

Abstract
Sodium meta-arsenite (SA) is implicated in the regulation of hepatic gluconeogenesis-related genes in vitro; however, the effects in vivo have not been studied. We investigated whether SA has antidiabetic effects in a type 2 diabetic mouse model. Diabetic db/db mice were orally intubated with SA (10 mg kg−1 body weight/day) for 8 weeks. We examined hemoglobin A1c (HbA1c), blood glucose levels, food intake, and body weight. We performed glucose, insulin, and pyruvate tolerance tests and analyzed glucose production and the expression of gluconeogenesis-related genes in hepatocytes. We analyzed energy metabolism using a comprehensive animal metabolic monitoring system. SA-treated diabetic db/db mice had reduced concentrations of HbA1c and blood glucose levels. Exogenous glucose was quickly cleared in glucose tolerance tests. The mRNA expressions of genes for gluconeogenesis-related enzymes, glucose 6-phosphatase (G6Pase), and phosphoenolpyruvate carboxykinase (PEPCK) were significantly reduced in the liver of SA-treated diabetic db/db mice. In primary hepatocytes, SA treatment decreased glucose production and the expression of G6Pase, PEPCK, and hepatocyte nuclear factor 4 alpha (HNF-4α) mRNA. Small heterodimer partner (SHP) mRNA expression was increased in hepatocytes dependent upon the SA concentration. The expression of Sirt1 mRNA and protein was reduced, and acetylated forkhead box protein O1 (FoxO1) was induced by SA treatment in hepatocytes. In addition, SA-treated diabetic db/db mice showed reduced energy expenditure. Oral intubation of SA ameliorates hyperglycemia in db/db mice by reducing hepatic gluconeogenesis through the decrease of Sirt1 expression and increase in acetylated FoxO1.
1. Introduction
Diabetes mellitus is a metabolic disease characterized by hyperglycemia, which results from defects in insulin secretion from pancreatic beta cells, insulin resistance in peripheral tissues, and increased glucose production by the liver [1–3]. The liver plays a critical role in the maintenance of glucose homeostasis by balancing the uptake, storage, and release of glucose [4]. Fasting or starvation induces glucose synthesis in the liver through glycogenolysis and gluconeogenesis [5]. However, elevated hepatic glucose production is associated with the pathogenesis of type 2 diabetes [6, 7]. In this process, glucose-6-phosphatase (G6Pase) catalyzes the terminal step in the glycogenolytic and gluconeogenic pathways, and phosphoenolpyruvate carboxykinase (PEPCK) is a key regulatory enzyme driving gluconeogenesis [8, 9]. Insulin suppresses gluconeogenesis by inhibiting the transcription of PEPCK and G6Pase [10, 11].
Arsenic trioxide has a long history as biomedical interest and is approved by the Food and Drug Administration (FDA) for treatment of certain leukemias [12, 13]. Sodium meta-arsenite (NaAsO2) is produced by dissolving arsenic trioxide. Sodium meta-arsenite (SA, KML001) has entered phase II clinical trials for the treatment of solid tumors and hematopoietic malignancies. In addition, sodium meta-arsenite (SA) is reported to have insulin-mimetic effects on glucose homeostasis. SA inhibits forskolin/dexamethasone-induced PEPCK and G6Pase gene expression in hepatic cell lines and rat primary hepatocytes [14, 15]. SA activates AMP-activated protein kinase [15, 16], which in turn induces small heterodimer partner (SHP) which inhibits the expression of hepatic gluconeogenic genes, and this repression is abolished by SHP inhibition [15]. SA also suppresses dexamethasone-induced PEPCK transcription in 14-day chick embryo livers in vivo [17].
Despite the demonstrated effects of SA in reducing the expression of gluconeogenesis genes, the antidiabetic effect of SA in type 2 diabetes has not yet been evaluated in vivo. In this study, we examined the therapeutic effect of SA in diabetic db/db mice, an animal model of human type 2 diabetes, as well as the mechanisms involved in the improvement of hepatic gluconeogenesis.
2. Materials and Methods
2.1. Animals
db/db mice were obtained from the Korea Research Institute of Bioscience and Biotechnology (Daejeon, Korea) and C57BL/6 mice were obtained from the Orient Bio Inc. (Gyeonggi, Korea) and maintained in specific pathogen-free conditions at the animal facility at Gachon University of Medicine and Science under a 12 h light : 12 h dark photoperiod. Animals were fed ad libitum on a standard rodent diet. The db/db male mice (aged 6–8 weeks) were monitored for the development of hyperglycemia using a glucometer (One Touch Ultra; LifeScan Inc., Milpitas, CA, USA). Pair-fed diabetic db/db mice were given the same daily amount of food as that eaten by the corresponding SA-treated group during the previous day. All animal experiments were carried out under a protocol approved by the Institutional Animal Care and Use Committee at the Gachon University of Medicine and Science. A total of 68 animals were used in the experiments described here.
2.2. Treatment with SA
Six- to eight-week-old diabetic male db/db mice (random blood glucose levels > 300 mg dL−1 for 3 consecutive days) were orally intubated with SA (10 mg kg−1 body weight/day; Komipharm, Seoul, Korea) or phosphate buffered saline (PBS) for 8 weeks. Food consumption was measured weekly. After 4 and 8 weeks of treatment, glucose levels were measured following the removal of food for 14 h. All animal groups were body weight-matched with the SA-treated group at the beginning of each experiment.
2.3. Blood Analysis
After 8 weeks of SA treatment, mice were not fed for 4 h, and blood samples were drawn into heparinized capillary tubes from the periorbital veins. The whole blood was used for HbA1c measurements, and serum was used for alanine aminotransferase (ALT) and aspartate aminotransferase (AST) measurements using the AU480 Chemistry System (Beckman Coulter Life Sciences, California, USA).
