

Abstract
Inflammatory myofibroblastic tumors (IMTs), also known as inflammatory pseudotumors and inflammatory fibrosarcomas, are uncommon mesenchymal tumors composed of myofibroblastic spindle cells admixed with lymphocytes, plasma cells and eosinophils. Once thought to be reactive, these lesions are now considered to be neoplastic. These tumors can occur throughout the body, most commonly in the lung, mesentery and omentum. Patients commonly present with painless abdominal mass or with intestinal obstruction. IMTs may be multicentric, have a high local recurrence rate and may metastasize in rare cases. The lesions show wide variability in their histologic features and cellularity, and marked inflammatory infiltration, predominantly of plasmatocytes and lymphocytes, and occasionally neutrophils and eosinophils. Anaplastic lymphoma kinase (ALK) rearrangements and/or ALK1 and p80 immunoreactivity are reported in 33-67% of the tumors. Owing to the rarity of these lesions, there are no specific imaging findings that distinguish IMTs from other mesenteric masses. Complete surgical resection is the treatment of choice. Local recurrence rates are high, and re-excision is the preferred therapy for local recurrences. ALK-positive tumors show good response to ALK inhibitors. Current knowledge and comprehensive review of the available literature on IMTs is herein presented.
Keywords: Keywords Inflammatory myofibroblastic tumors, ALK rearrangements, difficult diagnosis, surgery
Introduction
Inflammatory myofibroblastic tumors (IMTs), also known as inflammatory pseudotumors and inflammatory fibrosarcomas, are uncommon mesenchymal tumors composed of myofibroblastic spindle cells admixed with lymphocytes, plasma cells and eosinophils. IMTs and inflammatory fibrosarcomas were originally described as separate entities; they are now recognized as ends of the spectrum of tumors unified by a common molecular profile and are grouped together. Once thought to be reactive, these lesions are now considered to be neoplastic [1] and the terminology of these lesions has shifted from inflammatory myofibrohistiocytic proliferations or pseudosarcomatous myofibroblastic proliferations to IMTs.
Etiopathogenesis
These tumors were first described in the lungs and later became recognized in extrapulmonary locations. IMTs can occur throughout the body, most commonly in the lung, mesentery and omentum. IMTs have also been described in the intestines, rectum, appendix, retroperitoneum, gastroesophageal junction, and occasionally in the mediastinum, liver, and abdominal wall. In mesentery, they present as well-circumscribed, lobular, multinodular firm masses. The lesions range in size from 0.4 to 36 cm. lesions arising in the mesentery tend to be large, may be sessile or polypoid; some are pedunculated, and some are large and ulcerated, mimicking gastrointestinal stromal tumors (GISTs). It is believed that this tumor derives from myofibroblastic cells, although it may represent a proliferative lesion of fibroblastic reticulum cells. The tumors may be multicentric. Cytogenetic and molecular evidence supports a clonal origin, implying that the process is neoplastic. IMTs have been reported in patients 3 months to 46 years, but mostly in the childhood and young adults with a mean age of approximately 10 years [2], with a slight male predominance, but no race predominance. The etiology of these tumors is unclear. Some have described them to be related to Epstein-Barr virus infection, others have found human herpesvirus-8 viral sequences in the tumor cells [3]. The tumors have also been described in patients with human immunodeficiency virus infection [4], and chronic granulomatous disease [5]. IMTs have also been reported as a complication of ventriculoperitoneal shunt [6] and schistosomiasis [7].
Clinical presentation
Patients commonly present with a painless abdominal mass. They may remain completely asymptomatic until the mass reaches a considerable size to cause complications. Patients may also present with abdominal pain without a palpable lump [8]. Occasionally, may present with diarrhea or intestinal obstruction [2]. There are case reports in which the first presentation of IMTs was intestinal obstruction [4,9]. Perforation peritonitis has also been reported as the initial presentation in many cases [10]. About 33% of patients have associated systemic symptoms such as fever, night sweats, weight loss, malaise, anemia, and growth failure in children [11]. When the mass is excised the syndrome disappears, and reappearance of symptoms is typically associated with tumor recurrence. Various other atypical presentations have been reported in the literature such as portal venous thrombosis [12], intussusception [13], anemia, pyrexia of unknown origin, and leukemoid reaction [14] as initial presentation of IMTs (Table 1). Meis and Enzinger [15] in a series of 38 cases of intra as well as extra-abdominal IMTs reported that 17 of 38 cases presented with symptoms of abdominal pain, 21 with anemia, 14 with fever, 16 with asymptomatic mass, and 7 with gastrointestinal obstruction. Abnormal laboratory tests may include raised erythrocyte sedimentation rate, hypergammaglobulinemia, and thrombocytosis and these also resolve when the lesion is removed [16].
