

Abstract
Metastasis is responsible for the greatest number of cancer deaths. Metastatic disease, or the movement of cancer cells from one site to another, is a complex process requiring dramatic remodelling of the cell cytoskeleton. The various components of the cytoskeleton, actin (microfilaments), microtubules (MTs) and intermediate filaments, are highly integrated and their functions are well orchestrated in normal cells. In contrast, mutations and abnormal expression of cytoskeletal and cytoskeletal-associated proteins play an important role in the ability of cancer cells to resist chemotherapy and metastasize. Studies on the role of actin and its interacting partners have highlighted key signalling pathways, such as the Rho GTPases, and downstream effector proteins that, through the cytoskeleton, mediate tumour cell migration, invasion and metastasis. An emerging role for MTs in tumour cell metastasis is being unravelled and there is increasing interest in the crosstalk between key MT interacting proteins and the actin cytoskeleton, which may provide novel treatment avenues for metastatic disease. Improved understanding of how the cytoskeleton and its interacting partners influence tumour cell migration and metastasis has led to the development of novel therapeutics against aggressive and metastatic disease.
Linked Articles
This article is part of a themed section on Cytoskeleton, Extracellular Matrix, Cell Migration, Wound Healing and Related Topics. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2014.171.issue-24
Introduction
Metastasis accounts for the vast majority of cancer deaths (Schroeder et al., 2012) and is the result of movement of cancer cells from the primary site (site of origin of cancer) to a distant site or organ. The metastatic spread of cancer cells is a highly selective process consisting of a series of discrete, sequential steps, which have been modelled into a ‘metastatic cascade’. To generate metastatic lesions, tumour cells must successfully complete all steps of this process: detachment from the primary tumour → cell migration and invasion → intravasation – transport through vessels and anoikis (cell detachment-induced cell death) evasion – extravasation → growth of secondary tumour (Figure 1; reviewed in Steeg, 2006). Throughout this complex process, cell movements and alterations in cell shape require dramatic spatial and temporal reorganization of the cell cytoskeleton. The three major cytoskeletal filaments are the microfilaments (actin), intermediate filaments (IFs) and microtubules (MTs), and collectively, these provide and maintain cell shape and structure, and are key to important cellular events, including cell division, movement and vesicular transport. Although the three key components of the cytoskeleton are often studied independently, there is mounting evidence that clearly shows crosstalk and interdependency of the components. Consequently, a global view of the cytoskeleton in tumour cell migration and metastasis is of great interest. This review will provide an overview of cell migration, with a focus on the alterations in the cytoskeleton associated with metastasis. In addition, opportunities for the development of therapeutic inhibitors to target metastasis will be highlighted.
Figure 1.
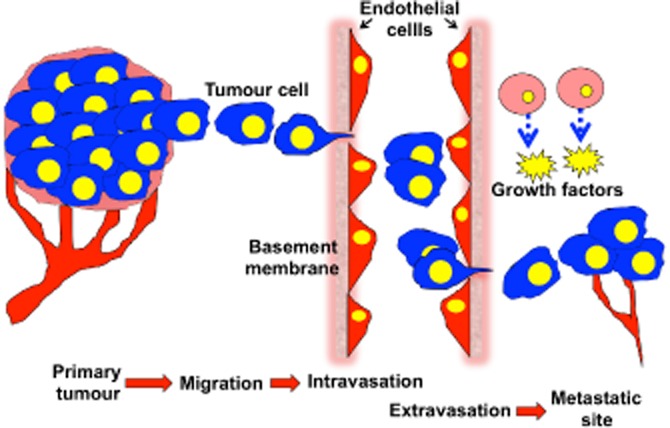
The metastatic cascade. For cancer cells to metastasize, they must successfully complete all of the steps of the metastatic cascade. (i) Cancer cells in the primary tumour acquire the ability to detach from the primary tumour and migrate through the surrounding ECM and stroma. (ii) Degradation of the vascular basement membrane and travel across the endothelium, termed intravasation. (iii) Tumour cells transport through the vasculature, arrest in a capillary bed and cross the vasculature (termed extravasation). (iv) Disseminated cells grow and interact with the extracellular environment to form metastatic tumours. Image modified from (Ara and DeClerck, 2006).
Cytoskeleton structure/function
MTs and microfilaments are polymer structures that orchestrate cellular movement, cell division, intracellular transport and signalling via an intricate cross-talking network that involves interaction with a diverse range of proteins and signalling molecules.
