08~0.22 nmol/mg of protein, but only one-fifth of this amount is present in the mitochondrial inner membrane (101,102). The concentration of vitamin E in membrane phospholipids is calculated to be 2 × 10−4 M (103). Whether vitamin C is present in the mitochondrial matrix is yet to be determined (103); its concentration in the mitochondria, however, is estimated to be approximately 1 mM (91,104). The classical actions of vitamin E (α-tocopherol: α-TOH) and vitamin C (ascorbic acid: AscH−) are that of chain-breaking antioxidants, although their site-specificities differ; α-TOH is lipophilic, whereas AscH- is hydrophilic (100,102).
Once generated through reactions with oxidative radicals, antioxidant radicals, for example, α-tocopherol radical (α-TO•) or ascorbyl free radical (Asc•−), must be terminated both to regenerate and to prevent pro-oxidant reactivity towards biomolecules. As co-antioxidants efficiently renewing α-TO• in vivo, ubiquinol-10 (CoQ10H2: the reduced form of coenzyme Q10), vitamin C, α-tocopheryl hydroquinone, and 3-hydroxyanthranilate are important, although the latter two compounds are less important as preventive antioxidants (99,105-107). CoQ10H2 and vitamin C are similar in their reduction potentials, and equally efficient as co-antioxidants for α-TOH (98); however, CoQ10H2 is likely to be the most effective co-antioxidant for α-TOH in mitochondria (99) for the following reasons: 1) Its abundance in mitochondria at approximately 35 times over α-TOH content (105), 2) Its incorporation into the lipid bilayer and/or association with lipoproteins (108), and 3) the presence of NADH and succinate dehydrogenase in mitochondria, which keep CoQ10 in a reduced form (CoQ10H2) (105).
The renewal processes of Asc•− lead to harmless products at the expense of various ‘indirectly working antioxidants’, thereby eliminating mobile electrons from the cell. Asc•− is produced by one-electron oxidation of AscH−, and further oxidation or dismutation of Asc•− produces dehydroascorbic acid (DHA) (109). In turn, DHA regenerates to AscH− through a two-electron reduction reaction in which GSH, dihydrolipoic acid (DHLA), or NADPH largely provide electrons (89,100,110). The regeneration processes of AscH−, using NADPH as an electron source, are catalyzed by thioredoxin reductase (TrxnR), which, without the involvement of thioredoxin (Trxn), promotes not only the twoelectron reduction of DHA, but also the one-electron reduction of Asc•− (109,111).
Electron-deprived co-antioxidants generated from α-TO• and Asc•− renewal processes are ultimately replenished with electrons in a catabolic metabolism-dependent manner. As mentioned above, CoQ10 is reduced to CoQ10H2 by mitochondrial NADH and succinate dehydrogenase (105). Cellular NADPH is mainly regenerated through ‘the oxidative pentose phosphate pathway’ and, to a smaller extent, through the oxidation of malate during fatty acid synthesis (112). Oxidized glutathione (GSSG) is catalytically regenerated by glutathione reductase (GR), with NADPH as a source of electrons (100). Lipoic acid (LA) is reduced to DHLA through the catalytic reaction of lipoamide dehydrogenase (LipDH), with NADH as a source of electrons (89,110).
Reduction of peroxides. Antioxidants are clearly a first-line of defense against reactive free radicals, as well as against oxidants, since their modes of action are direct and they often reside at/near the source of radical formation. However, their actions are neither controllable nor specific. Therefore, it is not surprising to find that antioxidant enzyme systems have evolved with high substrate specificities. Considering that their substrates have the characteristics of containing mobile electrons or having oxidative reduction potential, and that their modes of action involve the movement of electrons, the role of chalcogen-containing amino acids in antioxidant proteins is important.
The chalcogen elements are oxygen, sulfur, selenium, and tellurium, in group 16 of the periodic table. As a constituent of cysteine, methionine, selenocysteine, selenomethionine, and tyrosine amino acids, they provide antioxidant proteins with versatile redox mechanisms through their various oxidation states (113). The sulfur element in cysteine is of significance because of its 10 different oxidation states (i.e. from +6 to −2 and a fractional oxidation state of +0.5) (114). Many antioxidant proteins utilize cysteine, and to a lesser extent, selenocysteine. Examples include GSH, GR, glutathione peroxidase (GPx), glutaredoxin (Glrx), Txn, TxnR, and peroxiredoxin (Prxn) (89,113).
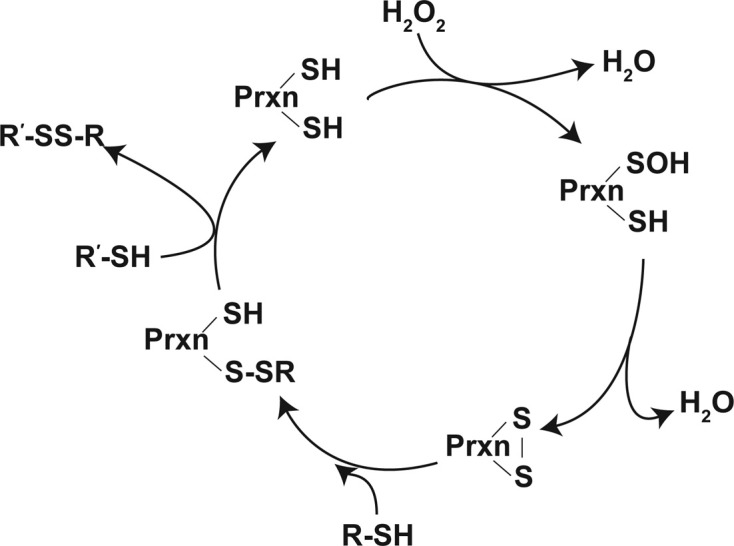
Two major antioxidant enzyme (AOE) systems, whose substrate specificities are mainly towards various forms of peroxides, are ‘the glutathione system’ and ‘the thioredoxin system’ (89). The glutathione system involves GSH, GPx, and GR, while the thioredoxin system consists of Txn, Prxn, and TxnR. In principle, both systems function analogously. GPx and Prxn reduce their substrates, i.e. H2O2 or alkyl hydroperoxides, to H2O or corresponding alcohols. In the process, a disulfide bond forms intermolecularly from two GSHs, or intramolecularly in Txn (Fig. 4).
