

Abstract
Mitochondrial integrity is critical for maintaining proper cellular functions. A key aspect of regulating mitochondrial homeostasis is removing damaged mitochondria through autophagy, a process called mitophagy. Autophagy dysfunction in various disease states can inactivate mitophagy and cause cell death, and defects in mitophagy are becoming increasingly recognized in a wide range of diseases from liver injuries to neurodegenerative diseases. Here we highlight our current knowledge on the mechanisms of mitophagy, and discuss how alterations in mitophagy contribute to disease pathogenesis. We also discuss mitochondrial dynamics and potential interactions between mitochondrial fusion, fission and mitophagy.
Keywords: Autophagy, Mitochondria, Mitophagy, Liver, Brain
INTRODUCTION
Macroauotphagy (referred to as autophagy hereafter) is a major catabolic pathway to remove surplus or unnecessary cytoplasmic contents and dysfunctional organelles, and defects in this pathway negatively impact the cellular environment by accumulating potentially toxic moieties. Autophagy is essential for sustaining cells during various physiological and pathological stresses, including nutrient deprivation, oxidative stress, ischemia/reperfusion (I/R) and pathogen invasion. However, depletion of essential cellular components due to excessive autophagy could also trigger autophagic cell death. In cancer, the role of autophagy is more complicated, as autophagy can either prevent or promote cancer based on the developmental stage of the disease. During initial stages of tumorigenesis when cells undergo hyperproliferation, activating autophagy inhibits cell division and suppresses tumor growth (1). However, in more established tumors at later stages, cancer cells depend on autophagy to supply essential nutrients and thus inhibiting autophagy has a therapeutic potential to suppress metastasis and malignancy of cancer (2,3).
How does autophagy impact mitochondrial health? Mitochondria are dynamic organelles that generate ATP through a proton motive force across inner membranes and are responsible for supplying the majority of energy needed by cells. Mitochondria are also the central regulators for apoptosis and the major source for the production of reactive oxygen species (ROS). Environmental toxins as well as chemotherapeutic drugs can damage mitochondria. Mitochondrial dysfunction is also a key causative factor inducing cell death after I/R, aging and many neurodegenerative conditions such as Alzheimer’s and Parkinson’s disease. Therefore, it is important to timely eliminate abnormal, aged and dysfunctional mitochondria to sustain cell viability. The process of eliminating mitochondria through a selective autophagy is termed mitophagy, which not only facilitates turnover of normal mitochondria but also removes unnecessary or damaged mitochondria to prevent the accumulation of dysfunctional mitochondria and potentially cytotoxic mitochondrial byproducts in the cytosol. De Duve first observed this process of mitochondrial sequestration into lysosomes in hepatocytes, after stimulating autophagy with glucagon (4). A strong positive link between autophagic capacity and mitochondrial integrity was observed in autophagy-deficient transgenic mice that exhibit swelling and loss of structural integrity in mitochondria (5).
To date, mitophagy is the only known mechanism to remove aged or damaged mitochondria. Depending on the tissue, normal mitochondria are turned over with a half-life range of 9~25 days, with livers having a relatively faster turnover than brain, heart or kidney (6). Active mitophagy in livers may be attributable to the high content of mitochondria in this tissue that reflects the high energy demand for operating multiple hepatic functions. For the same reason, hepatocytes are frequently used to characterize the features of mitophagy.
MITOPHAGY MECHANISMS
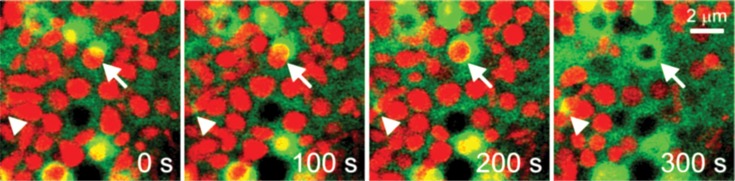
Although mitophagy is evolutionary conserved in all types of cells, the mechanisms triggering the onset of mitophagy differ depending on the type of stresses and cells. Using a fluorescent marker for the autophagosome [GFP-labeled microtubule-associated protein-1 light chain-3 (GFP-LC3)] and a mitochondrial fluorophore, activation of mitophagy can be visualized upon nutrient starvation of hepatocytes; whereby small pre-autophagic structures near the mitochondria grow into a cup-shaped phagophore that engulfs individual mitochondrion within 5 min after nutrient depletion (7,8) (Fig. 1). Mitochondria maintain their membrane potential during sequestration, as evidenced by red fluorescing tetramethylrhodamine methylester (TMRM), but become depolarized after either onset of the mitochondrial permeability transition (MPT) or acidification of luminal pH in the mitophagosome. Sequestration of polarized mitochondria also occurs to ischemic hepatocytes after reperfusion (9). Additional features of this type of mitophagy include the requirement for phosphatidylinositol-3-kinase (PI3K), and coordination with mitochondrial fission (7).