2.4. Glucose, Insulin, and Pyruvate Tolerance Tests
After 4 and 8 weeks of SA treatment, mice were not fed for 14 h, and then a glucose solution (2 g kg−1 body weight) was injected intraperitoneally. Blood glucose levels were measured at 0, 30, 60, 90, 150, and 180 min after glucose injection at 9:00 a.m. For insulin tolerance tests, mice were not fed for 4 h and were injected with insulin (2 units kg−1 body weight, i.p.), and blood glucose levels were measured at 0, 30, 60, and 90 min after insulin injection at 1:00 p.m. For pyruvate tolerance tests, C57BL/6 mice were orally intubated with SA (10 mg kg−1 body weight) or PBS and then fasted overnight. Fifteen hours after oral intubation, mice were injected i.p. with 1 g kg−1 body weight of sodium pyruvate in PBS, and blood glucose levels were measured at 0, 15, 30, 60, 90, and 120 min after pyruvate injection at 9:00 a.m.
2.5. Real-Time Quantitative PCR (RT-qPCR)
Total RNA was isolated from the liver of SA-treated mice or hepatocytes from C57BL/6 mice, and cDNA was synthesized using the PrimeScript First-Strand cDNA Synthesis Kit (TaKaRa Bio, Inc., Otsu, Japan). PCR was carried out in a 7900HT fast real-time PCR system (Applied Biosystems, Carlsbad, CA) at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s, 60°C for 1 min. As an internal control, cyclophilin mRNA was amplified. The specific PCR primers were G6Pase: sense 5′-GTGTTGACATCGGCCC-3′, antisense 5′-AACTGAAGCCGGTTAG-3′; PEPCK: sense 5′-CGCAAGCTGAAGAAATATGACAA3′, antisense 5′-TCGATCCTGGCCACATCTC-3′; hepatocyte nuclear factor-4α (HNF-4α): sense 5′-CCAACCTCAATTCATCCAACA-3′, antisense 5′-CCCGGTCCGCCACAGAT-3′; SHP: sense 5′-AGG AACCTGCCGTTCCTTCTG-3, antisense 5′-TGG CTT CCT CTA GCA GGA TC-3′; Sirt1: sense 5′-TTGGTGGTACAAACAGGTATTGA-3′, antisense 5′-CAGTGAGAAAATGCTGGCCTA-3′; AgRP: sense 5′-TGCTACTGCCGCTTCTTCAA-3′, antisense 5′-CTTTGCCCAAACAACATCCA-3′; POMC: sense 5′-GAGGCCACTGAACATCTTTGTC-3′, antisense 5′-GCAGAGGCAAACAAGATTGG; MC4R: sense 5′-TGCTGGTGAGCGTTTCGA-3′, antisense 5′-GGCATCCGTATCCGTACT-3′; and cyclophilin: sense 5′-TGGAGAGCACCAAGACAGACA-3′, antisense 5′-TGCCGGAGTCGACAATGAT-3′. The relative copy number was calculated using the threshold crossing point (Ct) as calculated by the 7900HT fast real-time PCR software combined with the delta delta Ct calculations.
2.6. Glucose Production Assay
Primary hepatocytes were isolated from C57BL/6 mice by collagenase perfusion. For the hepatic glucose production assay, hepatocytes were seeded in 6-well plates at a density of 2 × 105 cells 2 mL−1/well and cultured in Hepatozyme SFM media (Gibco) for 24 h; unattached cells were discarded. Hepatocytes were then treated for 24 h with SA (5 or 10 μM). After 24 h of culture, cells were washed with PBS and then cultured in glucose-free DMEM supplemented with 20 mM sodium lactate and 2 mM sodium pyruvate for 2 h. Glucose concentration in the media was measured by a glucose assay kit (BioVision Research Products, Mountain View, CA).
2.7. Western Blot Analysis
Hepatocyte cell lysates were preparedand subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were electrotransferred onto nitrocellulose membranes. Membranes were incubated with anti-Sirt1, anti-acetyl-FoxO1, or anti-FoxO1 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. After washing, membranes were incubated with a horseradish peroxidase-conjugated secondary antibody (anti-rabbit IgG, Chemicon International, Temecula, CA) for 1 h at room temperature. Reactive bands were detected by enhanced chemiluminescence (Thermo Fisher Scientific, Rockford, IL).
2.8. Indirect Calorimetry
A comprehensive animal metabolic monitoring system (CLAMS; Columbus Instruments, Columbus, OH) was used to evaluate energy expenditure, respiratory exchange ratio, and locomotor activity. Energy expenditure was measured by assessing oxygen consumption with indirect calorimetry. The respiratory exchange ratio was computed as carbon dioxide output (VCO2) divided by oxygen consumption (VO2). Locomotor activity was measured on x- and z-axis by using infrared beams to count the beam breaks during 72 h.
2.9. Cytotoxicity and Proliferation Assays
For cytotoxicity and proliferation assays, hepatocytes were seeded in 96-well plates at a density of 2 × 104 cells 100 μL−1/well and incubated for 24 h. The cells were then incubated in culture medium containing SA (5 or 10 μM). Following 24 h of incubation, cell viability was determined using a Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer's protocol. For the 3H-thymidine incorporation assay, hepatocytes were seeded and incubated as described above with the addition of 3H-thymidine (1 μCi/well). After 24 h of incubation, the cells were washed with PBS and then analyzed for 3H-thymidine incorporation using a scintillation beta-counter, 1450 LSC and Luminescence Counter MicroBeta TriLux (Perkin Elmer).