Fig. 4. Elimination of by antioxidants or AOE systems. , superoxide anion; O2, oxygen; H2O2, hydrogen peroxide; α-TOH, α-tocopherol; AscH−, ascorbic acid; SOD, superoxide dismutase; GSH, glutathione; GSSG, oxidized glutathione; GPx, glutathione peroxidase; GR, glutathione reductase; Prxn, peroxiredoxin; Txn, thioredoxin; Txn(S-S), oxidized Txn.

In these systems, upon interaction with the peroxides, the catalytic reaction is promoted by generating very reactive selenenic acid in the selenocysteine of GPx, or sulfenic acid in the cysteine of Prxn. The reduction of selenenic acid in GPx occurs through a nucleophilic substitution of HO- by RS- from GSH, forming a mixed selenylsulfide that is subsequently reduced to a selenol moiety through a thiol/selenylsulfide-exchange with GSH. One isoform of Prxn contains one cysteine in the catalytic site (1-Cys-Prxn), whereas other Prxns had two cysteines involved in the catalytic reactions (2-Cys-Prxns). Prxns exhibit very similar redox mechanisms as GPx, except that the catalytic cysteine of Prxn forms a sulfenic acid upon interacting with the peroxides. The sulfenic acid moiety of an atypical 2-Cys-Prxn forms a disulfide bond intramolecularly, whereas a disulfide bond in typical 2-Cys-Prxns is formed between subunits. The disulfide bonds formed in Prxns are subsequently reduced through a thiol/disulfide exchange mechanism, mainly with a Txn (Fig. 5) (113,115).
Fig. 5. Catalytic cycle of Prxns. For detailed information, see the text. Adapted from (104) with modifications.
GR is a flavin protein containing two cysteines in the redox-active site. GSSG regenerates into two GSHs by GR, oxidizing NADPH in the process. Initially, a mixed disulfide is formed between a thiol in the active sites of GR and GSSG through a thiol/disulfide exchange mechanism; then, this is converted into an intramolecular disulfide between two GR cysteines. By direct two-electron transfer from FADH2, the intramolecular disulfide bond in GR is reduced (113).
The reduction of oxidized Txn is often catalyzed by TxnR. TxnR is a flavoprotein containing a selenocysteine and a cysteine in its active site. Through ‘a (thiol, selenol)/disulfide-exchange mechanism’, a disulfide bond in the oxidized Txn is reduced, and in the process, TxnR forms a selenylsulfide at its redox-active site. Interestingly, this selenylsulfide bond is converted through ‘a (thiol, thiol)/selenylsulfide-exchange reaction’ into a disulfide bond between two additional cysteine residues at the redox center. This disulfide is subsequently reduced to thiols by a direct twoelectron transfer from FADH2 (Fig. 6) (113).
Fig. 6. Regeneration of Prxn by Trxn and TrxnR system. For detailed information, see the text. Adapted from (104) with modifications.

In addition to these two cellular peroxidase systems, catalase is another AOE with H2O2-substrate specificity and a rate constant of approximately 107 M−1 S−1 (116,117). Catalase is a monofunctional, heme-containing hydroperoxidase. Catalase is localized primarily in the peroxisome (116,118), and only at low levels in some mitochondria (119). Therefore, the GSH and Txn systems are the most important antioxidant systems in mitochondria.
All antioxidant protein components of the GSH and Txn systems have been identified in mitochondria. Mitochondrial GR and GPx4 are expressed, through alternative splicing, as an isoform of cytosolic GR and GPx4, respectively, whereas Txn2, TxnR2, and Prxn3 are mitochondrial forms expressed from nuclear genes (89,115,120,121). GPx and Prxn have differences in their rate constants with hydroperoxides, i.e. GPx is approximately 108 M−1 S−1 and Prxn is approximately 105 M−1 S−1 (117). Therefore, the Prxn system has been suggested to be a back-up or emergency peroxidase system compared to the GSH system. However, recent evidence supports a potential role of Prxn in regulatory functions (122). The latter seems to be very plausible since the Prxn system has been postulated to form ‘the cellular protein disulfide reduction machinery’ (113). Reduced Txn is shown to interact with other proteins, resulting in the regulation of key cellular functions (123,124).
Ultimate reducing power, NADPH. The significance of the GSH system as a first line of defense against oxidative stress partially arises from the importance of GSH in maintaining cellular redox balance. As the most abundant redox active compound, GSH levels are maintained at 1~10 mM concentrations in cytosol and the nucleus, often with higher concentrations in mitochondria (90,125). The calculated half-cell reduction potential of the GSSG/GSH halfcell ranges from −250 mV to −220 mV at 1~10 mM of GSH and a [GSH]/[GSSG] ratio of 100 : 1, whereas another important redox pair, NADP+/NADPH, has a reduction potential of −374 mV at a [NADPH]/[NADP+] ratio of 100 : 1 (90). Although the NADP+/NADPH half-cell is more reducing than is the GSSG/GSH half-cell, the higher reducing capacity of GSH, i.e. 1~10 mM vs. 0.1 mM respectively, makes the GSSG/GSH couple the most significant contributor to the cellular redox environment (90).
Approximately 80% of nicotinamide nucleotides are NAD(H), although levels of this compound vary between different cell types and tissues (126). However, cells keep the [NADPH]/[NADP+] ratio at approximately 100 : 1 and the [NADH]/[NAD+] ratio at approximately 1 : 1000; their levels show significant deviation from equilibrium with each other. Through compartmentalization and thermodynamic buffering, NAD is predominantly localized extramitochondrially as the NAD+ form, whereas NADP is compartmentalized to mitochondria as the NADPH form (126,127).
It has been suggested that cytosolic NADH may preferentially bind to various binding sites, thereby keeping the ratio of the free [NADH]/[NAD+] couple small (126). Consequently, the calculated reduction potential of the NAD+/NADH half-cell redox pair would increase. The reduction potential of the [NADH]/[NAD+] couple has therefore been suggested to be more oxidizing than that of the [NADPH]/[NADP+] couple. Thermodynamic buffering of the [NADH]/[NAD+] or [NADPH]/[NADP+] couples made the former an efficient electron acceptor and the latter an excellent electron donor (126). Also, due to the thermodynamic buffering of the [NADPH]/[NADP+] couple, NADPH may serve as the primary reducing equivalent for GSH (90).
