

Abstract
Background
Mutations in ABCC8 and KCNJ11 are the most common cause of congenital hyperinsulinism (CHI). Recessive as well as dominant acting ABCC8/KCNJ11 mutations have been described. Diazoxide, which is the first line medication for CHI, is usually ineffective in recessive ABCC8 mutations. We describe the clinical and molecular characterisation of a recessive ABCC8 mutation in a CHI patient that is diazoxide response.
Clinical case
A term macrosomic female infant presented with symptomatic persistent hypoglycaemia confirmed to be secondary to CHI. She exhibited an excellent response to moderate doses of diazoxide (10 mg/kg/day). Molecular genetic analysis of the proband confirmed a biallelic ABCC8 mutation – missense R526C inherited from an unaffected mother and a frameshift c.1879delC mutation (H627Mfs*20) inherited from an unaffected father. Follow-up highlighted persistent requirement for diazoxide to control CHI. Functional analysis of mutants confirmed them to result in diazoxide-responsive CHI, consistent with the clinical phenotype.
Conclusion
Biallelic ABCC8 mutations may result in diazoxide-responsive CHI. Irrespective of the molecular genetic analysis results, accurate assessment of the response to diazoxide should be undertaken before classifying a patient as diazoxide-responsive or unresponsive CHI.
Keywords: Congenital hyperinsulinism, Hypoglycaemia, ABCC8, Diazoxide
Background
Congenital hyperinsulinism (CHI) is due to an inappropriate insulin secretion by the β-cells of the islets of Langerhans [1]. It usually presents with severe hypoketotic hypofattyacidaemic hypoglycaemia [2]. The majority of the affected newborns are macrosomic at birth and require high intravenous glucose administration to maintain plasma glucose above 3.5 mmol/l [3].
Mutations in ABCC8 and KCNJ11, which encode the SUR1 and Kir6.2 subunits of pancreatic ATP-sensitive potassium channel (KATP), are by far the most common cause of CHI and are estimated to account for 36%-69% of all cases [4–6]. KATP channels are octameric protein complexes composed of four pore-forming Kir6.2 subunits and four sulfonylurea receptor 1 (SUR1) subunits, and form a link between cellular metabolism and membrane excitability [7, 8]. It is thought that the β-cells in patients with CHI are persistently depolarized because of abnormally modulated or absent KATP channels. This depolarisation opens the voltage-gated calcium channels and leads to unregulated insulin exocytosis. Although dominant acting ABCC8/KCNJ11 mutations have been reported, recessively inherited mutations are more common [4, 5, 9, 10].
Diazoxide, which binds to the SUR1 subunit of the KATP channel and reduces insulin secretion by hyperpolarisation of the pancreatic β-cell plasma membrane, is the first line of treatment for CHI [1]. However, recessive inactivating mutations in ABCC8 and KCNJ11 usually cause severe diazoxide-unresponsive CHI due to defects in channel biogenesis, turnover, trafficking or regulation [11].
We describe a unique genotype-phenotype correlation with diazoxide responsive CHI in a patient with compound heterozygous ABCC8 mutation. Functional work on the mutants was consistent with the observed clinical phenotype.
Case presentation
Clinical case
A term large-for-gestational age (birth weight 4500 g at 39 weeks gestation) female infant born to non-consanguineous Caucasian parents presented with symptomatic hypoglycaemia on first day of life. There was no history of gestational diabetes mellitus in the mother. The proband developed tonic clonic seizures associated with laboratory blood glucose of 0.4 mmol/l at 22 hours of age. The infection and metabolic screen was negative. She required infusion of high concentration glucose (glucose infusion rate 16 mg/kg/minute) to maintain blood glucose above 3.5 mmol/l.