Table 1.
Various presentations of inflammatory myofibroblastic tumors
Pathology
Macroscopic features
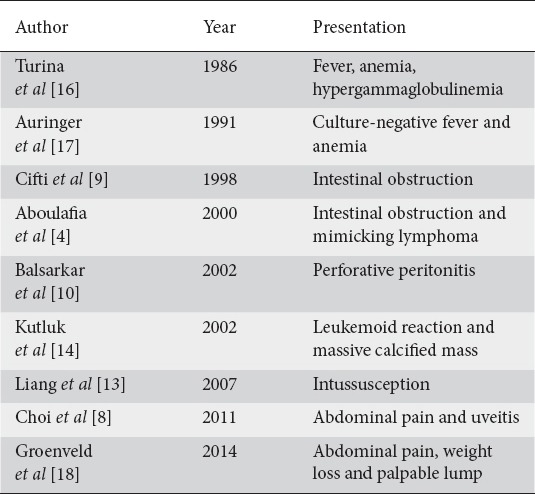
These tumors form whorled firm white or yellow colored fleshy infiltrative masses (Fig. 1A, B). Secondary changes include hemorrhage, necrosis, calcification, ulceration and ossification. The lesions may be sessile or polypoid.
Figure 1.
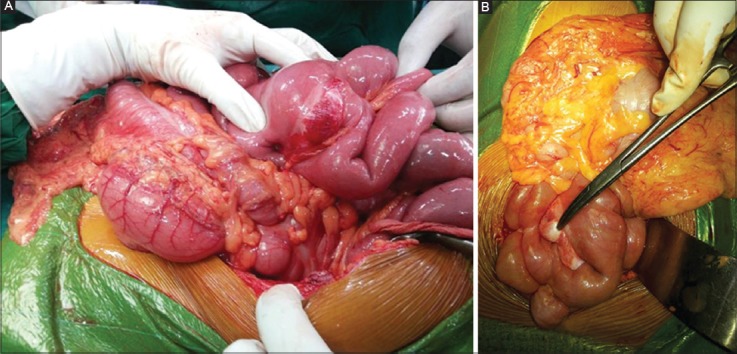
(A,B) Mesenteric inflammatory myofibroblastic tumor involving mesentery from its base to antimesenteric border
Microscopic features
The lesions show wide variability in their histologic features and cellularity and contain a mixture of inflammatory cells and myofibroblasts, and fibroblasts in fascicles or whorls. There is variable, but marked inflammatory infiltrate, predominantly plasmacytic, but with some lymphocytes and occasionally neutrophils or eosinophils. The proportion of spindle cells to inflammatory cells varies in different areas. Lymphoid follicles may be present. Pleomorphism is moderate, but mitoses are infrequently seen. Fibrosis and calcification can be seen in the stroma [2].
Three major histologic patterns are identified: 1) fibromyxoid/vascular pattern shows fasciitis-like, with vascular, myxoid and inflamed stroma, including plasma cells, this pattern predominates in approximately 24% of cases; 2) proliferating pattern shows compact proliferations of spindle-shaped cells arranged in a storiform or fascicular growth pattern with inflammation, mitoses can be seen and rare tumors contain cells with mild cytologic atypia; and 3) sclerosing pattern shows sclerosed desmoid-like areas with calcification [2].
Factors associated with unfavorable outcome are increased numbers of mitoses, high cellularity [19], and proliferative histologic pattern [2]. Cellular atypia is found in 100% of IMTs with an adverse outcome. In patients who die of their disease, high cellularity is present and is characterized by prominent atypical bizarre-shaped cells intermixed with rounded cells rather than the spindle cell proliferation, marked nuclear pleomorphism, and infiltrating borders [2,19]. In a study by Coffin et al [2], in IMTs with a favorable prognosis, mitoses ranged from 0-2 per 50 high power field, and in those with an adverse outcome mitoses ranged from 1-7 per 50 high power field, and suggested that IMT is a benign, nonmetastasizing proliferation of myofibroblasts with a potential for recurrence and persistent local growth, similar in some respects to fibromatoses.