The compartmentalization of NAD(H) and NADP(H) into mitochondria regulates the cellular redox reactions, but these compounds also serve as cellular signaling molecules (128-130). The origin of mitochondrial NAD(H) and NADP(H) is yet to be fully uncovered. However, a putative human mitochondrial isoform of a nicotinamide mononucleotide adenylyltransferase (NMNAT) [hNMNAT3] has been identified, suggesting the possibility of an autonomous mitochondrial NAD metabolism (130,131). In addition, humans have cytosolic forms of NAD kinase that catalyze the phosphorylation reaction of NAD+ to NADP+ (132). The elusive mitochondrial NAD kinase has recently been identified (133).
In contrast to the recent progress in understanding the biochemical mechanisms by which the mitochondrial compartmentalization of NAD(H)/NADP(H) occurs, there has been little progress in elucidating the mechanism(s) of mitochondrial GSH compartmentalization (134). It is generally accepted that mitochondrial GSH up-take may depend on an energy-dependent transport system, although its identity is still controversial (125,135-137).
In both ‘the GSH system’ and ‘the Txn system’, the ultimate reducing equivalents to reduce ![]() is supplied by NADPH. Due to the compartmentalization of NAD/NADP and GSH into the mitochondria, the local mitochondrial redox state may serve as a regulator of cell physiology, and may also modulate the intricate relationships between glycolysis, the TCA cycle, and the ETC (138).
is supplied by NADPH. Due to the compartmentalization of NAD/NADP and GSH into the mitochondria, the local mitochondrial redox state may serve as a regulator of cell physiology, and may also modulate the intricate relationships between glycolysis, the TCA cycle, and the ETC (138).
CONCLUSION
From a toxicological point of view, mitochondria and mitochondrial redox biology are unmethodically connected to bioenergetics and oxidative stress/damage. Mitochondria involve in pathophysiological cellular processes through other than ATP production. In addition, mitochondrial redox biology influences cellular processes through redox modulation, which is now described as ‘eustress,’ in contrast to ‘oxidative stress’.
Now, we should place mitochondria in the center of the cellular response to environmental stress. Pathophysiological responses may transpire through not only ATP production, but also through catabolic/anabolic metabolism and retrograde-signaling, albeit the exact nature has not yet been clarified. The retrograde-signaling influences the mitochondrial morphology, biogenesis, and kinetics. In addition, experimental approaches and novel technologies must be devised to adequately evaluate the environmental stressor-induced toxicity manifested through the mitochondria.
Acknowledgments
This manuscript was revised from the part of Aekyong Kim’s doctoral dissertation, under the supervision of the late Terry D. Oberley, MD, PhD, at the University of Wisconsin-Madison in 2004. This work is supported by grants from the National Research Foundation of Korea (NRF) funded by the Korea government (no. 2010-0024474 & no. 2011-002179) to Aekyong Kim.
References
- 1.Zimorski V., Ku C., Martin W.F., Gould S.B. Endosymbiotic theory for organelle origins. Curr. Opin. Microbiol. (2014);22C:38–48. doi: 10.1016/j.mib.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Vogtle F.N., Burkhart J.M., Rao S., Gerbeth C., Hinrichs J., Martinou J.C., Chacinska A., Sickmann A., Zahedi R.P., Meisinger C. Intermembrane space proteome of yeast mitochondria. Mol. Cell. Proteomics. (2012);11:1840–1852. doi: 10.1074/mcp.M112.021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herrmann J.M., Longen S., Weckbecker D., Depuydt M. Biogenesis of mitochondrial proteins. Adv. Exp. Med. Biol. (2012);748:41–64. doi: 10.1007/978-1-4614-3573-0_3. [DOI] [PubMed] [Google Scholar]
- 4.Tzagoloff A. Mitochondria. Plenum Press; New York: (1982). pp. 1–342. [Google Scholar]
- 5.Burger G., Gray M.W., Lang B.F. Mitochondrial genomes: anything goes. Trends Genet. (2003);19:709–716. doi: 10.1016/j.tig.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 6.Burger G., Forget L., Zhu Y., Gray M.W., Lang B.F. Unique mitochondrial genome architecture in unicellular relatives of animals. Proc. Natl. Acad. Sci. U.S.A. (2003);100:892–897. doi: 10.1073/pnas.0336115100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bendich A.J. Structural Analysis of mitochondrial DNA molecules from fungi and plants using moving pictures and pulsed-field gel electrophoresis. J. Mol. Biol. (1996);255:564–588. doi: 10.1006/jmbi.1996.0048. [DOI] [PubMed] [Google Scholar]
- 8.Berdanier C.D., Everts H.B. Mitochondrial DNA in aging and degenerative disease. Mutat. Res. (2001);475:169–183. doi: 10.1016/S0027-5107(01)00068-9. [DOI] [PubMed] [Google Scholar]
- 9.Ju Y.S., Alexandrov L.B., Gerstung M., Martincorena I., Nik-Zainal S., Ramakrishna M., Davies H.R., Papaemmanuil E., Gundem G., Shlien A., Bolli N., Behjati S., Tarpey P.S., Nangalia J., Massie C.E., Butler A.P., Teague J.W., Vassiliou G.S., Green A.R., Du M.Q., Unnikrishnan A., Pimanda J.E., Teh B.T., Munshi N., Greaves M., Vyas P., El-Naggar A.K., Santarius T., Collins V.P., Grundy R., Taylor J.A., Hayes D.N., Malkin D., Foster C.S., Warren A.Y., Whitaker H.C., Brewer D., Eeles R., Cooper C., Neal D., Visakorpi T., Isaacs W.B., Bova G.S., Flanagan A.M., Futreal P.A., Lynch A.G., Chinnery P.F., McDermott U., Stratton M.R., Campbell P.J. Oriins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife. (2014);3:eLife.02935. doi: 10.7554/eLife.02935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernández-Silva P., Enriquez J.A., Montoya J. Replication and transcription of mammalian mitochondrial DNA. Exp. Physiol. (2003);88:41–56. doi: 10.1113/eph8802514. [DOI] [PubMed] [Google Scholar]