Fig. 1. Visualization of mitophagy in primary mouse hepatocytes. Time-lapse images of confocal microscopy with GFP-LC3 and TMRM. Cells were maintained in amino acid- and serum-free Krebs-Ringer-HEPES (pH 7.4) solution for 2 hrs to stimulate autophagy. Arrow indicates a progress of mitophagy wherein polarized mitochondrion (red) is surrounding by an elongating autophagic membrane (green). Note a depolarized mitochondrion in the lumen of autophagosome at 300 sec. Arrowhead displays an initiation of new mitophagy.
In contrast to PI3K-dependent mitophagy, mitochondrial depolarization can induce a different type of mitophagy in a PI3K-independent manner (Fig. 2). Global mitochondrial injury from widespread depolarization induces a strong autophagy response (10,11). For instance, photodamage to hepatocytes has demonstrated a sequential chain of mitochondrial depolarization and subsequent mitophagy (7). The specific proteins that have known roles in mitophagy are summarized in Table 1. In yeast, ATG32 is a mitophagy receptor that specifically targets mitochondria for autophagy and has no impact on macroautophagy (12). For mammalian cells, FUN14 domain-containing protein-1 (FUNDC1) was recently identified as a mitochondrial autophagy receptor, whose exogenous expression induces mitophagy, in the absence of any mitochondrial damage or general autophagystimulating conditions (13). Activating mitophagy through elevated FUNDC1 expression has been reported to reduce the total amount of both outer and inner mitochondrial membrane proteins, without reducing other organelle markers. Following mitochondrial damage by hypoxia or mitochondrial uncoupler treatment, FUNDC1 is phosphorylated by ULK1, and promotes mitophagy (14).
Fig. 2. Scheme of mitochondrial dynamics and mitophagy. Under the conditions of nutrient depletion and I/R in the liver, a polarized mitochondrion is initially recognized by a U-shaped phagophore and later sequestered completely by an autophagosome. The mitochondrion entrapped in the autophagosome loses its membrane potential, presumably due to onset of the MPT or luminal acidification after fusion with a lysosome. When mitochondria undergo fission, a mitochondrion is divided into two daughter mitochondria with disparate membrane potential. Whereas the mitochondrion with the higher membrane potential returns to the cycle of fusion, the other with the lower or depolarized membrane potential is engulfed into an autophagosome and subsequently cleared by an autolysosome.
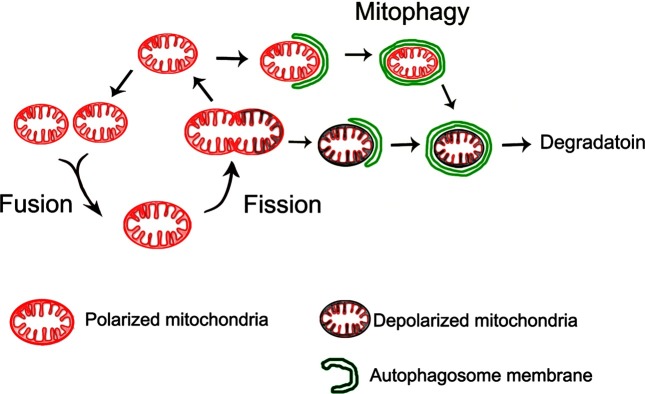
Table 1. Summary of mitophagy mechanisms.
| Protein | Function | Reference |
|---|---|---|
|
| ||
| ATG32 | • Yeast mitochondrial protein for recognition as autophagy cargo. | (12) |
| • Interacts with Atg8 and Atg11. | ||
| FUNDC1 | • Regulates Ulk1 recruitment to damaged mitochondria. | (13,14) |
| • Interaction with LC3 is regulated by phosphorylation. | ||
| PINK1 | • Normally undergoes rapid processing/degradation on mitochondria | (11,16,17,22) |
| • Full length protein recruits Parkin | ||
| Parkin | • E3 ubiquitin ligase | (11,16-18,22) |
| • Endogenous Parkin ubiquitinates MFN1 and MFN2, overepxressed Parkin ubiquitinates many other outer membrane proteins | ||
| BNIP3/NIX | • Mediates mitophagy during red blood cell maturation | (15,20,21) |
| • Activates mitophagy induced by hypoxia or mitochondria uncoupling. | ||
| • Homodimerization required for mitophagy | ||
| • Interacts with LC3 and OPA1 | ||
In mammalian cells, mitophagy of depolarized mitochondria has also been shown to involve BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3)/BNIP3L (also known as NIX), and PTEN-induced putative kinase protein 1 (PINK1)/Parkin (13,15). Among these two systems, PINK1 and Parkin have been the most actively researched. PINK1 and Parkin are proteins in which mutations are linked to familial forms of Parkinson’s disease. Pink1 recruits Parkin to the outer membrane of dysfunctional mitochondria and subsequent ubiquitination of proteins on the mitochondrial outer membrane induces mitophagy (11,16). The mitochondrial accumulation of Parkin is voltage-dependent, and does not depend on changes in pH or ATP levels (17), and suggests that Parkin may act as a sensor for mitochondrial integrity and trigger mitophagy upon dysfunction. Parkin-mediated ubiquitination is opposed by the deubiquitinase USP30, whereby reducing USP30 activity enhances mitophagy (18). Parkin also participates in the maturation of mitophago-somes through the endosomallysosomal protein CLEC16A, which interacts with the E3 ligase NRDP1 to regulate Parkin levels. Reduced CLEC16A expression elevates Parkin levels and increases mitophagosomes, however, the mitophagosomes fail to undergo fusion with lysosomes, which results in the net increase of abnormal and dysfunctional mitochondria. In hepatitis C virus, its core protein was found to bind directly to Parkin, and suppress its translocation to mitochondria, thereby inhibiting mitophagy (19).