A controlled hypoglycaemia screen established the diagnosis of CHI (serum Insulin 44.5 mU/l associated with lab glucose of 2.3 mmol/l and undetectable non-esterified fatty acids and β-hydroxybutyrate). Her serum cortisol was 570 nmol/l during the hypoglycaemia screen. The rest of the hypoglycaemic screen including insulin like growth factor-1 (IGF1), and insulin-like growth factor binding protein-3 (IGFBP3), serum ammonia, lactate, acylcarnitine profile, plasma amino acids and urine organic acids was within normal reference range (results not shown).
Molecular genetic analysis for CHI was performed after informed consent from the parents (see below). She was commenced on diazoxide (5 mg/kg/day in three divided doses) and the dose was gradually increased to 10 mg/kg/day. Chlorothiazide was given along with diazoxide to counteract the side effect of fluid retention. On 10 mg/kg/day of diazoxide, she was successfully weaned off intravenous glucose administration. She demonstrated age appropriate fasting tolerance on diazoxide before discharge.
Blood glucose monitoring performed at home demonstrated satisfactory glycaemic control on diazoxide. No adjustment in diazoxide dose was required with her growth. At 9 months of age, a trial off diazoxide therapy resulted in recurrence of hypoglycaemia with fasting tolerance of only 3½ hours. Diazoxide was recommenced at 5 mg/kg/day, which led to disappearance of hypoglycaemia and age appropriate fasting tolerance.
At the time of writing, the proband is 15 months old and is able to fast for 12 hours without developing hypoglycaemia on a low dose of diazoxide (5 mg/kg/day). Neurodevelopmental assessment did not identify any abnormality.
Genetic analysis
Methods
Genomic DNA was extracted from peripheral blood leukocytes using standard procedures. The single exon of the KCNJ11 gene was amplified in 3 overlapping fragments and sequenced. When no mutations were identified in KCNJ11, the 39 exons of the ABCC8 gene were amplified by polymerase chain reaction (PCR). The products were sequenced using Big Dye Terminator cycler sequencing Kit v3.1 (Applied Biosystems, Warrington, UK) and sequencing reactions were analysed on an ABI3730 (Applied Biosystems, Warrington, UK). Sequences were compared to the reference sequence (NM_000352.2) using Mutation Surveyor software (SoftGenetics, Pa., USA).
Results
Sequence analysis identified biallelic ABCC8 mutation in the proband – a missense mutation, R526C, inherited from an unaffected mother and a frameshift mutation, c.1879delC (H627Mfs*20), inherited from an unaffected father. Both R526C and c.1879delC (H627Mfs*20) mutations have previously been reported as recessive acting mutations in patients with focal CHI [5].
Functional analysis of mutant channels
Methods
Single point mutations (R526C and c.1879delC) were introduced into the hamster SUR1 clone by PCR using the Strategene XL Mutagenesis Kit according to the manufacturer’s instructions.
Human Embryonic Kidney (HEK) 293 cells were transiently transfected with Kir6.2/SUR1, Kir6.2/SUR1c1879delC, Kir6.2/SUR1R526C or Kir6.2/SUR1c1879C + SUR1R526C (together with a small amount of eGFP (Green Fluorescence Protein) expressing plasmid to enable identification of transfected cells using epifluorescence) using FuGENE HD (Roche Diagnostics, UK) as per the manufacturers’ instructions and cells were subjected to whole cell patch-clamp 48 hours after transfection.
Whole-cell patch-clamp recordings were performed as previously described [12]. Capacitance transients and series resistance in whole-cell recordings were compensated electronically by using amplifier circuitry (Multiclamp 700B). Data were filtered at 1 kHz using the filter provided with the Multiclamp 700B (4 pole Bessel) and sampled at 5 kHz using a Digidata 1440 (Axon Instruments). Currents were acquired and analysed using pClamp 10.4 (Axon Instruments). The intracellular (pipette) solution contained (mM); 140 KCl, 1.2 MgCl2, 1 CaCl2, 10 EGTA and 5 HEPES, 0.1 mM Na.ATP and 1 mM Na.ADP, pH 7.2 using KOH. The bath solution contained (mM); 5 KCl, 140 NaCl, 2.6 CaCl2, 1.2 MgCl2 and 5 HEPES (pH 7.4). Pipette resistances were between 2–4 mΩ. Diazoxide and Tolbutamide were obtained from Sigma Aldrich (Poole, UK). Agents were applied to the bath using a gravity-driven perfusion system.