- 11.Brenmoehl J., Hoeflich A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion. (2013);13:755–761. doi: 10.1016/j.mito.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 12.Suliman H.B., Carraway M.S., Welty-Wolf K.E., Whorton A.R., Piantadosi C.A. Lipopolysaccharide Stimulates Mitochondrial Biogenesis via Activation of Nuclear Respiratory Factor-1. J. Biol. Chem. (2003);278:41510–41518. doi: 10.1074/jbc.M304719200. [DOI] [PubMed] [Google Scholar]
- 13.Kelly D.P., Scarpulla R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. (2004);18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 14.Cotter D., Guda P., Fahy E., Subramaniam S. MitoProteome: mitochondrial protein sequence database and annotation system. Nucleic Acids Res. (2004);32:D463–467. doi: 10.1093/nar/gkh048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McDonald T.G., Van Eyk J.E. Mitochondrial proteomics. Undercover in the lipid bilayer. Basic Res. Cardiol. (2003);98:219–227. doi: 10.1007/s00395-003-0417-8. [DOI] [PubMed] [Google Scholar]
- 16.Herrmann J.M. Converting bacteria to organelles: evolution of mitochondrial protein sorting. Trends Microbiol. (2003);11:74–79. doi: 10.1016/S0966-842X(02)00033-1. [DOI] [PubMed] [Google Scholar]
- 17.Cohen I., Guillerault F., Girard J., Prip-Buus C. The N-terminal domain of rat liver carnitine palmitoyltransferase 1 contains an internal mitochondrial import signal and residues essential for folding of its C-terminal catalytic domain. J. Biol. Chem. (2001);276:5403–5411. doi: 10.1074/jbc.M009555200. [DOI] [PubMed] [Google Scholar]
- 18.Addya S., Anandatheerthavarada H.K., Biswas G., Bhagwat S.V., Mullick J., Avadhani N.G. Targeting of NH2-terminal-processed microsomal protein to mitochondria: a novel pathway for the biogenesis of hepatic mitochondrial P450MT2. J. Cell. Biol. (1997);139:589–599. doi: 10.1083/jcb.139.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rojo E.E., Stuart R.A., Neupert W. Conservative sorting of F0-ATPase subunit 9: export from matrix requires delta pH across inner membrane and matrix ATP. EMBO J. (1995);14:3445–3451. doi: 10.1002/j.1460-2075.1995.tb07350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Truscott K.N., Kovermann P., Geissler A., Merlin A., Meijer M., Driessen A.J., Rassow J., Pfanner N., Wagner R. A presequence- and voltage-sensitive channel of the mitochondrial preprotein translocase formed by Tim23. Nat. Struct. Biol. (2001);8:1074–1082. doi: 10.1038/nsb726. [DOI] [PubMed] [Google Scholar]
- 21.van Loon A.P., Schatz G. Transport of proteins to the mitochondrial intermembrane space:the ‘sorting’ domain of the cytochrome c1 presequence is a stop-transfer sequence specific for the mitochondrial inner membrane. EMBO J. (1987);6:2441–2448. doi: 10.1002/j.1460-2075.1987.tb02523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koehler C.M., Jarosch E., Tokatlidis K., Schmid K., Schweyen R.J., Schatz G. Import of mitochondrial carriers mediated by essential proteins of the intermembrane space. Science. (1998);279:369–373. doi: 10.1126/science.279.5349.369. [DOI] [PubMed] [Google Scholar]
- 23.Sirrenberg C., Endres M., Folsch H., Stuart R.A., Neupert W., Brunner M. Carrier protein import into mitochondria mediated by the intermembrane proteins Tim10/Mrs11 and Tim12/Mrs5. Nature. (1998);391:912–915. doi: 10.1038/36136. [DOI] [PubMed] [Google Scholar]
- 24.Baumann F., Neupert W., Herrmann J.M. Insertion of bitopic membrane proteins into the inner membrane of mitochondria involves an export step from the matrix. J. Biol. Chem. (2002);277:21405–21413. doi: 10.1074/jbc.M201670200. [DOI] [PubMed] [Google Scholar]
- 25.Vo T.D., Greenberg H.J., Palsson B.O. Reconstruction and functional characterization of the human mitochondrial metabolic network based on proteomic and biochemical data. J. Biol. Chem. (2004);279:39532–39540. doi: 10.1074/jbc.M403782200. [DOI] [PubMed] [Google Scholar]
- 26.Colombini M., Yeung C.L., Tung J., König T. The mitochondrial outer membrane channel, VDAC, is regulated by a synthetic polyanion. Biochim. Biophys. Acta. (1987);905:279–286. doi: 10.1016/0005-2736(87)90456-1. [DOI] [PubMed] [Google Scholar]
- 27.Mannella C.A., Forte M., Colombini M. Toward the molecular structure of the mitochondrial channel, VDAC. J. Bioenerg. Biomembr. (1992);24:7–19. doi: 10.1007/BF00769525. [DOI] [PubMed] [Google Scholar]
- 28.Shoshan-Barmatz V., Ben-Hail D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion. (2012);12:24–34. doi: 10.1016/j.mito.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 29.Brierley G.P., Baysal K., Jung D.W. Cation transport systems in mitochondria: Na+ and K+ uniports and exchangers. J. Bioenerg. Biomembr. (1994);26:519–526. doi: 10.1007/BF00762736. [DOI] [PubMed] [Google Scholar]
- 30.O’Rourke B. Pathophysiological and protective roles of mitochondrial ion channels. J. Physiol. (2000);529:23–36. doi: 10.1111/j.1469-7793.2000.00023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garlid K.D., Paucek P. Mitochondrial potassium transport: the K(+) cycle. Biochim. Biophys. Acta. (2003);1606:23–41. doi: 10.1016/S0005-2728(03)00108-7. [DOI] [PubMed] [Google Scholar]
- 32.Tang X.D., Santarelli L.C., Heinemann S.H., Hoshi T. Metabolic regulation of potassium channels. Annu. Rev. Physiol. (2004);66:131–159. doi: 10.1146/annurev.physiol.66.041002.142720. [DOI] [PubMed] [Google Scholar]
- 33.Kunji E.R. The role and structure of mitochondrial carriers. FEBS Lett. (2004);564:239–244. doi: 10.1016/S0014-5793(04)00242-X. [DOI] [PubMed] [Google Scholar]