Immunohistochemistry
Immunostaining is positive for smooth muscle actin (86%), muscle-specific actin (82%), desmin (41%), calponin, cytokeratin (26%), and vimentin, factor XIII A, CD68, CD117 may be present in submesothelial areas of the tumor [20].
Gene fusions involving the anaplastic lymphoma kinase (ALK) gene at chromosome 2p23 have been described in these lesions. ALK is a tyrosine kinase oncogene initially found to be rearranged in anaplastic large-cell lymphomas. The fusion partners have included ATIC, CARS, TMP3, TMP4, CLTC, RANB2, and SEC31L. ALK rearrangements and/or ALK1 and p80 immunoreactivity are reported in 33-67% of the tumors [21-23]. IMT in children and young adults often contains clonal cytogenetic rearrangements, whereas ALK rearrangements are uncommon in IMTs diagnosed in adults over 40 years in age. The search for rearrangement of ALK by fluorescence in situ hybridization is a useful complimentary tool for diagnosis of IMT. Cook et al [1] suggested that immunohistochemistry using anti-ALK staining is helpful in the differential diagnosis of these lesions. Cessna et al [24] recommended that immunohistochemistry for ALK1 and p80 is useful as an indicator of 2p23 abnormality, but it must be interpreted in the context of histologic and other clinicopathologic data if used as an adjunct to differential diagnosis as anaplastic large-cell lymphoma also expresses the same abnormality.
Role of fine-needle aspiration biopsy
Cytology shows spindle cells, eosiophils, neutrophils and plasma cells but in most of the cases fine-needle aspiration cytology can be extremely difficult and frustrating due to difficulty in sampling as these tumors are well collagenized.
Role of imaging studies
Owing to the rarity of these lesions, there are no specific imaging findings that distinguish IMT from other mesenteric masses. Uysal et al [25] suggested that recognition of this rare entity is important because the clinical manifestations and radiological features may be indistinguishable from a malignant lymphoproliferative disorder. In the abdomen and retroperitoneum, IMT can often appear much more diffuse and infiltrative, making initial diagnosis difficult if not impossible. Brown et al [26] concluded that the rarity of these lesions has resulted in poor documentation of its radiological manifestations. A plain abdominal radiograph will usually reveal no more than a diffuse haziness. Barium contrast studies demonstrate displacement of the intestine around the tumor, with possible narrowing of the lumen. Ultrasonography will determine the solid nature of the lesion, and contrast-enhanced computed tomography (CT) provides the information regarding the shape, extent of involvement and anatomical relationship to adjacent structures. IMT has variable and non-specific imaging characteristics, with most appearing as a lobulated solid mass that may appear heterogeneous. Brown et al [26] suggested that sonography depicts these lesions as well-defined masses with homogenous echo patterns, and CT as well-circumscribed masses of soft tissue density producing displacement of surrounding structures rather than local invasion (Fig. 2A, B). Kutluk et al [14] and Liang et al [13] encountered a massively calcified mass on ultrasonography and CT scan, which, after excision, on histopathological examination was diagnosed as IMT. On magnetic resonance imaging, they may appear as an infiltrative mesenteric mass that can be difficult to distinguish from malignant neoplasm. Auringer et al [17] documented the gallium-avid nature of these lesions in his report. Hirose et al [27] suggested that when abnormal high (18) F-FDG uptake is observed in the mesentery incidentally in clinical routine examination, IMT should be included in the differential diagnosis and (18) F-FDG may be useful in detecting local recurrence and follow up after operation.
Figure 2.