Results
Whole-cell patch-clamp recordings from HEK 293 cells transfected with wild-type (WT) Kir6.2/SUR1 cDNA showed normal KATP currents which was activated by the KATP channel opener diazoxide (100 μM) and inhibited by the KATP blocker tolbutamide (100 μM) (control, 144.77 ± 25.57 pA/pF; diazoxide, 382.7 ± 37.67 pA/pF; diazoxide + tolbutamide, 98.05 ± 28.05 pA/pF, n = 5 cells, P < 0.05). Currents from cells transfected with the frameshift mutation SUR1c1879delC were unresponsive to diazoxide and tolbutamide (control, 85.26 ± 12.7 pA/pF; diazoxide, 91.98 ± 14.36 pA/pF; tolbutamide, 79.57 ± 11.57 pA/pF, n = 7 cells, P > 0.05). In contrast, currents from cells transfected with the missense mutation, SUR1R526C, activated in the presence of diazoxide albeit to a lesser extent when compared to WT (control, 76.55 ± 8.2 pA/pF; diazoxide, 175.2 ± 16.47 pA/pF; tolbutamide, 64.55 ± 8.88 pA/pF, n = 5 cells, P < 0.05).
Interestingly, co-expression of SUR1c1879delC and SUR1R526C (to mimic the compound heterozygous ABCC8 (R256C/H627Mfs*20) mutation) rescued the diazoxide-sensitive KATP current which was absent when SUR1c1879delC was expressed alone (control, 62.9 ± 9.3 pA/pF; diazoxide, 223.11 ± 60.21 pA/pF; tolbutamide, 52.26 ± 11.36 pA/pF, n = 5 cells, P < 0.05). These data are shown in Figure 1. The diazoxide-sensitive KATP current in the double mutant transfected cells was significantly greater as compared to SUR1c1879delC transfected cells (160.2 ± 52.9 vs 8.72 ± 4.41 pA/pF, P = 0.01) and equivalent to WT Kir6.2/SUR1 transfected cells (160.2 ± 52.9 vs 237.9 ± 59.1 pA/pF, P = 0.33).
Figure 1.
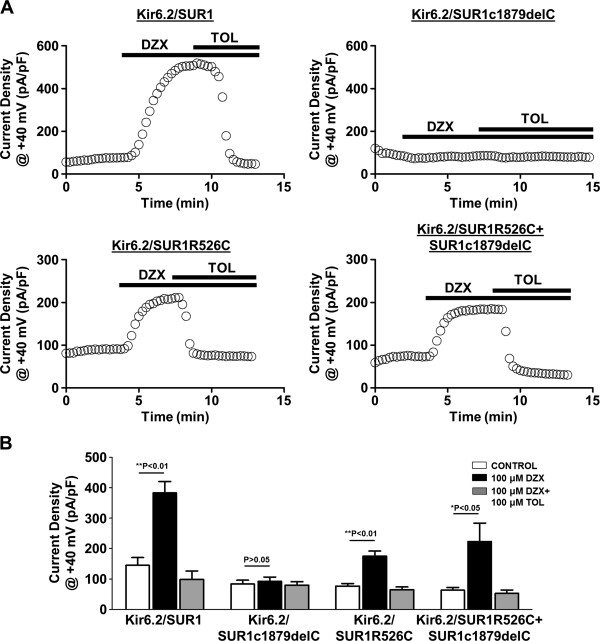
Functional characterisation of K ATP channels with a heterozygous ABCC8 R526C/H627Mfs*20 compound mutation. A, Representative whole-cell time-current density traces at +40 mV recorded from HEK293 cells expressing Kir6.2/SUR1, Kir6.2/SUR1c1879delC, Kir6.2/SUR1R526C and Kir6.2/SUR1R526C + SUR1C1879delC showing the effects of diazoxide (DZX) and tolbutamide (in the presence of diazoxide) (TOL). Current were recorded using a 1 s ramp protocol (-150 mV to 50 mV). B, Summary of the mean current-densities at +40 mV. Values are mean ± S.E.M from 5–7 cells, *P < 0.05, **P < 0.01 compared to control
There was no statistically significant difference in the KATP current seen in cells transfected with SUR1R256C alone and cells co-transfected with SUR1R256C and SUR1c1879delC (p = 0.291).