Actin
Microfilaments are composed of actin polymers and a large array of actin-binding proteins (ABPs). In cells, actin exists either in monomeric (G-actin) or polymeric forms (F-actin). Each actin subunit is able to bind ATP, which is hydrolysed to ADP during incorporation of the actin into a growing filament. Actin filaments consist of double helical polymers that are arranged head to tail. Most filaments also contain a tropomyosin (Tm) polymer that runs along the major groove in the microfilament (Gunning et al., 2008). The Tm does not have any van der Waals interactions with actin and, hence, ‘floats’ over the surface of the actin filament (Gunning et al., 2008). The actin cytoskeleton plays an important role in cell events such as motility, differentiation, division and membrane organization, all of which require the coordinated turnover and remodelling of actin filaments. The ability of actin filaments to contribute to this remarkable variety of activities derives from its selective interactions with ABPs, which, in turn, are regulated in part by the actin and Tm isoform composition of the filament (Gunning et al., 2008).
Microtubules
MTs are composed of α/β-tubulin heterodimers that self-associate into polymers. There are multiple α- and β-tubulin isotypes that display tissue- and developmental-specific expression (Luduena, 2013). The various β-tubulin isotypes are evolutionarily conserved across species and differ from each other predominantly in their carboxy-terminal region (Luduena, 2013). This region binds distinct microtubule-associated proteins and is therefore thought to influence MT stability and functionality.
MTs are highly dynamic structures that play an important role in cellular growth, vesicular transport and mitosis. The ability of MTs to polymerize and depolymerize in a regulated manner is essential for the segregation of chromosomes during mitosis. The assembly and disassembly of MTs occurs via GTP hydrolysis on the β-tubulin subunit of the α/β-tubulin heterodimer. In an interphase cell, MTs initiate from the centrosome, forming a hub and spoke-type network. This MT network is responsible for vesicular transport. During cell division, this network is completely remodelled to form the mitotic spindles, across which duplicate sets of chromosomes line up and are divided equally into two daughter cells. Upon completion of mitosis, the spindle is disassembled and the interphase MT network reforms (reviewed in Jordan and Wilson, 2004).
Intermediate filaments
IFs are the principal structural determinants within cells. Unlike the filament structures of F-actin and MTs, which are composed of highly conserved globular proteins, IFs can be formed from 40 different subunit proteins. They can be subdivided into five classes: keratins, neurofilaments, desmin, laminin and vimentin. The different types of IFs can be distinguished according to their localization and protein composition. IFs are linked to the extracellular matrix (ECM) and extend to the cytoplasmic interior that surrounds the nucleus. This extensive network allows IFs to coordinate cytoskeletal activities by relaying information from the cell surface to the inner compartments of the cell (Chang and Goldman, 2004).
Cell migration and metastasis
Metastasis is a complex and multifaceted process. Fundamental to the understanding of tumour cell metastasis is an appreciation of the process of cell motility, different manners in which tumour cells migrate and invade, and the role of the tumour microenvironment. While cell migration is necessary for numerous biological processes, such as embryonic morphogenesis, immune surveillance and tissue repair, aberrant control of cell migration promotes progression of many diseases, notably, cancer invasion and metastasis (reviewed in Yamaguchi and Condeelis, 2007). Broadly speaking, and as depicted in Figure 2, the process of cell motility can be broken down into four steps: protrusion, adhesion, contraction and retraction. Cells initiate cell motility, in response to an extracellular gradient of growth factors or chemokines, by polarizing and extending actin polymerization-driven cell membrane protrusions towards the extracellular cue (Small et al., 2002; Pollard and Borisy, 2003). The protrusions are then stabilized by adhesions linking the actin cytoskeleton to the ECM proteins and actomyosin contraction produces forces on the substratum. Contraction encourages the disassembly of adhesions at the rear of the cell, allowing retraction of the trailing cell body towards the direction of cell movement (Ridley et al., 2003). Cell motility (or migration), encompassing the above four steps, is often studied in a two-dimensional, planar manner where cells move towards a chemoattractant on an ECM utilizing filopodial and lamellipodial structures, whereas cell invasion is the movement of cells into or in a three-dimensional (3D) ECM matrix. Successfully crossing many of the physiological barriers to tumour cell metastasis (such as basement membranes) requires specialized structures, such as invadopodia and podosomes (reviewed in Yamaguchi and Condeelis, 2007). There exist different modes of cell migration, such as mesenchymal and amoeboid movement. Mesenchymal motility is associated with F-actin-rich protrusions where cell morphology is elongated, whereas amoeboid motility is a rounded bleb-associated mode of motility (Sahai and Marshall, 2003). Although aberrant cell migration promotes metastatic progression, the tumour microenvironment surrounding the tumour cells plays a key role in metastatic disease development. It is generally accepted that tumours above certain sizes contain populations of hypoxic cells. While hypoxia can influence the primary tumour response to drug treatment and subsequent acquired drug resistance, hypoxia plays a key role in tumour progression by modulating gene regulation and expression, such as hypoxia-inducible factor 1 (reviewed in Chaudary and Hill, 2007). Another aspect of the tumour microenvironment that has a role in tumour metastasis is inflammation. Dysregulation of the normal wound healing processes in cancer can result in an influx of angiogenic cytokines from nearby immune cells contributing to metastatic spread (reviewed in Finger and Giaccia, 2010). While hypoxia and inflammation play key roles in metastasis from within the primary tumour, the microenvironment of the ‘suitable’ metastatic site plays a critical role in regulating metastasis (reviewed in Langley and Fidler, 2011). Given the complicated nature of tumour cell migration and metastasis, it is not surprising that a complex array of regulatory processes exists, which enables the potential for treatment of metastatic disease by pharmacological intervention.