2.10. Glucose-Stimulated Insulin Secretion
After 8 weeks of SA treatment, mice were not fed for 14 h, and then a glucose solution (2 g kg−1 body weight) was injected intraperitoneally. Blood glucose levels were measured at 0 and 30 min after glucose injection. The concentration of serum insulin was measured by an ultrasensitive mouse insulin enzyme immunosorbent assay kit (ALPCO, Windham, NH).
2.11. Statistical Analysis
Data are presented as mean ± SE. Statistical significance of the difference between two groups was analyzed by unpaired Student's t-test for comparison of two groups or ANOVA followed by Fisher's protected least significant difference test for multiple groups. P < 0.05 was accepted as significant.
3. Results
3.1. Reduction of Blood Glucose Level and Food Intake in SA-Treated db/db Mice
To investigate the effects of SA on a type 2 diabetic mouse model, we orally intubated diabetic db/db mice with SA daily and measured HbA1c levels, glucose levels, food intake, and body weight. We found that HbA1c levels of SA-treated mice were significantly lower compared with the untreated, PBS-treated, and pair-fed control groups at 8 weeks of treatment (Figure 1(a)). Blood glucose levels were unchanged among groups at 4 weeks of treatment but were significantly lower in the SA-treated group at 8 weeks, compared with the control groups (Figure 1(b)). Fasting blood glucose levels were also significantly decreased in SA-treated mice compared with untreated, PBS-treated mice, and pair-fed mice at 8 weeks of treatment (Figure 1(c)). Interestingly, food intake in SA-treated mice was significantly decreased compared with PBS-treated mice (Figure 1(d)), but body weight gain was significantly increased at 8 weeks of SA treatment (Figure 1(e)). In contrast, the pair-fed group had significantly lower body weights as compared with the PBS-treated group (Figure 1(e)). In addition, the blood concentrations of ALT and AST, indicators of liver damage, were not different between SA- and PBS-treated db/db mice (see Supplementary Figure 1 in Supplementary Material available online at http://dx.doi.org/10.1155/2014/961732). These results suggest that treatment with 10 mg kg−1 of SA for 8 weeks may not have toxic effects in db/db mice. In low concentrations, SA has been reported to cause transient stimulation of cell growth [18]. In our study, we also found that SA in low doses (5 μM) stimulated hepatocyte cell growth and did not show cell toxicity, even at 10 μM (Supplementary Figure 2).
Figure 1.

Decrease of blood glucose levels and food intake in SA-treated db/db mice. Diabetic db/db mice were orally intubated with SA (10 mg kg−1 body weight/day) or PBS for 8 weeks. (a) Blood levels of HbA1c, (b) nonfasting blood glucose levels, (c) fasting blood glucose levels, (d) food intake, and (e) body weights were measured. Untreated diabetic db/db mice and pair-fed diabetic db/db mice served as controls. n = 5–11 per group. * P < 0.05, ** P < 0.01 compared with PBS-treated mice; # P < 0.05, ## P < 0.01 compared with the pair-fed group; § P < 0.01, compared with untreated mice.
3.2. Improvement of Glucose Tolerance in SA-Treated db/db Mice
To determine whether blood glucose levels are properly controlled in SA-treated db/db mice, we performed intraperitoneal glucose tolerance tests at 4 and 8 weeks of SA treatment. Blood glucose levels in SA-treated mice were significantly lower at all time points following glucose injection compared with pair-fed mice and significantly lower after 90 min compared with PBS-treated mice at 4 weeks (Figure 2(a)). At 8 weeks of treatment, blood glucose levels of SA-treated mice were significantly reduced at all time points compared with PBS-treated and pair-fed mice (Figure 2(b)). Blood glucose levels in the pair-fed group were not significantly different from the untreated or PBS-treated groups at any time point, except at 60 min after glucose loading (Figure 2(b)). The area under the curve was significantly reduced in SA-treated mice compared with PBS-treated and pair-fed mice at both 4 and 8 weeks of treatment (Figures 2(c) and 2(d)).
Figure 2.

SA improved glucose tolerance in diabetic db/db mice. Diabetic db/db mice were orally intubated with SA (10 mg kg−1 body weight/day) or PBS. (a) Four weeks and (b) 8 weeks later, mice were not fed for 4 h and were injected with glucose, and blood glucose levels were measured (n = 5–10 per group). The area under the curve measured at (c) 4 weeks and (d) 8 weeks. Untreated diabetic db/db mice and pair-fed diabetic db/db mice served as controls. * P < 0.05, ** P < 0.01, and *** P < 0.0001 compared with PBS-treated mice; # P < 0.05, ## P < 0.01, and ### P < 0.005 compared with the pair-fed group; † P < 0.05 pair-fed mice compared with PBS-treated mice; § P < 0.01, compared with untreated mice.
To address whether SA treatment improves insulin sensitivity, we performed insulin tolerance tests at 4 and 8 weeks of SA treatment. Glucose reduction in SA-treated mice was not different from untreated, PBS-treated, or pair-fed mice at 4 weeks or 8 weeks of treatment (Figures 3(a) and 3(b)). Similarly, the area under the curves was not different among groups (Figures 3(c) and 3(d)). To measure insulin secretion in SA-treated mice, we analyzed glucose-stimulated insulin secretion after 8 weeks of SA treatment. Insulin secretion was not different between the SA- and PBS-treated groups (Supplementary Figure 3).
Figure 3.