Discussion
We describe a patient with diazoxide responsive CHI due to compound heterozygous ABCC8 mutation. The proband was macrosomic at birth (consistent with foetal hyperinsulinism) and presented with severe hypoketotic hypoglycaemia requiring high glucose infusion (16 mg/kg/minute). Investigations confirmed CHI and compound heterozygous ABCC8 (R256C/H627Mfs*20) mutation. Surprisingly, the proband showed an excellent response to moderate doses of diazoxide (10 mg/kg/day). Subsequent follow-up revealed persistent requirement for diazoxide to control CHI. Functional analysis of the mutant KATP channel subunits confirmed a phenotype of diazoxide-responsive CHI in association with ABCC8 R526C/H627Mfs*20 compound heterozygous mutation.
Mutations in ABCC8 and KCNJ11, both monoallelic and biallelic, account for the majority of CHI patients [4, 5]. Although monoallelic ABCC8/KCNJ11 mutations can cause both diazoxide-responsive as well as diazoxide-unresponsive CHI, nearly all biallelic ABCC8/KCNJ11 mutations result in diazoxide unresponsive CHI [9, 10, 13, 14]. In two recent large studies comprising more than 700 patients with CHI, there was no patient reported with diazoxide responsive CHI due to biallelic ABCC8/KCNJ11 mutation [4, 5].
However, Dekel et al. reported a patient with a compound heterozygous ABCC8 mutation (c.3992-9G > A/F1388del) who responded to diazoxide [15]. However, no functional work was done to correlate with the clinical observations.
Previous functional work on compound heterozygous ABCC8 mutations has shown that mutations may interact to modify KATP channel function and influence disease severity. Muzyamba et al. showed that single SUR1 mutants (D1193V or R1436Q) trafficked to the plasma membrane whereas the double mutant (SUR1D1193V/R1436Q) was retained in the endoplasmic reticulum [16].
Both SUR1 R526C and H627Mfs*20 mutations have been described previously as presumed recessive acting mutations in association with CHI. Snider et al. reported one patient each with both these mutations in association with focal CHI [5]. No functional work had been done on either of these mutations. The H627Mfs*20 mutation is a frameshift mutation and is severely damaging to the protein function as shown by our functional work. When the SUR1H627Mfs*20 mutant was co-expressed with Kir6.2, there was no increase in current with the application of diazoxide indicating either absence of KATP channels on the plasma membrane surface and\or severely dysfunctional KATP channel. With the SUR1R526C mutant, there was reduced current flow under basal conditions as compared to wild-type. However, importantly, there was an increase in current with the application of diazoxide which persisted when the two mutants (SUR1R526C/H627Mfs*20) were expressed in combination, suggesting this variant to be diazoxide-responsive.
As the frameshift mutation H627Mfs*20 results in a premature termination codon and is likely to be degraded by non-sense mediated decay, it is possible that the KATP channels in double mutant SUR1R256C/H627Mfs*20 will contain SUR1 subunits produced by allele carrying R526C mutation only and hence the response shown by the SUR1R256C/H627Mfs*20 resembles that of SUR1R256C mutant. However non-sense mediated decay cannot be reproduced in the expression studies as that requires replication of intronic and exonic structure of the gene whereas the plasmids can only accommodate cDNA of the gene.