Figure 2.
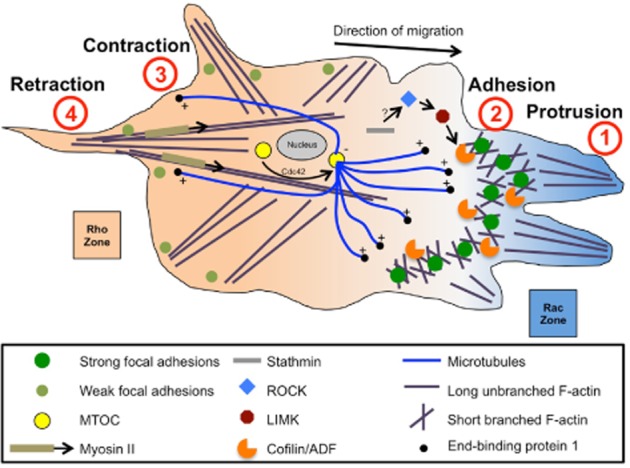
The cell cytoskeleton and four steps of cell migration. A cartoon schematic shows a migrating cell (direction of migration is indicated by the arrow). Four steps of cell migration shown: protrusion, adhesion, contraction, retraction. F-actin is shown in purple (short, branched F-actin at the leading edge, and long, unbranched F-actin stress fibres at the rear). Microtubules are shown in blue, with ends emanating from the MTOC (yellow circle) near the nucleus (grey ellipse). Strong and weak focal adhesions are shown as either dark or light green circles respectively. Cytoskeletal regulatory and associated proteins are shown (myosin II, cofilin/ADF, LIMK, ROCK, stathmin). The gradients of active Rho (orange) and Rac (blue) are shown. Adapted from Akhshi et al. (2014).
Cytoskeletal proteins in cell migration and metastasis
Actin and actin-regulating proteins in metastasis
Altered cellular motility is a hallmark feature of metastasis, facilitating the advancement of tumour cells to both local and distant sites in the body (reviewed in Hanahan and Weinberg, 2011). A key requirement of this process is the dynamic reorganization of the actin cytoskeleton. Reorganization of the actin cytoskeleton is critical for transdifferentiation of epithelial-like cells into motile mesenchymal-like cells, a process known as epithelial-mesenchymal transition (EMT), which is important in development, wound healing and cancer progression (Thiery et al., 2009). During EMT, cells reorganize their actin cytoskeleton, which enables dynamic cell elongation and directional motility, altogether increasing the migratory phenotype (reviewed in Lamouille et al., 2014). The leading edge of the mesenchymal-like migrating cells contains flat membranous lamellipodial protrusions, where protrusive force is generated by localized actin polymerization at the plasma membrane. Spatial and temporal regulation of this process is mediated by key cellular signalling events (Figure 3).
Figure 3.