SA treatment did not improve insulin sensitivity in diabetic db/db mice. Diabetic db/db mice were orally intubated with SA (10 mg kg−1 body weight/day) or PBS. (a) Four weeks and (b) 8 weeks later, insulin was injected and blood glucose levels were measured (n = 4–10 per group). Data are expressed as a percentage of the initial blood glucose level before insulin injection. The area under the curve measured at (c) 4 weeks and (d) 8 weeks. Untreated diabetic db/db mice and pair-fed diabetic db/db mice served as controls.
3.3. Decreased Expression of Gluconeogenic Genes in SA-Treated db/db Mice and Reduction of Glucose Production and Expression of Gluconeogenesis-Related Genes in SA-Treated Hepatocytes
To determine whether SA treatment affects the expression of genes involved in glucose production, we examined the expression of G6Pase and PEPCK mRNA, which are involved in gluconeogenesis in the liver. The expression of both G6Pase and PEPCK mRNA was significantly decreased in the liver of SA-treated db/db mice compared with PBS-treated mice at 8 weeks (Figures 4(a) and 4(b)). To investigate gluconeogenic fluxes in vivo, C57BL/6 mice were orally intubated with SA or PBS. After 15 h later from oral intubation, mice were injected i.p. with sodium pyruvate, and blood glucose levels were measured. SA treatment significantly inhibited blood glucose level at 15 min after pyruvate injection (Figure 4(c)). To investigate the direct effects of SA on hepatic gluconeogenesis in vitro, we treated hepatocytes isolated from C57BL/6 mice with SA and then measured glucose production. SA treatment significantly decreased glucose production approximately 70% and 90% at 5 and 10 μM, respectively, as compared with untreated hepatocytes (Figure 4(d)). In addition, the expression of G6Pase and PEPCK mRNA was significantly decreased in SA-treated hepatocytes (Figures 4(e) and 4(f)). The promoter activity of G6Pase and PEPCK is regulated by the transcription factors HNF-4α, HNF-3β, and FoxO1 [9, 19]. Small heterodimer partner (SHP) has been shown to downregulate G6Pase and PEPCK via the repression of HNF-4α, HNF-3β, and FoxO1 [20, 21]. Thus, we measured the expression of HNF-4α and SHP mRNA in SA-treated hepatocytes. SA treatment significantly decreased the expression of HNF-4α mRNA and significantly increased SHP mRNA (Figures 4(g) and 4(h)).
Figure 4.

SA suppressed gluconeogenic gene expression in the liver of SA-treated db/db mice and inhibited glucose production and gluconeogenic gene expression in hepatocytes. Diabetic db/db mice were orally intubated with SA (10 mg kg−1 body weight/day) or PBS. Eight weeks later, the liver tissue was removed. The mRNA expression of (a) G6Pase and (b) PEPCK was analyzed by RT-qPCR and normalized by cyclophilin expression. The fold change was calculated as a ratio of the expression level in PBS-treated diabetic db/db mice. * P < 0.005, ** P < 0.0001 compared with PBS-treated group (n = 4–9/group). (c) For pyruvate tolerance tests, C57BL/6 mice were orally intubated with SA (10 mg kg−1 body weight) or PBS and then fasted overnight. Fifteen hours after SA oral intubation, mice were injected i.p. with 1 g kg−1 body weight of sodium pyruvate in PBS, and blood glucose levels were measured at 0, 15, 30, 60, 90, and 120 min after pyruvate injection (n = 3-4 per group). The area under the curve (AUC) was calculated. * P < 0.05 and ** P < 0.01, compared with PBS-treated mice. (d) Primary hepatocytes from C57BL/6 mice were incubated with 5 or 10 μM SA for 48 h and cultured in glucose-free DMEM supplemented with 20 mM sodium lactate and 2 mM sodium pyruvate for 2 h. Glucose production in media was measured and normalized by the amount of total protein. Relative glucose production was calculated as a percent of glucose production from untreated hepatocytes. Primary hepatocytes from C57BL/6 mice were incubated with 5 or 10 μM SA for 24 h, and the expression of (e) G6Pase, (f) PEPCK, (g) HNF-4α, and (h) SHP mRNA was analyzed by RT-qPCR, with values normalized to cyclophilin expression. The fold change was calculated as ratio of the expression in untreated hepatocytes. Data are mean ± SE from three to four independent experiments. * P < 0.05, ** P < 0.01, and *** P < 0.001 compared with untreated hepatocytes.
3.4. Decreased Expression of Sirt1 mRNA and Protein and Increased Acetylation of FoxO1 in SA-Treated Hepatocytes
FoxO1 activity is known to be regulated by phosphorylation and acetylation [22, 23]. Sirt1 increases FoxO1 DNA-binding ability by deacetylating FoxO1 and potentiating its transcription activity [24]. Thus, we determined the expression of Sirt1 and acetylated FoxO1 in SA-treated hepatocytes. The expression of Sirt1 mRNA and protein in SA-treated hepatocytes was significantly decreased compared with untreated control hepatocytes (Figures 5(a) and 5(b)). In addition, acetylated FoxO1 was increased in SA-treated hepatocytes compared with untreated cells (Figure 5(b)).
Figure 5.

SA inhibited the expression of Sirt1 and increased acetylated FoxO1. (a) Hepatocytes isolated from C57BL/6 mice were incubated with 5 or 10 μM SA for 24 h, and the expression of Sirt1 mRNA was analyzed by RT-qPCR, with values normalized to cyclophilin expression. The fold change was calculated as ratio of the expression in untreated hepatocytes. (b) Hepatocytes were cultured for 48 h with 5 or 10 μM SA. Western blot was performed with antibodies against Sirt1, FoxO1, and acetylated FoxO1 proteins. Data are mean ± SE from three independent experiments. * P < 0.05 compared with untreated hepatocytes.