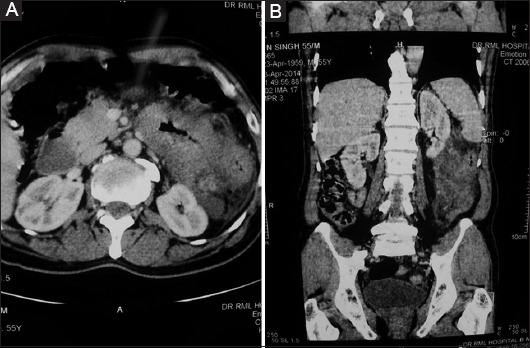
(A,B) Contrast-enhanced computed tomography showing well-circumscribed mesenteric mass causing displacement of surrounding structure
Differential diagnosis
The differential diagnosis includes GISTs, mesenteric fibromatosis, sclerosing mesenteritis, fibrohistiocytic sarcomas, Hodgkin and non-Hodgkin lymphoma, and metastatic carcinoma. IMT should always be considered in the differential diagnosis for a mesenteric mass.
Treatment
IMTs are locally aggressive lesions, multicentricity may be a feature and metastases are rare, but do occur. Therapy of choice is non-aggressive surgical resection of the tumor. In cases where complete surgical excision is not possible, symptomatic treatment is preferred over other modalities of therapy. Ill-circumscribed tumors, particularly in the abdomino-pelvic area, may be difficult to completely resect and in such cases local recurrence is more common. Tothova et al [28], and Groenveld et al [18] have recommended radical surgery as the mainstay of therapy because of high local recurrence rate. Re-excision is the treatment of choice for local recurrence and in several studies [2,29] the majority of such patients remained disease-free with long-term follow up. Chemotherapy and radiation are reserved for progressive disease after complete excision. Ciftci et al [9] suggested that radical unnecessary surgical procedures or potentially harmful therapy should be avoided, and total excision of the tumor is the appropriate treatment in most of the cases. As IMTs are a rare lesion, most of our knowledge about these lesions comes from individual case reports or case series. Detailed literature and data about an appropriate form of therapy of IMTs is still lacking, and there are no clear-cut treatment guidelines for patients in whom complete surgical excision with curative intent is not possible. Furthermore, there are no defined prognostic factors for IMTs. Intra-abdominal and retroperitoneal tumors are more likely to be aggressive than similar tumors arising elsewhere [18]. Following complete excision, approximately 23% of IMTs recur locally and only rarely these lesions undergo malignant transformation and metastasize [30]. Risk of distant metastases is <5% and the 5-year survival is about 87% [30]. There is no specific histological or molecular marker that can predict the malignant transformation of IMT, and risk of recurrence does not appear to be affected by the presence of ALK rearrangements. Cases positive for ALK rearrangements have a decreased risk of metastases when compared with ALK-negative IMT [31].
ALK activation, a feature of IMTs, diagnosed in childhood, has also been found in subsets of patients with large-cell anaplastic lymphoma. ALK-positive IMTs may therefore show good response to ALK inhibitors. Murga-Zamalloa et al [32] documented that receptor tyrosine kinases have emerged as promising therapeutic targets for these tumors. The role of crizotinib, an ALK inhibitor, has been studied, and initial trials have shown good results in the majority of cases, although more studies are required to establish selective targeting of ALK as a definitive therapeutic option. In a study by Mossé et al [33], 7 patients with ALK rearrangements with IMT received crizotinib and none of the patients showed progressive disease, 3 showed a partial response, and the rest remained stable. Other than anaplastic large-cell lymphoma and IMT, tumors that exhibit overactivation of ALK gene are neuroblastoma, non-small-cell lung carcinoma, rhabdomyosarcoma, renal cell carcinoma, and inflammatory breast cancer. In a study by Butrynski et al [34], one patient with local extension and refractive for chemotherapy was enrolled in a clinical trial for crizotinib. The patient showed a good response with crizotinib and remained disease-free. In the study by Tothova et al [28] recommended the role of crizotinib in the management of IMTs and also advocated the development of more selective ALK inhibitors, which can overcome emergent crizotinib resistance mutations, as well as the development of combination treatments with drugs targeting compensatory pathways.
Other treatment modalities have been used in IMTs where complete surgical resection was not feasible. Corticosteroid monotherapy [35,36], non-steroidal anti-inflammatory agents (NSAID) [37,38] and radiation [29,39] alone have been tried with good results in ALK-negative IMTs at sites other than mesentery. Carswel et al [35] recommended corticosteroid monotherapy for rapid resolution of the disease and sustained remission for cases in which rapid resolution is not possible. Chavez et al [37] concluded that a trial of NSAID should be considered in any patient in whom complete surgical resection is not an option, although successful outcome may only be rarely achieved. Anecdotal response to chemotherapy has also been reported [29,40]. The conclusion of various studies is summarized in Table 2.