Typical protrusive structures in invasive cancer cells. Cancer cell invasive phenotypes involve the formation of typical protrusive structures, such as plasma membrane blebs, invadopodia or pseudopodia, which are dependent on the nucleation and assembly of filamentous actin. Non-apoptotic blebs are highly dynamic protrusions in which the plasma membrane bulks out owing to increased hydrostatic pressure on regions of weak cortical actin (Fackler and Grosse, 2008). The initial, protruding bleb is devoid of detectable F-actin, which becomes repolymerized during bleb retraction by unknown actin nucleation factors. Ezrin is recruited into the growing bleb, and formins seem to have a role in bleb formation through mechanisms that still need to be defined (Charras and Paluch, 2008). Invadopodia are actin-rich cellular protrusions that are tailored for the degradation of the extracellular matrix. The formation of invadopodia relies on N-WASP–Arp2/3-driven actin assembly (Figure 4) and requires cortactin for invadopodia initiation and stabilization. Pseudopodia of cancer cells are lamellipodia-like structures and depend on the polymerization and assembly of actin by the WAVE–Arp2/3 nucleation machinery.
Rho GTPase as cytoskeletal regulators of migration
One of the major cellular signalling pathways involved in regulating actin and MT cytoskeletons is the Rho family GTPase signalling pathway. At the cell membrane, integrins, receptor tyrosine kinases, GPCRs (see Alexander et al., 2013a) and cadherins receive extracellular signals, which then influence the activity of Rho GTPase guanine nucleotide exchange factors (GEFs), which, in turn, influence the activity of the Rho GTPases (Figure 4 ).
Figure 4.
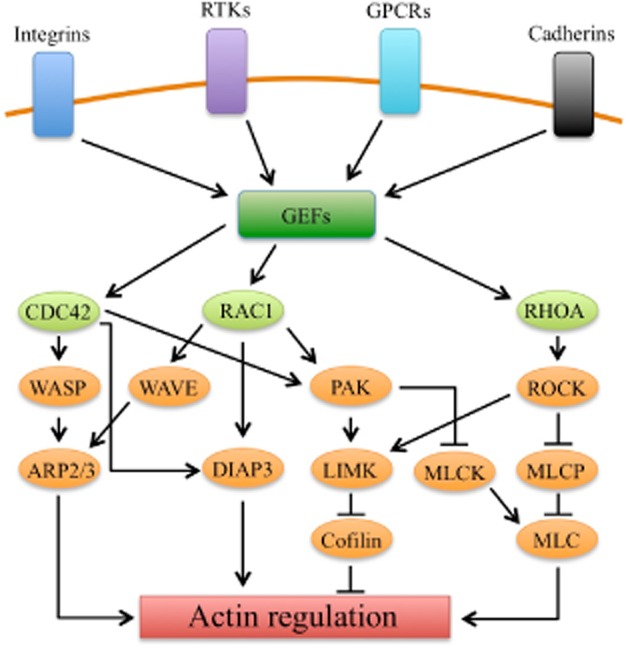
Rho GTPases and their effector proteins that mediate actin cytoskeletal regulation. Actin cytoskeletal regulation downstream of the Rho GTPases CDC42, RAC1 and Rho A (RHOA) is facilitated by numerous effector proteins. CDC42, via activation of WASP (Wiskott-Aldrich syndrome protein), activates the Arp2/3 complex result in actin polymerization and branched actin structures. RAC1 also activates the Arp2/3 via the WASP-relate WAVE (WASP family verprolin homologous protein) family of proteins. Both CDC42 and RAC1 activate DIAP3 (or mDIA2), resulting in unbranched actin filament nucleation. Additionally, CDC42 and RAC1 activate the PAK family kinases, which, via phosphorylation, activate LIMK (LIM domain kinase), which subsequently, via phosphorylation, inhibits cofilin. Cofilin facilitates actin filament severing and depolymerization; therefore, its inhibition results in elevated polymerized actin stability. Additionally, LIMK is also activated by ROCK (Rho-associated coiled-coil-containing protein kinase), which is a downstream kinase effector of Rho A. ROCK elevated MLC phosphorylation via the inhibition of myosin light chain phosphatase (MLCP). MLC phosphorylation results in its increased association with actin filaments. Lastly, PAK lessens ROCK function via MLC kinase (MLCK) inhibition, thus reducing MLC phosphorylation. Adapted from Tybulewicz and Henderson (2009).
Rho GTPases have strongly emerged as fundamental players in the control of several biochemical pathways underlying migration, such as cytoskeletal dynamics, directional sensing, cell–cell junction assembly/disassembly and integrin-matrix adhesion. Rho GTPases are a family of 20 small G proteins that, through the action of their downstream effector proteins, regulate the cytoskeleton influencing the cell cycle, cell polarity and cell migration (Jaffe and Hall, 2005). Importantly, human tumours from numerous cancer types show elevated expression of Rho GTPase genes, which correlate with an increased invasive and metastatic phenotype (reviewed in Karlsson et al., 2009).