3.5. Decreased Energy Expenditure in SA-Treated db/db Mice
SA-treated mice showed a significant increase in body weight in spite of a significant reduction in food intake (Figures 1(d) and 1(e)). To investigate whether the body weight gain is due to metabolic changes induced by SA treatment, we measured food intake, oxygen consumption, carbon dioxide output, energy expenditure, and respiratory exchange ratio by a metabolic monitoring system over 72 h at 8 weeks of SA treatment. Food intake was significantly decreased in SA-treated mice compared with untreated mice (Figure 6(a)). Oxygen consumption (Figure 6(b)), carbon dioxide output (Figure 6(c)), and energy expenditure (Figure 6(d)) were significantly decreased in SA-treated mice compared with untreated or pair-fed groups. The respiratory exchange ratio was not different in SA-treated or pair-fed mice compared with untreated mice (Figure 6(e)).
Figure 6.

Maintenance of a low level of energy expenditure in SA-treated db/db mice. Diabetic db/db mice were treated with SA. (a) Food intake, (b) oxygen consumption (VO2), (c) carbon dioxide output (VCO2), (d) energy expenditure (EE), and (e) respiratory exchange ratio (RER) were measured by indirect calorimetry analysis for 72 h at 8 weeks of SA treatment (n = 7-8/group). * P < 0.05, ** P < 0.01, and *** P < 0.005 compared with untreated mice; # P < 0.05, ## P < 0.01 compared with the pair-fed group.
Feeding behavior is regulated by a system with the hypothalamus at the centre, and energy homeostasis is maintained through regulation of food intake and energy expenditure [25, 26]. Therefore, we investigated the expression of hypothalamic molecules known to be involved in appetite regulation, AgRP, proopiomelanocortin (POMC), and a POMC receptor, melanocortin 4 receptor (MC4R), in SA-treated mice. Interestingly, the expression of AgRP mRNA, but not POMC and MC4R mRNA, was significantly increased in SA-treated mice compared with PBS-treated controls and pair-fed group (Figures 7(a)–7(c)).
Figure 7.
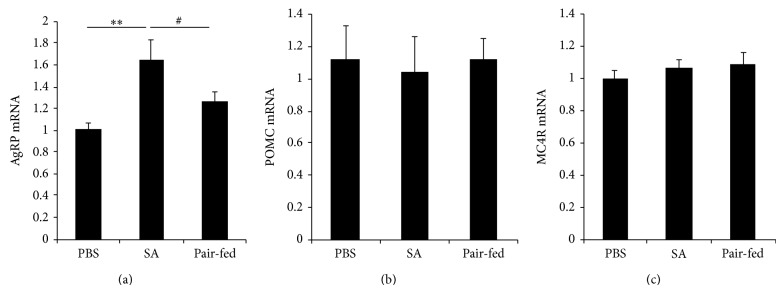
Increase of AgRP gene expression in hypothalamus of SA-treated db/db mice. Diabetic db/db mice were orally intubated with SA (10 mg kg−1 body weight/day) or PBS. Eight weeks later, the hypothalamus was removed, and the expression of (a) AgRP, (b) POMC, and (c) MC4R mRNA was analyzed by RT-qPCR and normalized by cyclophilin expression. The fold change was calculated as a ratio of the expression level in PBS-treated diabetic db/db mice. * P < 0.05, ** P < 0.01 compared with PBS-treated db/db mice (n = 5–10/group).
4. Discussion
The liver is an important organ in the regulation of glucose homeostasis in fed and fasting conditions [4]. Excess gluconeogenesis and glycogenolysis in the liver lead to elevated hepatic glucose production, which is a major cause of hyperglycemia in type 2 diabetes [6, 7]. SA has been previously shown to downregulate the expression of the hepatic gluconeogenic genes, G6Pase and PEPCK, in hepatic cell lines, rat hepatocytes, and embryonic chick liver [14, 15, 17]. In this study, we report that SA reduces hepatic gluconeogenesis in diabetic db/db mice, an animal model of human type 2 diabetes. SA-treated diabetic db/db mice showed decreased blood glucose levels under fed and fasting conditions and improved glucose tolerance. In addition, the expression of gluconeogenesis-related genes was downregulated in SA-treated db/db mice liver and mouse hepatocytes.
Gluconeogenesis is a metabolic pathway that generates glucose from noncarbohydrate carbon substrates. This pathway is catalyzed by several enzymes, first and last ones being PEPCK and G6Pase. SA suppressed the expression of PEPCK promoter-driven luciferase constructs in a rat hepatoma H4IIE cell line [14]. SA also repressed forskolin/dexamethasone-stimulated PEPCK and G6Pase gene expression and induced SHP gene expression via AMP-activated protein kinase to inhibit gluconeogenic enzyme gene expression in hepatic cell lines [15]. In our study, G6Pase and PEPCK mRNA levels were significantly reduced in SA-treated diabetic db/db mice liver. In addition, HbA1c and fed and fasting glucose levels were significantly decreased in SA-treated diabetic db/db mice. These results suggest that SA could improve hyperglycemia in type 2 diabetes through the reduction of gluconeogenesis.
SHP is a member of the nuclear receptor family of intracellular transcription factors [27]. SHP has been shown to inhibit numerous nuclear receptors and transcription factors as transcriptional corepressor [28]. The induction of SHP has been shown to downregulate G6Pase and PEPCK through the repression of HNF-4α-, HNF-3β-, and FoxO1-mediated transcriptional activity [20, 21]. In our study, the expression of SHP mRNA was significantly increased in SA-treated hepatocytes, and HNF-4α mRNA was decreased by SA in a dose-dependent manner in mouse hepatocytes. This suggests that SA-induced increases of SHP work as repressor to inhibit HNF-4α expression and subsequently G6Pase and PEPCK production.