BNIP3 and BNIP3L are proteins with homology to BCL2 in the BH3 domain, which induce both cell death and autophagy. In erythrocytes, the elimination of mitochondria during maturation requires BNIP3L (15). BNIP3L is also involved in mitochondrial depolarization-induced mitophagy. Upon treatment with a mitochondrial uncoupler [carbonyl cyanide m-chlorophenylhydrazone (CCCP)] to induce a short circuit between electron transfer and ATP generation, BNIP3L is required to activate mitophagy through mitochondrial depolarization and ROS generation (20). BNIP3 also regulates mitophagy in response to hypoxia. In cardiac myocytes, I/R leads to BNIP3-mediated upregulation of autophagy. This serves as a protective response to remove damaged mitochondria after hypoxia and reoxygenation (21).
MITOPHAGY IN LIVER DISEASE
Regarding systemic metabolism, the hepatocytes of livers serve a major role in regulating many integral metabolic functions. Hepatocytes also contain numerous mitochondria to meet the high energy demands of these metabolic activities. Thus, maintaining healthy mitochondria and, at the same time, clearing unhealthy mitochondria through efficient autophagy are critical for supporting liver functions. Indeed, in livers of autophagy-deficient mice, mitochondrial damage is evident such as swelling and ROS formation (5). Impaired autophagy and mitophagy can affect multiple aspects of hepatic functions and underlie many liver injuries including steatosis, hepatocellular carcinoma (HCC), and liver injuries caused by toxins, drugs and I/R (22). In HCC, autophagy may prevent or promote cancer process, depending on the disease stage itself as in other cancers. Autophagy initially functions as a tumor suppressor. On the contrary, in established tumors, autophagy fuels tumor survival by supplying its metabolic demands. Thus, the loss of autophagy can increase or decrease hepatic tumors at the early or late stage of HCC, respectively (5,23). In fatty liver disease, reduced autophagy can be causative in the accumulation of lipids, as autophagy degrades intracellular lipids through lipophagy (24). Moreover, the accumulation of excess lipids further depresses autophagy function by affecting autophagy protein expression and autophagy maturation (25). This perpetual worsening of autophagy function and lipid accumulation is implicated in the progression from simple steatosis to nonalcoholic steatohepatitis (NASH). In toxin-induced liver injuries including alcohol and acetaminophen (APAP), the disease process involves the metabolism of toxins that increase ROS production that damages mitochondria. In experimental models of alcohol and APAP-induced liver injury, enhancing autophagy and mitophagy has been shown to counteract insufficient autophagy and to improve liver function and mitochondrial bioenergetics (22,26,27).
ISCHEMIA/REPERFUSION (I/R) INJURY
Cellular respiration requires a continuous oxygen supply. For livers, hypoxic or anoxic stress can occur during hepatectomy, liver preservation for transplantation, cardiac failure and, hemorrhagic shock. Similar to the liver, organs such as the brain and the heart have a high metabolic demand and are also highly susceptible to ischemia-induced cellular damage. The pathogenic events during ischemia include loss of oxygen or anoxia, decreased ATP production, and acidosis. Acidosis results from the combination of high-energy phosphate hydrolysis, lactate buildup from anaerobic respiration, and proton release from acidic compartments. Interestingly, acidosis itself is not the culprit for cellular damage, as it may confer cytoprotection against ischemic necrosis (28,29). Instead, cellular damage paradoxically occurs upon reperfusion, when the oxygen supply is restored and cellular pH is recovered to normal.
Multiple events during reperfusion are likely to contribute to cell death, including ROS formation and oxidative damage to mitochondrial components, calcium overloading and activation of cytosolic calpains and other injurious proteases, impaired autophagy and loss of mitochondrial energy production (30). These events are likely to converge on the MPT. When ischemic cells undergo reperfusion, high conductance permeability transition pores in the mitochondrial inner membranes are opened, which triggers the onset of the MPT. These pores normally restrict the passage to particles less than 1,500 Da, and proper function of these inner membrane pores are critical to maintain mitochondrial polarization, oxidative phosphorylation cycle, and ATP production. Opening of the inner membrane pores leads to non-specific diffusion of proteins across the inner membrane, leading to mitochondrial swelling and subsequent loss of mitochondrial membrane integrity. The MPT also allows pro-apoptotic proteins such as cytochrome C, which is normally confined in the mitochondrial intermembrane space, to release into the cytosol and to activate caspases. ATP depletion that follows from uncoupling of oxidative phosphorylation, ultimately leads to either necrotic or apoptotic cell death, depending on glycolytic ATP availability (31,32).