- 34.Smith A.C., Robinson A.J. A metabolic model of the mitochondrion and its use in modelling diseases of the tricarboxylic acid cycle. BMC Syst. Biol. (2011);5:102. doi: 10.1186/1752-0509-5-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Divakaruni A.S., Murphy A.N. Cell biology. A mitochondrial mystery, solved. Science. (2012);337:41–43. doi: 10.1126/science.1225601. [DOI] [PubMed] [Google Scholar]
- 36.Fernie A.R., Carrari F., Sweetlove L.J. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. (2004);7:254–261. doi: 10.1016/j.pbi.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 37.Lichtor T., Dohrmann G.J. Respiratory patterns in human brain tumors. Neurosurgery. (1986);19:896–899. doi: 10.1227/00006123-198612000-00002. [DOI] [PubMed] [Google Scholar]
- 38.Verrier F., Mignotte B., Jan G., Brenner C. Study of PTPC composition during apoptosis for identification of viral protein target. Ann. N.Y. Acad. Sci. (2003);1010:126–142. doi: 10.1196/annals.1299.022. [DOI] [PubMed] [Google Scholar]
- 39.Tsujimoto Y. Cell death regulation by the Bcl-2 protein family in the mitochondria. J. Cell. Physiol. (2003);195:158–167. doi: 10.1002/jcp.10254. [DOI] [PubMed] [Google Scholar]
- 40.Donovan M., Cotter T.G. Control of mitochondrial integrity by Bcl-2 family members and caspase-independent cell death. Biochim. Biophys. Acta. (2004);1644:133–147. doi: 10.1016/j.bbamcr.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 41.Thornberry N.A., Lazebnik Y. Caspases: enemies within. Science. (1998);281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 42.Danial N.N., Korsmeyer S.J. Cell death: critical control points. Cell. (2004);116:205–219. doi: 10.1016/S0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 43.Debatin K.M., Krammer P.H. Death receptors in chemotherapy and cancer. Oncogene. (2004);23:2950–2966. doi: 10.1038/sj.onc.1207558. [DOI] [PubMed] [Google Scholar]
- 44.Hancock J.T., Desikan R., Neill S.J. Does the redox status of cytochrome C act as a fail-safe mechanism in the regulation of programmed cell death? Free Radical Biol. Med. (2001);31:697–703. doi: 10.1016/S0891-5849(01)00646-3. [DOI] [PubMed] [Google Scholar]
- 45.Yu S.W., Wang H., Poitras M.F., Coombs C., Bowers W.J., Federoff H.J., Poirier G.G., Dawson T.M., Dawson V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. (2002);297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 46.Li L.Y., Luo X., Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. (2001);412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 47.Hegde R., Srinivasula S.M., Zhang Z., Wassell R., Mukattash R., Cilenti L., DuBois G., Lazebnik Y., Zervos A.S., Fernandes-Alnemri T., Alnemri E.S. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. (2002);277:432–438. doi: 10.1074/jbc.M109721200. [DOI] [PubMed] [Google Scholar]
- 48.Vyssokikh M.Y., Brdiczka D. The function of complexes between the outer mitochondrial membrane pore (VDAC) and the adenine nucleotide translocase in regulation of energy metabolism and apoptosis. Acta Biochim. Pol. (2003);50:389–404. [PubMed] [Google Scholar]
- 49.Tarze A., Deniaud A., Le Bras M., Maillier E., Molle D., Larochette N., Zamzami N., Jan G., Kroemer G., Brenner C. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene. (2007);26:2606–2620. doi: 10.1038/sj.onc.1210074. [DOI] [PubMed] [Google Scholar]
- 50.Taylor S.W., Fahy E., Zhang B., Glenn G.M., Warnock D.E., Wiley S., Murphy A.N., Gaucher S.P., Capaldi R.A., Gibson B.W., Ghosh S.S. Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. (2003);21:281–286. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- 51.Cadenas E., Davies K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radical Biol. Med. (2000);29:222–230. doi: 10.1016/S0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 52.Gerschman R., Gilbert D.L., Nye S.W., Dwyer P., Fenn W.O. Oxygen poisoning and x-irradiation: a mechanism in common. Science. (1954);119:623–626. doi: 10.1126/science.119.3097.623. [DOI] [PubMed] [Google Scholar]
- 53.McCord J.M., Fridovich I. Superoxide Dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. (1969);244:6049–6055. [PubMed] [Google Scholar]
- 54.Halliwell B., Gutteridge J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. (1984);219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gutteridge J.M., Halliwell B. Free radicals and antioxidants in the year 2000. A historical look to the future. Ann. N.Y. Acad. Sci. (2000);899:136–147. doi: 10.1111/j.1749-6632.2000.tb06182.x. [DOI] [PubMed] [Google Scholar]
- 56.Fee J.A. Is superoxide important in oxygen poisoning? Trends Biochem. Sci. (1982);7:84–86. doi: 10.1016/0968-0004(82)90151-7. [DOI] [Google Scholar]
- 57.Picard V., Epsztejn S., Santambrogio P., Cabantchik Z.I., Beaumont C. Role of Ferritin in the Control of the Labile Iron Pool in Murine Erythroleukemia Cells. J. Biol. Chem. (1998);273:15382–15386. doi: 10.1074/jbc.273.25.15382. [DOI] [PubMed] [Google Scholar]
- 58.Tangerås A., Flatmark T., Bäckström D., Ehrenberg A. Mitochondrial iron not bound in heme and iron-sulfur centers. Estimation, compartmentation and redox state. Biochim. Biophys. Acta. (1980);589:162–175. doi: 10.1016/0005-2728(80)90035-3. [DOI] [PubMed] [Google Scholar]
- 59.Keyer K., Imlay J.A. Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. U.S.A. (1996);93:13635–13640. doi: 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mikkelsen R.B., Wardman P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene. (2003);22:5734–5754. doi: 10.1038/sj.onc.1206663. [DOI] [PubMed] [Google Scholar]
- 61.Fridovich I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. (1995);64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- 62.Rowley D.A., Halliwell B. Superoxide-dependent formation of hydroxyl radicals from NADH and NADPH in the presence of iron salts. FEBS Lett. (1982);142:39–41. doi: 10.1016/0014-5793(82)80214-7. [DOI] [PubMed] [Google Scholar]