FoxO1 is a transcription factor that has an important function in the regulation of gluconeogenesis [29]. Its transcriptional activity is regulated through phosphorylation or acetylation of multiple residues. When FoxO1 is phosphorylated, it is excluded from the nucleus, resulting in decreased transcription of G6Pase and decreased hepatic gluconeogenesis [22]. FoxO1 transcriptional activity is also regulated by acetylation, which inhibits the ability of FoxO1 to interact with the G6Pase promoter by decreasing the stability of the FoxO1-DNA complex [23].
Sirt1 reverses this acetylation process of FoxO1 and has been shown to play critical roles in glucose homeostasis in liver. However, Sirt1 has been shown to confer both positive and negative effects on hepatic gluconeogenesis. The transcriptional activity of FoxO1 is increased by Sirt1 through deacetylation [24]; on the other hand, Sirt1 suppresses the ability of HNF-4α to downregulate gluconeogenic gene expression. In our study, the expressions of Sirt1 mRNA and protein levels were significantly decreased depending on SA concentration. It was reported that SA induces NF-κB activation [30], which enhances the expression of miR-34a, a tumor suppressor gene [31]. And SA treatment increases miR-34a expression in TK-6 cells [32], which downregulates the expression of Sirt1 [33]. Thus, the decrease of Sirt1 mRNA by SA in our study may be mediated by the NF-κB/miR-34a pathway. Taken together, all these results suggest that SA decreases Sirt1, thus reducing the deacetylation of FoxO1 and reducing its transcriptional activity, which contributes to the reduced expression of hepatic gluconeogenic genes.
Interestingly, SA-treated db/db mice showed reduced food intake over the 8 weeks of the experiment; however, SA-treated mice gained more body weight compared with PBS-treated and pair-fed mice. In addition, SA-treated mice showed decreased consumption of oxygen and production of carbon dioxide, energy expenditure, and locomotor activity compared with pair-fed mice. In the brain, energy balance is regulated by neural and hormonal integrated signals. The hypothalamus is a primary center in which feeding behavior and energy metabolism are regulated [34, 35]. AgRP is a neuropeptide produced in the hypothalamus by the AgRP/neuropeptide Y (NPY) neuron. AgRP increases appetite and decreases metabolism and energy expenditure [36–38]. AgRP treatment for 7 days' intracerebroventricular administration in rat decreased oxygen consumption and energy expenditure [39]. In our study, the expression of AgRP mRNA was significantly increased in SA-treated mice compared with PBS-treated mice, whereas the pair-fed mice were not different from the PBS group. These results suggest that SA decreases energy expenditure and increases weight gain via the induction of AgRP expression in hypothalamus. It is known that FoxO1 regulates AgRP transcription in the hypothalamus, and decreased FoxO1 activity is associated with decreased AgRP expression [40]. However, we found that mRNA expression of AgRP was increased although FoxO1 transcriptional activity was decreased by SA treatment. The expression of AgRP may be regulated by several molecules as well as FoxO1 and further studies are needed for the detailed molecular mechanisms.
It is paradoxical that food intake decreased in SA-treated db/db mice in spite of an increase in AgRP, which might be expected to stimulate appetite. In addition, the appetite-stimulating effects of AgRP are inhibited by leptin [41], but db/db mice are deficient for the leptin receptor gene [42]. Therefore, the increase of AgRP expression may not affect the appetite in SA-treated db/db mice.
In conclusion, our studies show that SA improves glucose metabolism in a type 2 diabetic mouse model. SA directly inhibits hepatic glucose production via regulation of gluconeogenesis-related genes and protein expression, such as PEPCK, G6Pase, SHP, Sirt1, and acetylated FoxO1. In addition, SA increases body weight via decreasing energy expenditure due to induction of AgRP gene expression in hypothalamus. This may benefit end-state lean diabetes patients and increase their longevity.
Supplementary Material
Supplemental Figure 1: The concentration of ALT and AST in blood of SA-treated db/db mice.
Supplemental Figure 2: Proliferation induced in SA-treated mouse hepatocytes.
Supplemental Figure 3: Glucose-stimulated insulin secretion test in SA-treated db/db mice.
Acknowledgments
This study was supported by grants from Komipharm International Co. Ltd. and in part by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A102060). The authors thank Dr. Ann Kyle for editorial assistance.
Conflict of Interests
The authors declare that this work was partially supported by a grant from the Komipharm International Co. and that Sujong Kim is an employee of Komipharm International Co.
Authors’ Contribution
Young-Sun Lee and Eun-Kyu Lee contributed equally to this research.