During ischemia, anoxia depletes the liver of ATP. Since the execution of autophagy needs a large amount of ATP, the formation of autophagic vesicles is halted in ischemic cells. However, at the stage of reperfusion, the capacity to carry out autophagy and mitophagy are critical for cell survival. Upon restoration of oxygen and nutrients, a brief period of mitochondrial polarization is followed by the MPT and consequent depolarization of mitochondrial membrane potential. Importantly, key autophagy proteins, including ATG 7, BECN1, and ATG12-5, decrease over the periods of prolonged ischemia and further reduce at the early stage of reperfusion when the MPT has not yet to occur. Depletion of these autophagy proteins is partially attributable to calpain-mediated degradation (33). Loss of autophagy-related proteins and subsequent impaired autophagy leads to or potentiates a sequential chain of the MPT onset, widespread mitochondria failure and ultimately cell death. Conversely, activating autophagy either by pharmacological activators or by overexpression of key autophagy proteins is cytoprotective against reperfusion injury and restores the autophagic process (33,34) Recovering autophagy also suppresses the spread of toxic insults to neighboring healthy mitochondria through enhanced mitophagy, as the sequestration of damaged mitochondria prevents the propagation of injury signals that can originate from a small subset of mitochondria.
MITOCHONDRIA AND AUTOPHAGY DEFECTS
In response to persistent proteotoxic stress such as protein misfolding diseases, the autophagic machinery may become activated in attempts to clear aggregate proteins. Under such conditions, activating autophagy, or mitochondrial biogenesis, or both appears to be therapeutic. In Huntington’s disease, the mutant Huntingtin protein is associated with the formation of toxic aggregates, mitochondrial damage and oxidative stress. Interestingly, enhancing mitochondrial biogenesis by activating the peroxisome proliferatoractivated receptor-gamma coactivator 1-alpha (PGC-1α) ameliorates neuronal damage caused by mutant Huntingtin, through activating autophagy-regulating transcription factor EB (TFEB) (35), which assumes to be a master regulator of autophagy and lysosomal genes. Alternatively, diseasecausing proteins may interfere with transcriptional regulators of autophagy to inhibit autophagy activation. In the polyglutamine expansion of the androgen receptor of x-linked spinal and muscular dystrophy, polyglutamineexpanded mutant protein binds directly to TFEB and interferes with its nuclear transactivation (36). Additionally, prolonged autophagy impairment may result in the gradual buildup of damaged mitochondria. An example for this type of secondary mitochondria damage is the antitrypsin disease (ATD), a relatively common liver disease that is caused by a point mutation in the secretory protein alpha 1-antitrypsin. Whereas this protein is normally secreted by hepatocytes, the mutant protein is misfolded, and causes endoplasmic reticulum (ER) retention and proteotoxic stress (37). Using a genetic model for autophagy inhibition (ATG5 knockouts), the critical role of autophagy in the disposal of the disease protein has been demonstrated, as the turnover of mutant antitrypsin is significantly delayed when autophagosome synthesis is genetically impaired (38). The pivotal role of autophagy for antitrypsin clearance is confirmed in the yeast, as the yeast homolog of ATG6 or ATG16 was identified in a mutant library screening, and found that absence of either gene causes a marked delay in anti-trypsin clearance (39). This hypothesis that ineffective or defective autophagy contributes to ATD disease is further supported by studies where activating autophagy by pharmacological agents or liver-directed gene transfer of TFEB promotes degradation of the mutant protein and ameliorates hepatic pathologies (40,41). While the primary defect in this disease is a mutation of secretory proteins, extensive mitochondrial defect and accumulation of mitophagosomes have been reported in patient liver tissues (42). Thus, primary autophagy defects can, in turn, precipitate in mitochondrial damage.
MITOCHONDRIA DYNAMICS, AUTOPHAGY AND MITOPHAGY
Mitochondria are dynamic organelles that continuously undergo fusion and fission. Not only do fusion and fission regulate mitochondrial morphology, but also modulate intermixing mitochondrial contents. Mitochondrial fusion is mediated by three proteins: mitofusin-1 (MFN1) and mitofusin-2 (MFN2) for the outer membranes, and optic atrophy- 1 (OPA1) for the inner membranes (43,44). Among them, MFN2 localizes at the contact site between outer mitochondrial and ER membranes (45). Mitochondrial fusion facilitates intra-mitochondrial repair and exchange of mitochondrial DNA (mtDNA) (46), whereas fission can serve to temporarily isolate defective segments and/or promote their autophagic clearance (47,48). Growing evidence is indicating that mitophagy and mitochondrial dynamics are intimately connected, as fusion and fission maintain a healthy pool while segregating mitochondria that are destined for mitophagy (Fig. 2). In both yeast and mammalian cells, a certain type of mitophagy is preceded by mitochondrial fission, which divides elongated mitochondria into pieces of manageable size for encapsulation. Furthermore, the reduction of mitochondrial volume by fission can also assist mitochondrial quality control since smaller mitochondria can be readily cleared by mitophagy.