- 63.Rowley D.A., Halliwell B. Superoxide-dependent formation of hydroxyl radicals in the presence of thiol compounds. FEBS Lett. (1982);138:33–36. doi: 10.1016/0014-5793(82)80388-8. [DOI] [PubMed] [Google Scholar]
- 64.Nauser T., Koppenol W.H. The Rate Constant of the Reaction of Superoxide with Nitrogen Monoxide: Approaching the Diffusion Limit. J. Phys. Chem. A. (2002);106:4084–4086. doi: 10.1021/jp025518z. [DOI] [Google Scholar]
- 65.Kirsch M., de Groot H. Formation of peroxynitrite from reaction of nitroxyl anion with molecular oxygen. J. Biol. Chem. (2002);277:13379–13388. doi: 10.1074/jbc.M108079200. [DOI] [PubMed] [Google Scholar]
- 66.Beckman J.S., Koppenol W.H. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol. (1996);271:C1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 67.Halliwell B. Superoxide, iron, vascular endothelium and reperfusion injury. Free Radical Res. Commun. (1989);5:315–318. doi: 10.3109/10715768909073413. [DOI] [PubMed] [Google Scholar]
- 68.Denicola A., Souza J.M., Radi R. Diffusion of peroxynitrite across erythrocyte membranes. Proc. Natl. Acad. Sci. U.S.A. (1998);95:3566–3571. doi: 10.1073/pnas.95.7.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alderton W.K., Cooper C.E., Knowles R.G. Nitric oxide synthases: structure, function and inhibition. Biochem. J. (2001);357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tay Y.M., Lim K.S., Sheu F.S., Jenner A., Whiteman M., Wong K.P., Halliwell B. Do mitochondria make nitric oxide? no? Free Radical Res. (2004);38:591–599. doi: 10.1080/10715760410001694008. [DOI] [PubMed] [Google Scholar]
- 71.Lacza Z., Horn T.F., Snipes J.A., Zhang J., Roychowdhury S., Horváth E.M., Figueroa J.P., Kollai M., Szabó C., Busija D.W. Lack of mitochondrial nitric oxide production in the mouse brain. J. Neurochem. (2004);90:942–951. doi: 10.1111/j.1471-4159.2004.02553.x. [DOI] [PubMed] [Google Scholar]
- 72.Zanella B., Giordano E., Muscari C., Zini M., Guarnieri C. Nitric oxide synthase activity in rat cardiac mitochondria. Basic Res. Cardiol. (2004);99:159–164. doi: 10.1007/s00395-003-0454-3. [DOI] [PubMed] [Google Scholar]
- 73.Kanai A., Epperly M., Pearce L., Birder L., Zeidel M., Meyers S., Greenberger J., de Groat W., Apodaca G., Peterson J. Differing roles of mitochondrial nitric oxide synthase in cardiomyocytes and urothelial cells. Am. J. Physiol. Heart Circ. Physiol. (2004);286:H13–21. doi: 10.1152/ajpheart.00737.2003. [DOI] [PubMed] [Google Scholar]
- 74.Zaobornyj T., Valdez L.B. Heart mitochondrial nitric oxide synthase: a strategic enzyme in the regulation of cellular bioenergetics. Vitam. Horm. (2014);96:29–58. doi: 10.1016/B978-0-12-800254-4.00003-9. [DOI] [PubMed] [Google Scholar]
- 75.Jourd'heuil D., Jourd'heuil F.L., Kutchukian P.S., Musah R.A., Wink D.A., Grisham M.B. Reaction of superoxide and nitric oxide with peroxynitrite. Implications for peroxynitrite-mediated oxidation reactions in vivo. J. Biol. Chem. (2001);276:28799–28805. doi: 10.1074/jbc.M102341200. [DOI] [PubMed] [Google Scholar]
- 76.Radi R., Cassina A., Hodara R., Quijano C., Castro L. Peroxynitrite reactions and formation in mitochondria. Free Radical Biol. Med. (2002);33:1451–1464. doi: 10.1016/S0891-5849(02)01111-5. [DOI] [PubMed] [Google Scholar]
- 77.Stamler J.S., Jaraki O., Osborne J., Simon D.I., Keaney J., Vita J., Singel D., Valeri C.R., Loscalzo J. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc. Natl. Acad. Sci. U.S.A. (1992);89:7674–7677. doi: 10.1073/pnas.89.16.7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jourd'heuil D., Laroux F.S., Miles A.M., Wink D.A., Grisham M.B. Effect of superoxide dismutase on the stability of S-nitrosothiols. Arch. Biochem. Biophys. (1999);361:323–330. doi: 10.1006/abbi.1998.1010. [DOI] [PubMed] [Google Scholar]
- 79.Goldstein S., Czapski G. Direct and indirect oxidations by peroxynitrite. Inorg. Chem. (1995);34:4041–4048. doi: 10.1021/ic00120a006. [DOI] [Google Scholar]
- 80.Merényi G., Lind J. Free radical formation in the peroxynitrous acid (ONOOH)/peroxynitrite (ONOO−) system. Chem. Res. Toxicol. (1998);11:243–246. doi: 10.1021/tx980026s. [DOI] [PubMed] [Google Scholar]
- 81.Augusto O., Bonini M.G., Amanso A.M., Linares E., Santos C.C., De Menezes S.L. Nitrogen dioxide and carbonate radical anion: two emerging radicals in biology. Free Radical Biol. Med. (2002);32:841–859. doi: 10.1016/S0891-5849(02)00786-4. [DOI] [PubMed] [Google Scholar]
- 82.Lymar S.V., Hurst J.K. Rapid reaction between peroxonitrite ion and carbon dioxide: Implications for biological activity. J. Am. Chem. Soc. (1995);117:8867–8868. doi: 10.1021/ja00139a027. [DOI] [Google Scholar]
- 83.Bonini M.G., Radi R., Ferrer-Sueta G., Ferreira A.M., Augusto O. Direct EPR detection of the carbonate radical anion produced from peroxynitrite and carbon dioxide. J. Biol. Chem. (1999);274:10802–10806. doi: 10.1074/jbc.274.16.10802. [DOI] [PubMed] [Google Scholar]
- 84.Ono Y., Lin L., Storey B.T., Taguchi Y., Dodgson S.J., Forster R.E. Continuous measurement of 13C16O2 production from [13C]pyruvate by intact liver mitochondria: effect of HCO3-. Am. J. Physiol. Cell Physiol. (1996);270:C98–106. doi: 10.1152/ajpcell.1996.270.1.C98. [DOI] [PubMed] [Google Scholar]
- 85.Buxton G.V., Elliot A.J. Rate-constant for reaction of hydroxyl radical with bicarbonate ions. Radiat. Phys. Chem. (1986);27:241–243. [Google Scholar]
- 86.Santos C.X., Bonini M.G., Augusto O. Role of the carbonate radical anion in tyrosine nitration and hydroxylation by peroxynitrite. Arch. Biochem. Biophys. (2000);377:146–152. doi: 10.1006/abbi.2000.1751. [DOI] [PubMed] [Google Scholar]