References
- 1.Scheen A. J. Pathophysiology of insulin secretion. Annales d'Endocrinologie. 2004;65(1):29–36. doi: 10.1016/s0003-4266(04)95627-2. [DOI] [PubMed] [Google Scholar]
- 2.Pawlak J., Derlacz R. A. The mechanism of insulin resistance in peripheral tissues. Postepy Biochemii. 2011;57(2):200–206. [PubMed] [Google Scholar]
- 3.Edgerton D. S., Johnson K. M. S., Cherrington A. D. Current strategies for the inhibition of hepatic glucose production in type 2 diabetes. Frontiers in Bioscience. 2009;14(3):1169–1181. doi: 10.2741/3301. [DOI] [PubMed] [Google Scholar]
- 4.Oh K.-J., Han H.-S., Kim M.-J., Koo S.-H. Transcriptional regulators of hepatic gluconeogenesis. Archives of Pharmacal Research. 2013;36(2):189–200. doi: 10.1007/s12272-013-0018-5. [DOI] [PubMed] [Google Scholar]
- 5.Nordlie R. C., Foster J. D., Lange A. J. Regulation of glucose production by the liver. Annual Review of Nutrition. 1999;19:379–406. doi: 10.1146/annurev.nutr.19.1.379. [DOI] [PubMed] [Google Scholar]
- 6.Radziuk J., Pye S. Production and metabolic clearance of glucose under basal conditions in type II (non-insulin-dependent) diabetes mellitus. Diabetologia. 2001;44(8):983–991. doi: 10.1007/s001250100589. [DOI] [PubMed] [Google Scholar]
- 7.Magnusson I., Rothman D. L., Katz L. D., Shulman R. G., Shulman G. I. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. The Journal of Clinical Investigation. 1992;90(4):1323–1327. doi: 10.1172/jci115997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiong Y., Lei Q.-Y., Zhao S., Guan K.-L. Regulation of glycolysis and gluconeogenesis by acetylation of PKM and PEPCK. Cold Spring Harbor Symposia on Quantitative Biology. 2011;76:285–289. doi: 10.1101/sqb.2011.76.010942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yabaluri N., Bashyam M. D. Hormonal regulation of gluconeogenic gene transcription in the liver. Journal of Biosciences. 2010;35(3):473–484. doi: 10.1007/s12038-010-0052-0. [DOI] [PubMed] [Google Scholar]
- 10.Sutherland C., O'Brien R. M., Granner D. K. New connections in the regulation of PEPCK gene expression by insulin. Philosophical Transactions of the Royal Society B: Biological Sciences. 1996;351(1336):191–199. doi: 10.1098/rstb.1996.0016. [DOI] [PubMed] [Google Scholar]
- 11.Schmoll D., Allan B. B., Burchell A. Cloning and sequencing of the 5′ region of the human glucose-6-phosphatase gene: transcriptional regulation by cAMP, insulin and glucocorticoids in H4IIE hepatoma cells. FEBS Letters. 1996;383(1-2):63–66. doi: 10.1016/0014-5793(96)00224-4. [DOI] [PubMed] [Google Scholar]
- 12.List A., Beran M., DiPersio J., Slack J., Vey N., Rosenfeld C. S., Greenberg P. Opportunities for Trisenox (arsenic trioxide) in the treatment of myelodysplastic syndromes. Leukemia. 2003;17(8):1499–1507. doi: 10.1038/sj.leu.2403021. [DOI] [PubMed] [Google Scholar]
- 13.Zhu J., Chen Z., Lallemand-Breitenbach V., de Thé H. How acute promyclocytic leukaemia revived arsenic. Nature Reviews Cancer. 2002;2(9):705–713. doi: 10.1038/nrc887. [DOI] [PubMed] [Google Scholar]
- 14.Kaltreider R. C., Davis A. M., Lariviere J. P., Hamilton J. W. Arsenic alters the function of the glucocorticoid receptor as a transcription factor. Environmental Health Perspectives. 2001;109(3):245–251. doi: 10.1289/ehp.01109245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chanda D., Kim S.-J., Lee I.-K., Shong M., Choi H.-S. Sodium arsenite induces orphan nuclear receptor SHP gene expression via AMP-activated protein kinase to inhibit gluconeogenic enzyme gene expression. American Journal of Physiology—Endocrinology and Metabolism. 2008;295(2):E368–E379. doi: 10.1152/ajpendo.00800.2007. [DOI] [PubMed] [Google Scholar]
- 16.Lochhead P. A., Salt I. P., Walker K. S., Hardie D. G., Sutherland C. 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes. 2000;49(6):896–903. doi: 10.2337/diabetes.49.6.896. [DOI] [PubMed] [Google Scholar]
- 17.Hamilton J. W., Kaltreider R. C., Bajenova O. V., et al. Molecular basis for effects of carcinogenic heavy metals on inducible gene expression. Environmental Health Perspectives. 1998;106(supplement 4):1005–1015. doi: 10.2307/3434145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komissarova E. V., Saha S. K., Rossman T. G. Dead or dying: the importance of time in cytotoxicity assays using arsenite as an example. Toxicology and Applied Pharmacology. 2005;202(1):99–107. doi: 10.1016/j.taap.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 19.Sakamaki J.-I., Daitoku H., Kaneko Y., Hagiwara A., Ueno K., Fukamizu A. GSK3β regulates gluconeogenic gene expression through HNF4α and FOXO1. Journal of Receptors and Signal Transduction. 2012;32(2):96–101. doi: 10.3109/10799893.2012.660531. [DOI] [PubMed] [Google Scholar]
- 20.Kim J.-Y., Kim H.-J., Kyung T. K., Park Y.-Y., Seong H.-A., Ki C. P., Lee I.-K., Ha H., Shong M., Sang C. P., Choi H.-S. Orphan nuclear receptor small heterodimer partner represses hepatocyte nuclear factor 3/foxa transactivation via inhibition of its DNA binding. Molecular Endocrinology. 2004;18(12):2880–2894. doi: 10.1210/me.2004-0211. [DOI] [PubMed] [Google Scholar]
- 21.Yamagata K., Daitoku H., Shimamoto Y., et al. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. The Journal of Biological Chemistry. 2004;279(22):23158–23165. doi: 10.1074/jbc.m314322200. [DOI] [PubMed] [Google Scholar]