Mitochondria may also be a source of autophagic membranes. In starved mammalian cells, it was shown that autophagosomes are formed in the vicinity of mitochondria (49,50), especially at mitochondria-associated ER membranes, and that mitochondria supply phospholipids to growing autophagosomes via a transient interaction (50). Intriguingly, autophagosome formation is impaired by depletion of either MFN2 or phosphofurin acidic cluster sorting protein-2 (PACS-2) (49), later of which is a cytosolic protein implicated in ER–mitochondrion communication.
Evidence has been accumulating that either MFN1 or MFN2 or both can directly regulate mitophagy. Both MFN1 and MFN2 are targets for ubiquitination by Parkin (16), thus the ubiquitination status of these proteins likely acts as receptors for mitophagy. In cardiac myocytes, genetic ablation of MFN2 prevents depolarization-induced translocation of Parkin to the mitochondria, leading to suppression of mitophagy (51). In addition, mutations in MFN2 have been reported in patients of Charcot-Marie-Tooth neuropathy type 2A (CMT2A) (52,53), which causes degeneration of sensory neurons. Interestingly, both dopaminergic neurons in Parkinson’s disease and sensory neurons in CMT2A lack myelination, which makes electrical impulse propagation energetically more demanding. This suggests that cells with a higher energy demand are likely to be more reliant on efficient mitochondrial dynamics and mitophagy. Supporting this hypothesis, reduced MFN2 expression is observed in skeletal muscle of type 2 diabetes (54,55). Patients with this disease demonstrate characteristic muscle fatigue and weakness. In the liver, loss of MFN2 disrupts metabolic functions and displays characteristics of enhanced hepatic glucose production by gluconeogenesis and increased expression of gluconeogenesis (56). We have also observed that silencing of MFN2 abolishes both basal autophagy and sirtuin 1-dependent cytoprotection against I/R in the liver (57).
Besides the role in initiating mitophagy, the levels of PINK1 or Parkin regulate mitochondrial morphology, implying that PINK1/Parkin could regulate both mitochondrial dynamics and mitophagy. In hippocampal neurons, overexpression of either PINK1 or Parkin causes an overall smaller size of mitochondria due to prevailing fission, whereas inactivation of PINK1 produces elongated mitochondria or more incidences of mitochondrial fusion (58). Similarly, flies expressing PINK1 or Parkin mutants display the loss of mitochondrial integrity, which is derived directly from reduced mitochondrial fission (59). Thus mutations in PINK1/Parkin may impact autophagy or mitophagy through alterations in mitochondrial dynamics.
CLINICAL IMPLICATIONS OF MITOPHAGY
I/R injury is a causative factor of morbidity and mortality during liver resection, hemorrhagic shock, cardiac arrest and transplantation. During hepatectomy, ischemia is induced through clamping of the portal venous and hepatic arterial blood flow in an attempt to minimize blood loss. However, recovery of blood flow after the inflow occlusion causes reperfusion injury and severely damages the liver and other organs. Importantly, livers with preexisting diseases such as aging, steatosis and fibrosis from alcohol or hepatitis B or C infection tolerate I/R poorly. With increasing prevalence of obesity, diabetes, and metabolic syndrome, hepatic steatosis has become a major cause of chronic liver diseases and the prevalence of hepatic steatosis would substantially increase the likelihood of hepatectomy or liver transplantation and the associated I/R injury (22). At present, efforts to improve liver function after I/R have not been successful mostly due to an incomplete understanding of the pathogenesis of I/R injury. For instance, pharmacological treatment of mitochondria-related diseases with MPT blockers such as cyclosporin A (CSA) and their derivatives has been disappointing due to its narrow range of therapeutic efficacy and the onset of CSA-insensitive MPT (60). Gene targeting therapy has been heralded as a promising therapy, but technical and ethical issues must be addressed prior to its clinical application. A novel and previously unexplored avenue to the treatment of I/R injury to the liver may be up-regulation of endogenous cytoprotective events. Since mitophagy selectively targets and timely removes damaged or abnormal mitochondria, active enhancement of mitophagy could have a therapeutic potential for mitochondria-related diseases.
CONCLUSIONS
Mitophagy functions as an early protective response, favoring adaptation to stress by removing damaged mitochondria. Despite its importance and therapeutic potentials, our understanding of mitophagy remains limited. Future studies are warranted to identify new signaling proteins of mitophagy and unravel its interaction with other signaling pathways.
Acknowledgments
This work was supported in part by US National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases grant DK079879 and DK090115 (J.-S. Kim) and National Institute on Aging AG028740 (J.-S. Kim).