Rho GTPases work as molecular switches existing in either an inactive, GDP-bound form or an active, GTP-bound form (Jaffe and Hall, 2005). Regulation of GTPase activity is a complex affair with mammalian Rho GTPase inactivation regulated by a family of 67 GTPase activating proteins, while activation is mediated by a family of 82 GEFs, which act downstream of many cell surface receptors such as growth factor receptors, integrins, cytokine receptors and cadherins (Rossman et al., 2005). In their active state, they interact with various different effector proteins to mediate the functions of Rho GTPases. The three most extensively examined Rho GTPases, Rho, Rac and Cdc42, regulate the assembly and organization of the actin cytoskeleton, and Cdc42 the microtubule cytoskeleton, in eukaryotic cells (reviewed in Hall, 2012). Broadly speaking, Rho can recruit the ROCK (also known as Rho-associated coiled-coil forming protein kinase, or Rho kinase) family of kinases (see Alexander et al., 2013b) that phosphorylate various cytoskeletal proteins promoting actin stress fibre formation and the generation of contractile force (reviewed in Hall, 2012); Rac reorganizes the actin cytoskeleton to promote formation of large membrane protrusions, called lamellipodia, which drive motility in many cell types; and Cdc42 signalling promotes the formation of actin-rich microspikes to sense extracellular chemotactic gradients and initiate directed cell movement. A major downstream effector of the Rho GTPase family of proteins is Rho kinase. Rho kinase is an effector of the small GTPase Rho and plays a key role in the regulation of actin remodelling, through the phosphorylation of cofilin and myosin light chain (MLC).
Recently, Eitaki et al. (2012) identified a key role for microtubules in the regulation of tumour cell mode of motility. Altered microtubule dynamics in gastric adenocarcinoma cells increases RhoA activity and subsequent ROCK signalling resulting in alterations to cytoskeletal organization and ultimate shifting to a amoeboid-like cell motility (Eitaki et al., 2012). It is believed that altered microtubule dynamics influences the mode of motility by releasing the Rho GEF-H1, which acts on RhoA regulating downstream MLC phosphorylation and contractility. The influence of microtubule dynamics on actin organization during mode of motility regulation highlights the interactions of microtubules and actin filaments. Collectively, accumulating biological and clinical data reveal that disruption of this signalling cascade is a common feature of metastatic cancer (Amin et al., 2013; Fortin Ensign et al., 2013).
Cofilin, cell motility and metastasis
Downstream of the Rho GTPase pathway, the cofilin pathway consisting of the actin-related protein 2/3 (ARP2/3) complex and cofilin plays a principal role in the generation of free actin filament ends, resulting in actin filament remodelling by polymerization and depolymerization within the lamellipodia (Edwards et al., 1999). The Arp2/3 complex facilitated by nucleation promoting factors, through initiation of dendritic nucleation (filament formation on the sides of pre-existing actin filaments), generates a branched actin network (Rotty et al., 2013). The Arp2/3 complex is involved in both directional cell motility and vesicular transport of cells (Rotty et al., 2013). The expression of the Arp2/3 complex-regulator protein WAVE2 has been observed to be highly correlated with greater metastatic risk in several cancer types (Sahai and Marshall, 2002; Wang et al., 2004; Vega and Ridley, 2008). Moreover, RNAi-mediated gene silencing of WAVE2 resulted in significantly reduced invasion and pulmonary metastasis of melanoma cells (Takahashi, 2012), while increased expression of cofilin has been observed in numerous malignancies (e.g. glioma, ovarian and lung cancer, and oral squamous cell carcinoma; Sinha et al., 1999; Gunnersen et al., 2000; Martoglio et al., 2000; Wang et al., 2004; Keshamouni et al., 2006; Turhani et al., 2006; Dowling et al., 2007) and is associated with chemotherapy resistance, invasive and metastatic disease.
Cofilin activity is inactivated via phosphorylation by LIM kinase 1 (LIMK) (Mouneimne et al., 2006). Together, LIMK, through cofilin inactivation, and phospholipase C (PLC), through cofilin activation, regulate cofilin in a synchronized manner to spatially restrict its activity (Mouneimne et al., 2006). The spatial restriction of cofilin activity is required for chemotaxis (directed cell movement towards a chemoattractant) as it leads to reorganization of the actin filaments, which is required for movement towards a chemoattractant such as a growth factor (Mouneimne et al., 2004; 2006,). The role of cofilin and its regulatory counterparts in chemotaxis highlights a possible role for cofilin activity regulation in the metastatic process. In support of this, it was observed that components from both the activation and the inactivation arms of cofilin activity regulation are overexpressed in numerous invasive carcinomas (Wang et al., 2004; 2007,). Hence, it is apparent that the invasive and metastatic behaviour of tumour cells, underpinned by actin reorganization, requires an optimal balance between cofilin and its regulatory components (Wang et al., 2007), highlighting this as a potential therapeutic pathway.