Although we used 100 μM diazoxide to assess the responsiveness of SUR1 mutants, to our knowledge there is no data in the literature as to what concentration of diazoxide is achieved at the cellular level with the standard doses of diazoxide used for medical management of CHI. However, assuming a distribution volume of ~0.2 L/kg, the dose of diazoxide administered orally to this proband is likely to result in a concentration higher (~2×) than the 100 μM we used for our studies at the cellular level [17].
Our functional data, however, is not consistent with the previous observation of diazoxide-unresponsive focal CHI in association with paternally inherited heterozygous ABCC8 R526C mutation [5]. Marked intrafamilial clinical heterogeneity in four haploidentical siblings harbouring the identical ABCC8 homozygous c.3992-9G > A mutation was also highlighted by Kapoor et al. [18]. This difference in clinical expression may be due to background genetic factors and other unknown factors involved in regulating gene expression.
Conclusion
In conclusion, although the majority of biallelic ABCC8/KCNJ11 mutations result in diazoxide-unresponsive CHI, occasional biallelic and particularly compound heterozygous ABCC8 mutations may lead to a diazoxide-responsive phenotype. Accurate clinical assessment would avoid the need for near-total pancreatectomy in such cases.
Consent
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Acknowledgements
The authors would like to acknowledge Dr Sarah E. Flanagan and Prof Sian Ellard from University of Exeter Medical School for contribution towards genetic testing. There is no source of funding for this manuscript preparation.
Abbreviations
- CHI
Congenital hyperinsulinism
- KATP
Potassium sensitive ATP channel
- SUR1
Sulfonylurea receptor 1
- IGF1
Insulin-like growth factor 1
- IGFBP3
IGF binding protein 3
- PCR
Polymerase chain reaction
- HEK 293 cells
Human Embryonic Kidney 293 cells
- GFP
Green Fluorescence Protein
- WT
Wild type.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
VBA contributed to acquisition of data and drafted the manuscript. QA performed the experiments, analyzed and interpreted the data and helped in drafting the manuscript. AN contributed to interpretation of data and revising it critically for important intellectual content. AT contributed to analysis and interpretation of data and revising it critically for important intellectual content. KH contributed to analysis and interpretation of data and revising it critically for important intellectual content. All authors read and approved the final manuscript.
Contributor Information
Ved Bhushan Arya, Email: v.arya@ucl.ac.uk.
Qadeer Aziz, Email: q.h.aziz@qmul.ac.uk.
Azizun Nessa, Email: azizun.nessa.10@ucl.ac.uk.
Andrew Tinker, Email: a.tinker@qmul.ac.uk.
Khalid Hussain, Email: khalid.hussain@ucl.ac.uk.