References
- 1.Yang S., Wang X., Contino G., Liesa M., Sahin E., Ying H., Bause A., Li Y., Stommel J.M., Dell’antonio G., Mauther J., Tonon G., Haigis M., Shirihai O.S., Doglioni C., Bardeesy N., Kimmelman A.C. Pancreatic cancers require autophagy for tumor growth. Genes Dev. (2011);25:717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo J.Y., Chen H.Y., Mathew R., Fan J., Strohecker A.M., Karsli-Uzunbas G., Kamphorst J.J., Chen G., Lemons J.M., Karantza V., Coller H.A., Dipaola R.S., Gelinas C., Rabinowitz J.D., White E. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. (2011);25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer. (2012);12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Duve C., Wattiaux R. Functions of lysosomes. Annu. Rev. Physiol. (1966);28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 5.Takamura A., Komatsu M., Hara T., Sakamoto A., Kishi C., Waguri S., Eishi Y., Hino O., Tanaka K., Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. (2011);25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Menzies R.A., Gold P.H. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J. Biol. Chem. (1971);246:2425–2429. [PubMed] [Google Scholar]
- 7.Kim I., Lemasters J.J. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid. Redox Signaling. (2011);14:1919–1928. doi: 10.1089/ars.2010.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim I., Rodriguez-Enriquez S., Lemasters J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. (2007);462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J.S., Wang J.H., Lemasters J.J. Mitochondrial permeability transition in rat hepatocytes after anoxia/reoxygenation: role of Ca2+-dependent mitochondrial formation of reactive oxygen species. Am. J. Physiol. (2012);302:G723–731. doi: 10.1152/ajpcell.00202.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim E.H., Choi K.S. A critical role of superoxide anion in selenite-induced mitophagic cell death. Autophagy. (2008);4:76–78. doi: 10.4161/auto.5119. [DOI] [PubMed] [Google Scholar]
- 11.Narendra D., Tanaka A., Suen D.F., Youle R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. (2008);183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanki T., Wang K., Cao Y., Baba M., Klionsky D.J. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell. (2009);17:98–109. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu L., Feng D., Chen G., Chen M., Zheng Q., Song P., Ma Q., Zhu C., Wang R., Qi W., Huang L., Xue P., Li B., Wang X., Jin H., Wang J., Yang F., Liu P., Zhu Y., Sui S., Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. (2012);14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 14.Wu W., Tian W., Hu Z., Chen G., Huang L., Li W., Zhang C., Liu L., Zhu Y., Zhang X., Li L., Zhang L., Sui S., Zhao B., Feng D. ULK2 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. (2014);15:566–575. doi: 10.1002/embr.201438501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sandoval H., Thiagarajan P., Dasgupta S.K., Schumacher A., Prchal J.T., Chen M., Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. (2008);454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gegg M.E., Cooper J.M., Chau K.Y., Rojo M., Schapira A.H., Taanman J.W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. (2010);19:4861–4870. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Narendra D., Tanaka A., Suen D.F., Youle R.J. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy. (2009);5:706–708. doi: 10.4161/auto.5.5.8505. [DOI] [PubMed] [Google Scholar]
- 18.Bingol B., Tea J.S., Phu L., Reichelt M., Bakalarski C.E., Song Q., Foreman O., Kirkpatrick D.S., Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. (2014);510:370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- 19.Hara Y., Yanatori I., Ikeda M., Kiyokage E., Nishina S., Tomiyama Y., Toida K., Kishi F., Kato N., Imamura M., Chayama K., Hino K. Hepatitis C virus core protein suppresses mitophagy by interacting with parkin in the context of mitochondrial depolarization. Am. J. Pathol. (2014);184:3026–3039. doi: 10.1016/j.ajpath.2014.07.024. [DOI] [PubMed] [Google Scholar]
- 20.Ding W.X., Ni H.M., Li M., Liao Y., Chen X., Stolz D.B., Dorn G.W., Yin X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. (2010);285:27879–27890. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamacher-Brady A., Brady N.R., Logue S.E., Sayen M.R., Jinno M., Kirshenbaum L.A., Gottlieb R.A., Gustafsson A.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. (2007);14:146–157. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 22.Czaja M.J., Ding W.X., Donohue T.M., Friedman S.L., Kim J.S., Komatsu M., Lemasters J.J., Lemoine A., Lin J.D., Ou J.H., Perlmutter D.H., Randall G., Ray R.B., Tsung A., Yin X.M. Functions of autophagy in normal and diseased liver. Autophagy. (2013);9:1131–1158. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qu X., Yu J., Bhagat G., Furuya N., Hibshoosh H., Troxel A., Rosen J., Eskelinen E.L., Mizushima N., Ohsumi Y., Cattoretti G., Levine B. Promotion of turmorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. (2003);112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A.M., Czaja M.J. Autophagy regulates lipid metabolism. Nature. (2009);458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koga H., Kaushik S., Cuervo A.M. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. (2010);24:3052–3065. doi: 10.1096/fj.09-144519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harada M., Hanada S., Toivola D.M., Ghori N., Omary M.B. Autophagy activation by rapamycin elimates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology. (2008);47:2026–2035. doi: 10.1002/hep.22294. [DOI] [PubMed] [Google Scholar]
- 27.Ni H.M., Bockus A., Boggess N., Jaeschke H., Ding W.X. Activation of autophagy protects against acetaminophen- induced hepatotoxicity. Hepatology. (2012);55:222–232. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bond J.M., Herman B., Lemasters J.J. Protection by acidotic pH against anoxia/reoxygenation injury to rat neonatal cardiac myocytes. Biochem. Biophys. Res. Commun. (1991);179:798–803. doi: 10.1016/0006-291X(91)91887-I. [DOI] [PubMed] [Google Scholar]
- 29.Currin R.T., Gores G.J., Thurman R.G., Lemasters J.J. Protection by acidotic pH against anoxic cell killing in perfused rat liver: evidence for a pH paradox. FASEB J. (1991);5:207–210. doi: 10.1096/fasebj.5.2.2004664. [DOI] [PubMed] [Google Scholar]
- 30.Piper H.M. Energy deficiency, calcium overload or oxidative stress: possible causes of irreversible ischemic myocardial injury. Klin. Wochenschr. (1989);67:465–476. doi: 10.1007/BF01721672. [DOI] [PubMed] [Google Scholar]
- 31.Kim J.S., Qian T., Lemasters J.J. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology. (2003);124:494–503. doi: 10.1053/gast.2003.50059. [DOI] [PubMed] [Google Scholar]
- 32.Nishimura Y., Romer L.H., Lemasters J.J. Mitochondrial dysfunction and cytoskeletal disruption during chemical hypoxia to cultured rat hepatic sinusoidal endothelial cells: the pH paradox and cytoprotection by glucose, acidotic pH, and glycine. Hepatology. (1998);27:1039–1049. doi: 10.1002/hep.510270420. [DOI] [PubMed] [Google Scholar]
- 33.Kim J.S., Nitta T., Mohuczy D., O’Malley K.A., Moldawer L.L., Dunn W.A., Behrns K.E. Impaired autophagy: A mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology. (2008);47:1725–1736. doi: 10.1002/hep.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J.H., Behrns K.E., Leeuwenburgh C., Kim J.S. Critical role of autophagy in ischemia/reperfusion injury to aged livers. Autophagy. (2012);8:140–141. doi: 10.4161/auto.8.1.18391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsunemi T., Ashe T.D., Morrison B.E., Soriano K.R., Au J., Roque R.A., Lazarowski E.R., Damian V.A., Masliah E., La Spada A.R. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. (2012);4:142ra97. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cortes C.J., Miranda H.C., Frankowski H., Batlevi Y., Young J.E., Le A., Ivanov N., Sopher B.L., Carromeu C., Muotri A.R., Garden G.A., La Spada A.R. Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat. Neurosci. (2014);17:1180–1189. doi: 10.1038/nn.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perlmutter D.H. Autophagic disposal of the aggregation- prone protein that causes liver inflammation and carcinogenesis in alpha-1-antitrypsin deficiency. Cell Death Differ. (2009);16:39–45. doi: 10.1038/cdd.2008.103. [DOI] [PubMed] [Google Scholar]
- 38.Kamimoto T., Shoji S., Hidvegi T., Mizushima N., Umebayashi K., Perlmutter D.H., Yoshimori T. Intracellular inclusions containing mutant alpha1-antitrypsin Z are propagated in the absence of autophagic activity. J. Biol. Chem. (2006);281:4467–4476. doi: 10.1074/jbc.M509409200. [DOI] [PubMed] [Google Scholar]
- 39.Kruse K.B., Brodsky J.L., McCracken A.A. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol. Biol. Cell. (2006);17:203–212. doi: 10.1091/mbc.E04-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hidvegi T., Ewing M., Hale P., Dippold C., Beckett C., Kemp C., Maurice N., Mukherijee A., Goldbach C., Watkins S., Michalopoulos G., Perlmutter D.H. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. (2010);329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 41.Pastore N., Blomenkamp K., Annunziata F., Piccolo P., Mithbaokar P., Maria Sepe R., Vetrini F., Palmer D., Ng P., Polishchuk E., Iacobacci S., Polishchuk R., Teckman J., Ballabio A., Brunetti-Pierri N. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol. Med. (2013);5:397–412. doi: 10.1002/emmm.201202046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teckman J.H., An J.K., Blomenkamp K., Schmidt B., Perlmutter D. Mitochondrial autophagy and injury in the liver in alpha 1-antitrypsin deficiency. Am. J. Physiol. (2004);286:G851–862. doi: 10.1152/ajpgi.00175.2003. [DOI] [PubMed] [Google Scholar]