Congruous with the role of LIMK1 activity in the regulation of actin cytoskeletal dynamics, there is expanding evidence indicating an important role for LIMK1 in tumour cell invasion and metastasis. Elevated LIMK1 expression has been observed in numerous tumour types. Both in vitro and in vivo studies have confirmed a role for LIMK1 in this process, as down-regulation of LIMK1 activity is associated with a decreased invasive phenotype, and conversely, overexpression of LIMK1 activity is associated with increased invasion (Bagheri-Yarmand et al., 2006; Horita et al., 2008; Ross-Macdonald et al., 2008; Scott et al., 2010). Additionally, the influence of elevated LIMK1 levels on cell motility and invasion is reversed by overexpression of cofilin (Wang et al., 2006); therefore, it is apparent that the LIMK1/cofilin ratio determines the cellular response and that small changes in actin dynamics could either increase or decrease invasiveness (Scott et al., 2010). The potential to target LIMK activity, through inhibition of its kinase activity, may lead to reduced cancer cell motility and invasion.
The activity of LIMK proteins is controlled for the most part by phosphorylation of the Rho GTPase effector proteins Rho-associated kinase 1 and 2 (ROCK1 and ROCK2) (reviewed in Bernard, 2007). In addition to LIMK and cofilin phosphorylation, active ROCK exerts its influence on cell migration by affecting MLC phosphorylation and hence activity. ROCK phosphorylates MLC directly, although the principal effect of ROCK on MLC phosphorylation is its ability to block the dephosphorylation of MLC via MLC phosphatase (MLCP) inhibition (reviewed in Mierke et al., 2008). The increase in MLC phosphorylation induced by ROCK, contributes to actin reorganization and stress fibre formation (reviewed in Maekawa et al., 1999); these, in turn, are important for invasive cell behaviour during metastasis (Yee et al., 2001; Wyckoff et al., 2006; Mierke et al., 2008). Altogether, ROCK signalling influences various aspects of actin cytoskeletal remodelling during cell migration and metastasis. Disrupting ROCK signalling, either indirectly or via its downstream targets, represents a promising pharmacological target for treating metastatic disease.
Focal adhesions (FAs): connecting the cytoskeleton and extracellular matrix
Actomyosin is a contractile complex made up of actin and myosin that mediates cell spreading and adhesion to the ECM, typically through the formation of prominent bundles of F-actin, the so-called microfilament bundles or stress fibres. These terminate on the inner face of the membrane at certain contact areas known as focal contacts (FCs) or FAs to form a vital connection between the ECM and the cytoskeleton (reviewed in Pellegrin and Mellor, 2007). The interplay between actomyosin contractile forces and FA dynamics results in a balance between adhesion and contraction, which greatly influences migration velocity (Gupton and Waterman-Storer, 2006). It is believed that the asymmetry of the microtubule network coordinates the spatial regulation of FA dynamics at the cell front, centre and rear. Targeting of microtubule growth towards FAs behind the leading edge results in FA disassembly (reviewed in Broussard et al., 2008). Proteins that require microtubules for proper localization and activity, such as the tyrosine kinase Arg, can influence Rho GTPase activity, resulting in the inhibition of FA turnover at the cell front and rear (Peacock et al., 2007). Importantly, the tearing off of adhesions by cell retraction, controlled largely by myosin contractility, at the cell rear may be microtubule dependent.
FAs are sites of transmembrane integrin clustering consisting of many structural and signalling proteins (reviewed in Gupton and Waterman-Storer, 2006). FA formation is preceded by a cytoskeletal precursor consisting of a bundle of actin filaments, or microspike, orientated radially within the leading edge of a cell (reviewed in Nobes and Hall, 1995). During FA formation, several structural and regulatory proteins are recruited to the complex, such as vinculin and paxillin. Vimentin has been recognized as a key player in cell adhesion. For example, endothelial cells exhibit vimentin-rich focal contacts, and upon vimentin suppression, the size of focal contacts and concurrent adhesion strength is sharply reduced (Tsuruta and Jones, 2003). Interestingly, recent studies have highlighted a strong collaboration between the intermediate-filament protein vimentin and the ABP filamin in regulating adhesion supporting the notion that various cytoskeletal components interact to influence cellular processes such as cell adhesion (Kim et al., 2010a,b,). The regulatory proteins that have been identified in FAs have been the subject of extensive investigation, not only as models for studying the links between the ECM and the cytoskeleton but also as sites of transmembrane communication between the extracellular environment and the cytoplasm during tumour cell migration and metastasis.