Table 2.
Conclusion of various studies
Complete surgical resection is the treatment of choice. Local recurrence rates are high, and re-excision is the preferred therapy for local recurrences. ALK-positive tumors show good response to ALK inhibitors. Other treatment modalities have no proven role in mesenteric IMTs. The author recommends:
Complete surgical resection of IMTs is the treatment of choice because of high local recurrence rate. Radical surgery is not possible in the majority of cases because these cases present late and resection of large IMT warrants resection of a large length of gut which itself results in complications.
Re-excision is the treatment of choice for local recurrences.
Other modalities of therapy are indicated for non-resectable tumors or for progressive tumors after excision.
Concluding remarks
Inflammatory pseudotumors, also known as IMT, and inflammatory fibrosarcomas are uncommon mesenchymal tumors composed of myofibroblastic spindle cells admixed with lymphocytes, plasma cells and eosinophils. Most commonly, patients present with asymptomatic abdominal lump or with a complication like intestinal obstruction. It is very difficult to diagnose these tumors pre-operatively. Surgery is the therapy of choice for these tumors and also for local recurrences. ALK-positive tumors respond to ALK inhibitors.
Biography
Lady Hardinge Medical College and Associated Dr. Ram Manohar Lohia Hospital, New Delhi, India
Footnotes
Conflict of Interest: None
References
- 1.Cook JR, Dehner LP, Collins MH, et al. Anaplastic lymphoma kinase (ALK) expression in the inflammatory myofibroblastic tumor: a comparative immunohistochemical study. Am J Surg Pathol. 2001;25:1364–1371. doi: 10.1097/00000478-200111000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Coffin CM, Watterson J, Priest JR, Dehner LP. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol. 1995;19:859–872. doi: 10.1097/00000478-199508000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Gómez-Román JJ, Sánchez-Velasco P, Ocejo-Vinyals G, Hernández-Nieto E, Leyva-Cobián F, Val-Bernal JF. Human herpesvirus-8 genes are expressed in pulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor) Am J Surg Pathol. 2001;25:624–629. doi: 10.1097/00000478-200105000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Aboulafia DM. Inflammatory pseudotumor causing small bowel obstruction and mimicking lymphoma in a patient with AIDS: clinical improvement after initiation of thalidomide treatment. Clin Infect Dis. 2000;30:826–831. doi: 10.1086/313768. [DOI] [PubMed] [Google Scholar]
- 5.Purdy DJ, Levine EJ, Forsthoefel KJ, Fromkes JJ. Periampullary pseudotumor secondary to granulomatous disease. Am J Gastroenterol. 1994;89:2087–2088. [PubMed] [Google Scholar]
- 6.Keen PE, Weitzner S. Inflammatory pseudotumor of mesentery: a complication of ventriculoperitoneal shunt. Case report. J Neurosurg. 1973;38:371–373. doi: 10.3171/jns.1973.38.3.0371. [DOI] [PubMed] [Google Scholar]
- 7.Segun AO, Alebiosu CO, Agboola AO, Banjo AA. Schistosomiasis – An unusual cause of abdominal pseudotumor. J Natl Med Assoc. 2006;98:1365–1368. [PMC free article] [PubMed] [Google Scholar]
- 8.Choi AH, Bohn OL, Beddow TD, McHenry CR. Inflammatory myofibroblastic tumor of the small bowel mesentery: an unusual cause of abdominal pain and uveitis. J Gastrointest Surg. 2011;15:584–588. doi: 10.1007/s11605-010-1408-3. [DOI] [PubMed] [Google Scholar]
- 9.Ciftci AO, Akçören Z, Tanyel FC, Senocak ME, Caglar M, Hiçsönmez A. Inflammatory pseudotumor causing intestinal obstruction: diagnostic and therapeutic aspects. J Pediatr Surg. 1998;33:1843–1845. doi: 10.1016/s0022-3468(98)90303-7. [DOI] [PubMed] [Google Scholar]