- 43.Song Z., Ghochani M., McCaffery J.M., Frey T.G., Chan D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell. (2009);20:3525–3532. doi: 10.1091/mbc.E09-03-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meeusen S., DeVay R., Block J., Cassidy-Stone A., Wayson S., McCaffery J.M., Nunnari J. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. (2006);127:383–395. doi: 10.1016/j.cell.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 45.de Brito O.M., Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. (2008);456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 46.Ono T., Isobe K., Nakada K., Hayashi J.I. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat. Genet. (2001);28:272–275. doi: 10.1038/90116. [DOI] [PubMed] [Google Scholar]
- 47.Twig G., Elorza A., Molina A.J., Mohamed H., Wikstrom J.D., Walzer G., Stiles L., Haigh S.E., Katz S., Las G., Alroy J., Wu M., Py B.F., Yuan J., Deeney J.T., Corkey B.E., Shirihai O.S. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. (2008);27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dagda R.K., Cherra S.J., Kulich S.M., Tandon A., Park D., Chu C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. (2009);284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamasaki M., Furuta N., Matsuda A., Nezu A., Yamamoto A., Fujita N., Oomori H., Noda T., Haraguchi T., Hiraoka Y., Amano A., Yoshimori T. Autophagosomes form at ER-mitochondria contact sites. Nature. (2013);495:389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 50.Hailey D.W., Rambold A.S., Satpute-Krishnan P., Mitra K., Sougrat R., Kim P.K., Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. (2010);141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Y., Dorn G.W. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. (2013);340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verhoeven K., Claeys K.G., Züchner S., Schröder J.M., Weis J., Ceuterick C., Jordanova A., Nelis E., De Vriendt E., Van Hul M., Seeman P., Mazanec R., Saifi G.M., Szigeti K., Mancias P., Butler I.J., Kochanski A., Ryniewicz B., De Bleecker J., Van den Bergh P., Verellen C., Van Coster R., Goemans N., Auer-Grumbach M., Robberecht W., Milic Rasic V., Nevo Y., Tournev I., Guergueltcheva V., Roelens F., Vieregge P., Vinci P., Moreno M.T., Christen H.J., Shy M.E., Lupski J.R., Vance J.M., De Jonghe P., Timmerman V. MFN2 mutation distribution and genotype/ phenotype correlation in Charcot-Marie-Tooth type 2. J. Neurol. (2006);129:2093–2102. doi: 10.1093/brain/awl126. [DOI] [PubMed] [Google Scholar]
- 53.Chung K.W., Kim S.B., Park K.D., Choi K.G., Lee J.H., Eun H.W., Suh J.S., Hwang J.H., Kim W.K., Seo B.C., Kim S.H., Son I.H., Kim S.M., Sunwoo I.N., Choi B.O. Early onset severe and late-onset mild Charcot-Marie-Tooth disease with mitofusin 2 (MFN2) mutations. J. Neurol. (2006);129:2103–2118. doi: 10.1093/brain/awl174. [DOI] [PubMed] [Google Scholar]
- 54.Bach D., Naon D., Pich S., Soriano F.X., Vega N., Rieusset J., Laville M., Guillet C., Boirie Y., Wallberg-Henriksson H., Manco M., Calvani M., Castagneto M., Palacin M., Mingrone G., Zierath J.R., Vidal H., Zorzano A. Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes. (2005);54:2685–2693. doi: 10.2337/diabetes.54.9.2685. [DOI] [PubMed] [Google Scholar]
- 55.Hernández-Alvarez M.I., Thabit H., Burns N., Shah S., Brema I., Hatunic M., Finucane F., Liesa M., Chiellini C., Nano D., Zorzano A., Nolan J.J. Subjects with early-onset type 2 diabetes show defective activation of the skeletal muscle PGC-1{alpha}/Mitofusin-2 regulatory pathway in response to physical activity. Diabetes Care. (2010);33:645–651. doi: 10.2337/dc09-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sebastián D., Hernández-Alvarez M.I., Segalés J., Sorianello E., Muñoz J.P., Sala D., Waget A., Liesa M., Paz J.C., Gopalacharyulu P., Orešič M., Pich S., Burcelin R., Palacín M., Zorzano A. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. U.S.A. (2012);109:5523–5528. doi: 10.1073/pnas.1108220109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Biel T., Flores-Toro J.A., Dean J., Lee M.H., Lee S., Dunn W., Zendejas I., Behrns K.E., Kim J.S. Mitofusin- 2 is a novel target of sirtuin 1 that enhances autophagy and confers cytoprotection against ischemia/reperfusion injury in human and mouse livers. Hepatology. (2014);60:250A–251A. [Google Scholar]
- 58.Yu W., Sun Y., Guo S., Lu B. The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum. Mol. Genet. (2011);20:3227–3240. doi: 10.1093/hmg/ddr235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Poole A.C., Thomas R.E., Andrews L.A., McBride H.M., Whitworth A.J., Pallanck L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. U.S.A. (2008);105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim J.S., He L., Lemasters J.J. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun. (2003);304:463–470. doi: 10.1016/S0006-291X(03)00618-1. [DOI] [PubMed] [Google Scholar]