A widely examined FA regulatory protein is focal adhesion kinase (FAK). FAK is a tyrosine kinase that acts as an integrator protein, responding to several extracellular inputs and controlling numerous signalling pathway outputs. This role as an integrator can be seen in its activation by an array of different extracellular factors. FAK activity is controlled by integrin-mediated cell adhesion and by being a downstream target of growth factors and G-protein-linked receptors (Schlaepfer and Mitra, 2004). Migrating cells experience changes in mechanical force, which influence cytoskeletal organization and cell migration (Huang and Ingber, 1999). FAK is important in the sensing of these mechanical forces; therefore, FAK activation is involved in regulating cell responses to environmental stimuli to influence tumour cell migration (Katsumi et al., 2004). FAK does this, at least in part, through signal-mediated effects on actin organization. Namely, it is the involvement of FAK with activators and/or inhibitors of the small GTPase RhoA that enable changes in FAK activity to be connected to alterations in the polymerization or stabilization of actin filaments during tumour cell adhesion and motility (Rico et al., 2004; Lim et al., 2008). This role of FAK in controlling actin-remodelling dynamics during tumour cell adhesion and motility is congruous with the observations that perturbed FAK expression and activity correlate with increased clinical progression to highly malignant and metastatic phenotypes (de Vicente et al., 2013).
As tumour cells lose contact with the basement membrane during invasion and proceeding intravasation into blood and lymph vessels, and extravasation at distant sites (refer also to Figure 1), they hit a critical barrier against metastasis: anoikis (apoptotic cell death induced by loss or inadequate cell adhesion in anchorage-dependent cells). Normal cells embedded in an ECM undergo an apoptotic response upon loss of cell–cell and cell–matrix interactions (reviewed in Cordes and van Beuningen, 2003). In normal physiology, anoikis is thought to ensure homeostasis, for example, colon epithelial cells undergo apoptosis upon reaching the top of the villi, or during post-lactation mammary gland involution (reviewed in Geiger and Peeper, 2009). Similar to its roles in development and homeostasis, anoikis hinders metastasis by inducing apoptosis when tumour cells enter ‘foreign’ environments; therefore, suppression of anoikis is likely to be necessary for tumour cells to metastasize successfully. Consequently, the restoration of anoikis sensitivity may help limit the spread of metastatic tumours.
Tropomyosins (Tms) and metastasis
The Tms are a family of ABPs that form head-to-tail polymers in the major groove of polymerized actin filaments. Various Tm isoforms have been observed to inhibit both the rates of polymerization and depolymerization of actin (reviewed in Perry, 2001). Consequently, Tm suppress actin dynamics; this arises because Tms mechanically stabilize the filaments by inhibiting actin filament branching and nucleation, resulting in less depolymerization and in fewer actin filament ends available for polymerization (reviewed in Gunning et al., 2008). (Tm1), belonging to the Tm family of proteins, is an actin-associated protein that plays a pivotal role in the spatial and temporal regulation of actin filaments and loss of Tm1 in breast cancer confers resistance to anoikis (Bharadwaj et al., 2005). A number of other members of the tropomyosin family have been observed to play a role in various aspects of metastasis biology. The low MW tropomyosin 5 non-muscle isoform 1 (Tm5NM1), a predominant cytoskeletal tropomyosin isoform in primary tumours and tumour cell lines, is believed to be associated with regulating mesenchymal cell motility (Lees et al., 2011; Stehn et al., 2013). Tm5NM1 influences mesenchymal cell motility via its effects on actin filament dynamics and organization. A number of studies have shown that high Tm5NM1 expression inhibits cell migration and invasion, whereas a decrease in Tm5NM1 expression increases cell migration (reviewed in Gunning et al., 2008). In addition, Tm5NM1 is necessary for the motility of highly metastatic melanoma and increased expression of another Tm isoform, Tm4, is associated with breast cancer lymph node metastasis (reviewed in Gunning et al., 2008). There is prominent and concurrent stabilization of FAs with Tm5NM1-mediated stabilization of actin filaments (Bach et al., 2010). Tm5NM1 stabilizes FAs and promotes the alteration to fibrillar adhesions via actin filament stabilization. Tm5NM1 expression is a critical factor in paxillin phosphorylation, which is vital for FA disassembly. Taken together, it has been proposed that Tm5NM1 regulates FA assembly and disassembly at the leading edge, which is required for directed cell movement (Bach et al., 2009). Tropomyosin has recently been identified as a new therapeutic target for cancer (Stehn et al., 2013) and will be discussed in a latter section of this review.