- 10.Balsarkar DJ, Ranadive NU, Gore MA, Joshi MA. Recurrent inflammatory pseudotumor of small bowel mesentery presenting as perforative peritonitis. Indian J Gastroenterol. 2002;21:79–80. [PubMed] [Google Scholar]
- 11.Day DL, Sane S, Dehner LP. Inflammatory pseudotumor of the mesentery and small intestine. Pediatr Radiol. 1986;16:210–215. doi: 10.1007/BF02456289. [DOI] [PubMed] [Google Scholar]
- 12.Miyazaki H, Isada A, Ohira K, et al. Inflammatory pseudotumor of the mesentery causing portal venous thrombosis and cavernomatous transformation. Intern Med. 2002;41:633–637. doi: 10.2169/internalmedicine.41.633. [DOI] [PubMed] [Google Scholar]
- 13.Liang HH, Chai CY, Lin YH, Lee CH, Wu CH, Chang CC. Jejunal and multiple mesenteric calcifying fibrous pseudotumor induced jejunojejunal intussusception. J Formos Med Assoc. 2007;106:485–489. doi: 10.1016/S0929-6646(09)60298-9. [DOI] [PubMed] [Google Scholar]
- 14.Kutluk T, Emir S, Karnak I, Gaglar M, Büyükpamukçu M. Mesenteric inflammatory pseudotumor: unusual presentation with leukemoid reaction and massive calcified mass. J Pediatr Hematol Oncol. 2002;24:158–159. doi: 10.1097/00043426-200202000-00021. [DOI] [PubMed] [Google Scholar]
- 15.Meis JM, Enzinger FM. Inflammatory fibrosarcoma of the mesentery and retroperitoneum. A tumor closely simulating inflammatory pseudotumor. Am J Surg Pathol. 1991;15:1146–1156. doi: 10.1097/00000478-199112000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Turina J, Maurer R, Hollinger A, Goebel N, Siegenthaler W. Abdominal inflammatory pseudotumor (plasma cell granuloma) with anemia and hypergammaglobulinemia. Schweiz Med Wochenschr. 1986;116:473–478. [PubMed] [Google Scholar]
- 17.Auringer ST, Scott MD, Sumner TE. Inflammatory pseudotumor: a gallium-avid mobile mesenteric mass. J Nucl Med. 1991;32:1614–1616. [PubMed] [Google Scholar]
- 18.Groenveld RL, Raber MH, Oosterhof-Berktas R, Eijken E, Klaase JM. Abdominal inflammatory myofibroblastic tumor. Case Rep Gastroenterol. 2014;8:67–71. doi: 10.1159/000360843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makhlouf HR, Sobin LH. Inflammatory myofibroblastic tumors (inflammatory pseudotumors) of the gastrointestinal tract: how closely are they related to inflammatory fibroid polyps? Hum Pathol. 2002;33:307–315. doi: 10.1053/hupa.2002.32213. [DOI] [PubMed] [Google Scholar]
- 20.Meis-Kindblom JM, Kjellström C, Kindblom LG. Inflammatory fibrosarcoma: update, reappraisal, and perspective on its place in the spectrum of inflammatory myofibroblastic tumors. Semin Diagn Pathol. 1998;15:133–143. [PubMed] [Google Scholar]
- 21.Coffin CM, Patel A, Perkins S, Elenitoba-Johnson KS, Perlman E, Griffin CA. ALK1 and p80 expression and chromosomal rearrangements involving 2p23 in inflammatory myofibroblastic tumor. Mod Pathol. 2001;14:569–576. doi: 10.1038/modpathol.3880352. [DOI] [PubMed] [Google Scholar]
- 22.Lawrence B, Perez-Atayde A, Hibbard MK, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol. 2000;157:377–384. doi: 10.1016/S0002-9440(10)64550-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Debelenko LV, Arthur DC, Pack SD, Helman LJ, Schrump DS, Tsokos M. Identification of CARS-ALK fusion in primary and metastatic lesions of an inflammatory myofibroblastic tumor. Lab Invest. 2003;83:1255–1265. doi: 10.1097/01.lab.0000088856.49388.ea. [DOI] [PubMed] [Google Scholar]
- 24.Cessna MH, Zhou H, Sanger WG, et al. Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: a study of 135 cases. Mod Pathol. 2002;15:931–938. doi: 10.1097/01.MP.0000026615.04130.1F. [DOI] [PubMed] [Google Scholar]