- 22.Nakae J., Kitamura T., Silver D. L., Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. The Journal of Clinical Investigation. 2001;108(9):1359–1367. doi: 10.1172/jci200112876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsuzaki H., Daitoku H., Hatta M., Aoyama H., Yoshimochi K., Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(32):11278–11283. doi: 10.1073/pnas.0502738102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh B. K., Sinha R. A., Zhou J., et al. FoxO1 deacetylation regulates thyroid hormone-induced transcription of key hepatic gluconeogenic genes. The Journal of Biological Chemistry. 2013;288(42):30365–30372. doi: 10.1074/jbc.m113.504845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leibowitz S. F., Wortley K. E. Hypothalamic control of energy balance: different peptides, different functions. Peptides. 2004;25(3):473–504. doi: 10.1016/j.peptides.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Valassi E., Scacchi M., Cavagnini F. Neuroendocrine control of food intake. Nutrition, Metabolism and Cardiovascular Diseases. 2008;18(2):158–168. doi: 10.1016/j.numecd.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 27.Lee H.-K., Lee Y.-K., Park S.-H., et al. Structure and expression of the orphan nuclear receptor SHP gene. The Journal of Biological Chemistry. 1998;273(23):14398–14402. doi: 10.1074/jbc.273.23.14398. [DOI] [PubMed] [Google Scholar]
- 28.Båvner A., Sanyal S., Gustafsson J.-Å., Treuter E. Transcriptional corepression by SHP: molecular mechanisms and physiological consequences. Trends in Endocrinology & Metabolism. 2005;16(10):478–488. doi: 10.1016/j.tem.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Nakae J., Kitamura T., Kitamura Y., Biggs W. H., III, Arden K. C., Accili D. The forkhead transcription factor Fox01 regulates adipocyte differentiation. Developmental Cell. 2003;4(1):119–129. doi: 10.1016/S1534-5807(02)00401-X. [DOI] [PubMed] [Google Scholar]
- 30.Ruiz-Ramos R., Lopez-Carrillo L., Rios-Perez A. D., de Vizcaya-Ruíz A., Cebrian M. E. Sodium arsenite induces ROS generation, DNA oxidative damage, HO-1 and c-Myc proteins, NF-κB activation and cell proliferation in human breast cancer MCF-7 cells. Mutation Research. 2009;674(1-2):109–115. doi: 10.1016/j.mrgentox.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 31.Kauppinen A., Suuronen T., Ojala J., Kaarniranta K., Salminen A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cellular Signalling. 2013;25(10):1939–1948. doi: 10.1016/j.cellsig.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 32.Marsit C. J., Eddy K., Kelsey K. T. MicroRNA responses to cellular stress. Cancer Research. 2006;66(22):10843–10848. doi: 10.1158/0008-5472.CAN-06-1894. [DOI] [PubMed] [Google Scholar]
- 33.Yamakuchi M., Ferlito M., Lowenstein C. J. miR-34a repression of SIRT1 regulates apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(36):13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delgado J. M., Anand B. K. Increase of food intake induced by electrical stimulation of the lateral hypothalamus. American Journal of Physiology. 1953;172(1):162–168. doi: 10.1152/ajplegacy.1952.172.1.162. [DOI] [PubMed] [Google Scholar]
- 35.Nutrition classics. The Anatomical Record, Volume 78, 1940: hypothalamic lesions and adiposity in the rat. Nutrition Reviews. 1983;41(4):124–127. doi: 10.1111/j.1753-4887.1983.tb07169.x. [DOI] [PubMed] [Google Scholar]
- 36.Flier J. S. AgRP in energy balance: will the real AgRP please stand up? Cell Metabolism. 2006;3(2):83–85. doi: 10.1016/j.cmet.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 37.Shutter J. R., Graham M., Kinsey A. C., Scully S., Lüthy R., Stark K. L. Hypothalamic expression of ART, a novel gene related to agouti, is up-regulated in obese and diabetic mutant mice. Genes & Development. 1997;11(5):593–602. doi: 10.1101/gad.11.5.593. [DOI] [PubMed] [Google Scholar]
- 38.Ollmann M. M., Wilson B. D., Yang Y.-K., Kerns J. A., Chen Y., Gantz I., Barsh G. S. Antagonism of Central Melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278(5335):135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 39.Small C. J., Liu Y. L., Stanley S. A., et al. Chronic CNS administration of Agouti-related protein (Agrp) reduces energy expenditure. International Journal of Obesity. 2003;27(4):530–533. doi: 10.1038/sj.ijo.0802253. [DOI] [PubMed] [Google Scholar]
- 40.Kitamura T., Feng Y., Kitamura Y. I., et al. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nature Medicine. 2006;12(5):534–540. doi: 10.1038/nm1392. [DOI] [PubMed] [Google Scholar]
- 41.Morrison C. D., Morton G. J., Niswender K. D., Gelling R. W., Schwartz M. W. Leptin inhibits hypothalamic Npy and Agrp gene expression via a mechanism that requires phosphatidylinositol 3-OH-kinase signaling. The American Journal of Physiology—Endocrinology and Metabolism. 2005;289(6):E1051–E1057. doi: 10.1152/ajpendo.00094.2005. [DOI] [PubMed] [Google Scholar]
- 42.Leibel R. L. Molecular physiology of weight regulation in mice and humans. International Journal of Obesity. 2008;32(supplement 7):S98–S108. doi: 10.1038/ijo.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: The concentration of ALT and AST in blood of SA-treated db/db mice.
Supplemental Figure 2: Proliferation induced in SA-treated mouse hepatocytes.
Supplemental Figure 3: Glucose-stimulated insulin secretion test in SA-treated db/db mice.