- 87.Shafirovich V., Dourandin A., Huang W., Geacintov N.E. The carbonate radical is a site-selective oxidizing agent of guanine in double-stranded oligonucleotides. J. Biol. Chem. (2001);276:24621–24626. doi: 10.1074/jbc.M101131200. [DOI] [PubMed] [Google Scholar]
- 88.Yermilov V., Yoshie Y., Rubio J., Ohshima H. Effects of carbon dioxide/bicarbonate on induction of DNA single-strand breaks and formation of 8-nitroguanine, 8-oxoguanine and base-propenal mediated by peroxynitrite. FEBS Lett. (1996);399:67–70. doi: 10.1016/S0014-5793(96)01288-4. [DOI] [PubMed] [Google Scholar]
- 89.Nordberg J., Arnér E.S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radical Biol. Med. (2001);31:1287–1312. doi: 10.1016/S0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 90.Schafer F.Q., Buettner G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radical Biol. Med. (2001);30:1191–1212. doi: 10.1016/S0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 91.Kirsch M., De Groot H. NAD(P)H, a directly operating antioxidant? FASEB J. (2001);15:1569–1574. doi: 10.1096/fj.00-0823hyp. [DOI] [PubMed] [Google Scholar]
- 92.Gotoh N., Niki E. Rates of interactions of superoxide with vitamin E, vitamin C and related compounds as measured by chemiluminescence. Biochim. Biophys. Acta. (1992);1115:201–207. doi: 10.1016/0304-4165(92)90054-X. [DOI] [PubMed] [Google Scholar]
- 93.Buettner G.R., Jurkiewicz B.A. Ascorbate free radical as a marker of oxidative stress: an EPR study. Free Radical Biol. Med. (1993);14:49–55. doi: 10.1016/0891-5849(93)90508-R. [DOI] [PubMed] [Google Scholar]
- 94.Slot J.W., Geuze H.J., Freeman B.A., Crapo J.D. Intracellular localization of the copper-zinc and manganese superoxide dismutases in rat liver parenchymal cells. Lab. Invest. (1986);55:363–371. [PubMed] [Google Scholar]
- 95.Okado-Matsumoto A., Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. (2001);276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 96.Muller F.L., Liu Y., Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. (2004);279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 97.Lévêque V.J., Vance C.K., Nick H.S., Silverman D.N. Redox properties of human manganese superoxide dismutase and active-site mutants. Biochemistry. (2001);40:10586–10591. doi: 10.1021/bi010792p. [DOI] [PubMed] [Google Scholar]
- 98.Buettner G.R. The pecking order of free radicals and antioxidants: lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. (1993);300:535–543. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- 99.Thomas S.R., Stocker R. Molecular action of vitamin E in lipoprotein oxidation: Implications for atherosclerosis. Free Radical Biol. Med. (2000);28:1795–1805. doi: 10.1016/S0891-5849(00)00236-7. [DOI] [PubMed] [Google Scholar]
- 100.May J.M. How does ascorbic acid prevent endothelial dysfunction? Free Radical Biol. Med. (2000);28:1421–1429. doi: 10.1016/S0891-5849(00)00269-0. [DOI] [PubMed] [Google Scholar]
- 101.Lang J.K., Gohil K., Packer L. Simultaneous determination of tocopherols, ubiquinols, and ubiquinones in blood, plasma, tissue homogenates, and subcellular fractions. Anal. Biochem. (1986);157:106–116. doi: 10.1016/0003-2697(86)90203-4. [DOI] [PubMed] [Google Scholar]
- 102.Thomas S.M., Gebicki J.M., Dean R.T. Radical initiated alpha-tocopherol depletion and lipid peroxidation in mitochondrial membranes. Biochim. Biophys. Acta. (1989);1002:189–197. doi: 10.1016/0005-2760(89)90286-5. [DOI] [PubMed] [Google Scholar]
- 103.Antunes F., Salvador A., Marinho H.S., Alves R., Pinto R.E. Lipid peroxidation in mitochondrial inner membranes. I. An integrative kinetic model. Free Radical Biol. Med. (1996);21:917–943. doi: 10.1016/S0891-5849(96)00185-2. [DOI] [PubMed] [Google Scholar]
- 104.Vatassery G.T., Smith W.E., Quach H.T., Lai J.C. In vitro oxidation of vitamin E, vitamin C, thiols and cholesterol in rat brain mitochondria incubated with free radicals. Neurochem. Int. (1995);26:527–535. doi: 10.1016/0197-0186(94)00147-M. [DOI] [PubMed] [Google Scholar]
- 105.Crane F.L. Biochemical functions of coenzyme Q10. J. Am. Coll. Nutr. (2001);20:591–598. doi: 10.1080/07315724.2001.10719063. [DOI] [PubMed] [Google Scholar]
- 106.Neuzil J., Witting P.K., Stocker R. Alpha - tocopheryl hydroquinone is an efficient multifunctional inhibitor of radical-initiated oxidation of low density lipoprotein lipids. Proc. Natl. Acad. Sci. U.S.A. (1997);94:7885–7890. doi: 10.1073/pnas.94.15.7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Thomas S.R., Witting P.K., Stocker R. 3-Hydroxyanthranilic acid is an efficient, cell-derived co-antioxidant for alpha -tocopherol, inhibiting human low density lipoprotein and plasma lipid peroxidation. J. Biol. Chem. (1996);271:32714–32721. doi: 10.1074/jbc.271.51.32714. [DOI] [PubMed] [Google Scholar]
- 108.Lenaz G., Fato R., Di Bernardo S., Jarreta D., Costa A., Genova M.L., Parenti Castelli G. Localization and mobility of coenzyme Q in lipid bilayers and membranes. BioFactors. (1999);9:87–93. doi: 10.1002/biof.5520090202. [DOI] [PubMed] [Google Scholar]
- 109.May J.M., Cobb C.E., Mendiratta S., Hill K.E., Burk R.F. Reduction of the ascorbyl free radical to ascorbate by thioredoxin reductase. J. Biol. Chem. (1998);273:23039–23045. doi: 10.1074/jbc.273.36.23039. [DOI] [PubMed] [Google Scholar]
- 110.Packer L., Witt E.H., Tritschler H.J. Alphalipoic acid as a biological antioxidant. Free Radical Biol. Med. (1995);19:227–250. doi: 10.1016/0891-5849(95)00017-R. [DOI] [PubMed] [Google Scholar]
- 111.May J.M., Mendiratta S., Hill K.E., Burk R.F. Reduction of dehydroascorbate to ascorbate by the selenoenzyme thioredoxin reductase. J. Biol. Chem. (1997);272:22607–22610. doi: 10.1074/jbc.272.36.22607. [DOI] [PubMed] [Google Scholar]