- 25.Uysal S, Tunçbilek I, Unlübay D, Tiras U, Bilaloglu P, Kosar U. Inflammatory pseudotumor of the sigmoid colon mesentery: US and CT findings (2004:12b) Eur Radiol. 2005;15:633–635. doi: 10.1007/s00330-004-2535-6. [DOI] [PubMed] [Google Scholar]
- 26.Brown G, Shaw DG. Inflammatory pseudotumours in children: CT and ultrasound appearances with histopathological correlation. Clin Radiol. 1995;50:782–786. doi: 10.1016/s0009-9260(05)83220-9. [DOI] [PubMed] [Google Scholar]
- 27.Hirose Y, Kaida H, Kurata S, Okabe Y, Kage M, Ishibashi M. Incidental detection of rare mesenteric inflammatory pseudotumor by (18)F-FDG PET. Hell J Nucl Med. 2012;15:247–250. doi: 10.1967/s002449910060. [DOI] [PubMed] [Google Scholar]
- 28.Tothova Z, Wagner AJ. Anaplastic lymphoma kinase-directed therapy in inflammatory myofibroblastic tumors. Curr Opin Oncol. 2012;24:409–413. doi: 10.1097/CCO.0b013e328354c155. [DOI] [PubMed] [Google Scholar]
- 29.Kovach SJ, Fischer AC, Katzman PJ, et al. Inflammatory myofibroblastic tumors. J Surg Oncol. 2006;94:385–391. doi: 10.1002/jso.20516. [DOI] [PubMed] [Google Scholar]
- 30.Singer S, Neilsen T, Antonescu CR. Cancer Principles & Practice of Oncology. In: De Vita VT, Lawrence TS, Rosenberg SA, editors. 9th ed. International edition. Philadelphia, PA: Lippincott Williams and Wilkins; 2012. p. 1540. [Google Scholar]
- 31.Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007;31:509–520. doi: 10.1097/01.pas.0000213393.57322.c7. [DOI] [PubMed] [Google Scholar]
- 32.Murga-Zamalloa C, Lim MS. ALK-driven tumors and targeted therapy: focus on crizotinib. Pharmgenomics Pers Med. 2014;7:87–94. doi: 10.2147/PGPM.S37504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mossé YP, Balis FM, Lim MS, et al. Efficacy of crizotinib in children with relapsed/refractory ALK-driven tumors including anaplastic large cell lymphoma and neuroblastoma: a children's oncology group phase I consortium study. J Clin Oncol. 2012;30:9500. [Google Scholar]
- 34.Butrynski JE, D’Adamo DR, Hornick JL, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363:1727–1733. doi: 10.1056/NEJMoa1007056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carswell C, Chataway J. The successful long-term management of an intracranial inflammatory myofibroblastic tumor with corticosteroids. Clin Neurol Neurosurg. 2012;114:77–79. doi: 10.1016/j.clineuro.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 36.Lee MH, Lee HB, Lee YC, et al. Bilateral multiple inflammatory myofibroblastic tumors of the lung successfully treated with corticosteroids. Lung. 2011;189:433–435. doi: 10.1007/s00408-011-9314-3. [DOI] [PubMed] [Google Scholar]
- 37.Chavez C, Hoffman MA. Complete remission of ALK-negative plasma cell granuloma (inflammatory myofibroblastic tumor) of the lung induced by celecoxib: a case report and review of the literature. Oncol Lett. 2013;5:1672–1676. doi: 10.3892/ol.2013.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hakozaki Y, Katou M, Nakagawa K, Shirahama T, Matsumoto T. Improvement of inflammatory pseudotumor of the liver after nonsteroidal anti-inflammatory agent therapy. Am J Gastroenterol. 1993;88:1121–1122. [PubMed] [Google Scholar]
- 39.Imperato JP, Folkman J, Sagerman RH, Cassady JR. Treatment of plasma cell granuloma of the lung with radiation therapy. A report of two cases and a review of the literature. Cancer. 1986;57:2127–2129. doi: 10.1002/1097-0142(19860601)57:11<2127::aid-cncr2820571107>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 40.Bertocchini A, Lo Zupone C, Callea F, et al. Unresectable multifocal omental and peritoneal inflammatory myofibroblastic tumor in a child: revisiting the role of adjuvant therapy. J Pediatr Surg. 2011;46:e17–21. doi: 10.1016/j.jpedsurg.2011.01.007. [DOI] [PubMed] [Google Scholar]