References
- 1.Aynsley-Green A, Hussain K, Hall J, Saudubray JM, Nihoul-Fekete C, De Lonlay-Debeney P, Brunelle F, Otonkoski T, Thornton P, Lindley KJ. Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed. 2000;82:F98–F107. doi: 10.1136/fn.82.2.F98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hussain K, Blankenstein O, De Lonlay P, Christesen HT. Hyperinsulinaemic hypoglycaemia: biochemical basis and the importance of maintaining normoglycaemia during management. Arch Dis Child. 2007;92:568–570. doi: 10.1136/adc.2006.115543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arya VB, Mohammed Z, Blankenstein O, De Lonlay P, Hussain K. Hyperinsulinaemic hypoglycaemia. Horm Metab Res. 2014;46:157–170. doi: 10.1055/s-0034-1367063. [DOI] [PubMed] [Google Scholar]
- 4.Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur J Endocrinol. 2013;168:557–564. doi: 10.1530/EJE-12-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, Ganapathy K, Bhatti T, Stanley CA, Ganguly A. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab. 2013;98:E355–E363. doi: 10.1210/jc.2012-2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demirbilek H, Arya VB, Ozbek MN, Akinci A, Dogan M, Demirel F, Houghton J, Kaba S, Guzel F, Baran RT, Unal S, Tekkes S, Flanagan SE, Ellard S, Hussain K. Clinical characteristics and phenotype-genotype analysis in Turkish patients with congenital hyperinsulinism; predominance of recessive KATP channel mutations. Eur J Endocrinol. 2014;170(6):885–892. doi: 10.1530/EJE-14-0045. [DOI] [PubMed] [Google Scholar]
- 7.Shyng S, Nichols CG. Octameric stoichiometry of the KATP channel complex. J Gen Physiol. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest. 2005;115:2047–2058. doi: 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kapoor RR, Flanagan SE, James CT, McKiernan J, Thomas AM, Harmer SC, Shield JP, Tinker A, Ellard S, Hussain K. Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/KCNJ11 mutations. Diabetologia. 2011;54:2575–2583. doi: 10.1007/s00125-011-2207-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huopio H, Reimann F, Ashfield R, Komulainen J, Lenko HL, Rahier J, Vauhkonen I, Kere J, Laakso M, Ashcroft F, Otonkoski T. Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. J Clin Invest. 2000;106:897–906. doi: 10.1172/JCI9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.James C, Kapoor RR, Ismail D, Hussain K. The genetic basis of congenital hyperinsulinism. J Med Genet. 2009;46:289–299. doi: 10.1136/jmg.2008.064337. [DOI] [PubMed] [Google Scholar]
- 12.Aziz Q, Thomas AM, Khambra T, Tinker A. Regulation of the ATP-sensitive potassium channel subunit, Kir6.2, by a Ca2+-dependent protein kinase. C. J Biol Chem. 2012;287:6196–6207. doi: 10.1074/jbc.M111.243923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinney SE, MacMullen C, Becker S, Lin YW, Hanna C, Thornton P, Ganguly A, Shyng SL, Stanley CA. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest. 2008;118:2877–2886. doi: 10.1172/JCI35414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Macmullen CM, Zhou Q, Snider KE, Tewson PH, Becker SA, Aziz AR, Ganguly A, Shyng SL, Stanley CA. Diazoxide-unresponsive congenital hyperinsulinism in children with dominant mutations of the beta-cell sulfonylurea receptor SUR1. Diabetes. 2011;60:1797–1804. doi: 10.2337/db10-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dekel B, Lubin D, Modan-Moses D, Quint J, Glaser B, Meyerovitch J. Compound heterozygosity for the common sulfonylurea receptor mutations can cause mild diazoxide-sensitive hyperinsulinism. Clin Pediatr (Phila) 2002;41:183–186. doi: 10.1177/000992280204100310. [DOI] [PubMed] [Google Scholar]
- 16.Muzyamba M, Farzaneh T, Behe P, Thomas A, Christesen HB, Brusgaard K, Hussain K, Tinker A. Complex ABCC8 DNA variations in congenital hyperinsulinism: lessons from functional studies. Clin Endocrinol (Oxf) 2007;67:115–124. doi: 10.1111/j.1365-2265.2007.02847.x. [DOI] [PubMed] [Google Scholar]
- 17.Ogilvie RI, Nadeau JH, Sitar DS. Diazoxide concentration-response relation in hypertension. Hypertension. 1982;4:167–173. doi: 10.1161/01.HYP.4.1.167. [DOI] [PubMed] [Google Scholar]
- 18.Kapoor RR, Flanagan SE, Ellard S, Hussain K. Congenital hyperinsulinism: marked clinical heterogeneity in siblings with identical mutations in the ABCC8 gene. Clin Endocrinol (Oxf) 2012;76:312–313. doi: 10.1111/j.1365-2265.2011.04203.x. [DOI] [PubMed] [Google Scholar]