- 112.Kletzien R.F., Harris P.K., Foellmi L.A. Glucose-6-phosphate dehydrogenase: a “housekeeping” enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress. FASEB J. (1994);8:174–181. doi: 10.1096/fasebj.8.2.8119488. [DOI] [PubMed] [Google Scholar]
- 113.Jacob C., Giles G.I., Giles N.M., Sies H. Sulfur and selenium: the role of oxidation state in protein structure and function. Angew. Chem. Int. Ed. Engl. (2003);42:4742–4758. doi: 10.1002/anie.200300573. [DOI] [PubMed] [Google Scholar]
- 114.Torchinsky Y.M. Sulfur in proteins. Pergamon Press; New York: (1981). pp. 1–294. [Google Scholar]
- 115.Wood Z.A., Schröder E., Robin Harris J., Poole L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. (2003);28:32–40. doi: 10.1016/S0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 116.Chelikani P., Fita I., Loewen P.C. Diversity of structures and properties among catalases. Cell. Mol. Life Sci. (2004);61:192–208. doi: 10.1007/s00018-003-3206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hofmann B., Hecht H.J., Flohé L. Peroxiredoxins. Biol. Chem. (2002);383:347–364. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- 118.Nobumoto M., Yamada M., Song S., Inouye S., Nakazawa A. Mechanism of mitochondrial import of adenylate kinase isozymes. J. Biochem. (1998);123:128–135. doi: 10.1093/oxfordjournals.jbchem.a021899. [DOI] [PubMed] [Google Scholar]
- 119.Bai J., Cederbaum A.I. Mitochondrial catalase and oxidative injury. Biol. Signals Recept. (2001);10:189–199. doi: 10.1159/000046887. [DOI] [PubMed] [Google Scholar]
- 120.Hirota K., Nakamura H., Masutani H., Yodoi J. Thioredoxin superfamily and thioredoxin-inducing agents. Ann. N.Y. Acad. Sci. (2002);957:189–199. doi: 10.1111/j.1749-6632.2002.tb02916.x. [DOI] [PubMed] [Google Scholar]
- 121.Imai H., Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radical Biol. Med. (2003);34:145–169. doi: 10.1016/S0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- 122.Rhee S.G., Woo H.A., Kil I.S., Bae S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. (2012);287:4403–4410. doi: 10.1074/jbc.R111.283432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Powis G., Montfort W.R. Properties and biological activities of thioredoxins. Annu. Rev. Biophys. Biomol. Struct. (2001);30:421–455. doi: 10.1146/annurev.biophys.30.1.421. [DOI] [PubMed] [Google Scholar]
- 124.Nishiyama A., Masutani H., Nakamura H., Nishinaka Y., Yodoi J. Redox regulation by thioredoxin and thioredoxin-binding proteins. IUBMB Life. (2001);52:29–33. doi: 10.1080/15216540252774739. [DOI] [PubMed] [Google Scholar]
- 125.Smith C.V., Jones D.P., Guenthner T.M., Lash L.H., Lauterburg B.H. Compartmentation of glutathione: implications for the study of toxicity and disease. Toxicol. Appl. Pharmacol. (1996);140:1–12. doi: 10.1006/taap.1996.0191. [DOI] [PubMed] [Google Scholar]
- 126.Sies H. Nicotinamide nucleotide compartmentation in Metabolic compartmentation (Sies, H. Ed.). Academic Press; New York: (1982). pp. 205–231. [Google Scholar]
- 127.Reich J.G., Sel'kov E.E. Pools and pathways of energy metabolism in Energy metabolism of the cell: A theoretical trestise Ed.). Academic Press; New York: (1981). pp. 108–198. [Google Scholar]
- 128.Di Lisa F., Menabò R., Canton M., Barile M., Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. (2001);276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 129.Di Lisa F., Ziegler M. Pathophysiological relevance of mitochondria in NAD+ metabolism. FEBS Lett. (2001);492:4–8. doi: 10.1016/S0014-5793(01)02198-6. [DOI] [PubMed] [Google Scholar]
- 130.Berger F., Ramírez-Hernández M.H., Ziegler M. The new life of a centenarian: signalling functions of NAD(P). Trends Biochem. Sci. (2004);29:111–118. doi: 10.1016/j.tibs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 131.Zhang X., Kurnasov O.V., Karthikeyan S., Grishin N.V., Osterman A.L., Zhang H. Structural Characterization of a Human Cytosolic NMN/NaMN Adenylyltransferase and Implication in Human NAD Biosynthesis. J. Biol. Chem. (2003);278:13503–13511. doi: 10.1074/jbc.M300073200. [DOI] [PubMed] [Google Scholar]
- 132.Iwahashi Y., Hitoshio A., Tajima N., Nakamura T. Characterization of NADH kinase from saccharomyces cerevisiae. J. Biochem. (1989);105:588–593. doi: 10.1093/oxfordjournals.jbchem.a122709. [DOI] [PubMed] [Google Scholar]
- 133.Ohashi K., Kawai S., Murata K. Identification and characterization of a human mitochondrial NAD kinase. Nat. Commun. (2012);3:1248. doi: 10.1038/ncomms2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Toledano M.B., Delaunay-Moisan A., Outten C.E., Igbaria A. Functions and cellular compartmentation of the thioredoxin and glutathione pathways in yeast. Antioxid. Redox Signaling. (2013);18:1699–1711. doi: 10.1089/ars.2012.5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chen Z., Lash L.H. Evidence for mitochondrial uptake of glutathione by dicarboxylate and 2-oxoglutarate carriers. J. Pharmacol. Exp. Ther. (1998);285:608–618. [PubMed] [Google Scholar]
- 136.García-Ruiz C., Morales A., Colell A., Rodés J., Yi J.R., Kaplowitz N., Fernández-Checa J.C. Evidence that the rat hepatic mitochondrial carrier is distinct from the sinusoidal and canalicular transporters for reduced glutathione. J. Biol. Chem. (1995);270:15946–15949. doi: 10.1074/jbc.270.27.15946. [DOI] [PubMed] [Google Scholar]
- 137.Fernández-Checa J.C., Kaplowitz N., García-Ruiz C., Colell A. Mitochondrial glutathione: importance and transport. Semin. Liver Dis. (1998);18:389–401. doi: 10.1055/s-2007-1007172. [DOI] [PubMed] [Google Scholar]
- 138.Kembro J.M., Aon M.A., Winslow R.L., O’Rourke B., Cortassa S. Integrating mitochondrial energetics, redox and ROS metabolic networks: a two-compartment model. Biophys. J. (2013);104:332–343. doi: 10.1016/j.bpj.2012.11.3808. [DOI] [PMC free article] [PubMed] [Google Scholar]