Intermediate filaments and metastasis
IFs are 10-nm-thick cytoskeletal structures formed by the self-assembly of members of the IF superfamily of proteins that are encoded by about 70 genes and classified into 5 classes (reviewed in Sihag et al., 2007). While the vast majority of IF research focuses on diseases other than cancer, evidence exists indicating exciting insights into the involvement of IFs in tumour cell metastasis. Connections between the cytoskeleton and plasma membrane are vital in controlling cell migration (reviewed in Pan et al., 2008). IFs provide prominent connections in epithelial cells, where keratin IFs are anchored at the cell–cell junction desmosomes and hemidesmosomes, and in myocytes, where desmin IFs are secured at costameres (Green and Jones, 1996; Capetanaki et al., 2007). While many proteins (such as FAK and Tm5NM1) influence tumour cell migration largely via their control of cell–ECM interactions, cell–cell interactions are also imperative and are commonly aberrantly regulated during metastasis (Hanahan and Weinberg, 2011).
Nestin is an IF protein that has been reported as being overexpressed in various tumour types. Additionally, nestin expression correlates with aggressive growth and metastasis, and poor prognosis in pancreatic and prostate cancer, melanoma and glioblastoma (reviewed in Ishiwata et al., 2011). Despite these findings, nestin's role in cancer cells has not been well characterized. Another IF that has been observed to be overexpressed in various cancers and correlates with tumour invasion and poor prognosis is the IF vimentin (Satelli and Li, 2011). Although similar to nestin, the role of vimentin in cancer progression remains obscure. Despite this, Satelli and Li (2011) discovered a vimentin-binding mini-peptide, which has potential as a vimentin-targeted tumour-specific therapy. However, more knowledge is required before the suitability of this potential therapy can be assessed for metastatic disease. Little is known about the biology of IF proteins in tumour cell migration, invasion and metastasis. Further research is required to elucidate whether these proteins may prove viable targets for pharmacological intervention in cancer.
Microtubule system: emerging role in metastasis
In addition to the abundance of literature on the involvement of the actin cytoskeleton and its regulatory partners in tumour cell migration, invasion and metastasis, there is increasing evidence highlighting the microtubule system in these processes. Indeed, a number of signalling pathways such as the Rho GTPase pathway is common, or overlapping, in terms of regulation of microtubule and actin structures to mediate cell movement and other cellular events. In effect, although often studied independently, these two cytoskeletal systems are coordinately regulated. The functions of interphase and mitotic MTs are dependent on their assembly and stability, which are influenced by multiple factors including tubulin isotype composition, tubulin post-translational modifications, and interactions with various stabilizing and destabilizing proteins (reviewed in Luduena, 2013). As mentioned in the preceding sections, MTs are made up of α/β-tubulin heterodimers and there are numerous α- and β-tubulin isotypes. β-Tubulin is the cellular target of important anticancer agents such as the vinca alkaloids and taxanes (reviewed in Jordan and Wilson, 2004). Alterations in specific β-tubulin isotypes, particularly the neuronally expressed βΙΙΙ-tubulin, in epithelial cancers, are associated with resistance to tubulin-binding agent chemotherapy and more aggressive disease (Kavallaris, 2010). Our research has demonstrated direct functional roles for specific β-tubulin isotypes in sensitivity to chemotherapeutic agents (Gan et al., 2007; Gan and Kavallaris, 2008; McCarroll et al., 2010). Of interest is our recent finding that depletion of βΙΙΙ-tubulin using gene silencing with RNAi leads to decreased tumour incidence (McCarroll et al., 2010). It is possible that this tubulin isotype may also play a role in metastasis in epithelial cancer. Some support for this possibility comes from a recent clinical study that expression of βIII-tubulin in stage II non-small cell lung cancer patients was associated with overall survival and lymphatic metastasis (Jiang et al., 2013).
Interphase microtubules operate as tracks to carry vesicles and organelles to various sub-cellular compartments (reviewed in Wittmann and Waterman-Storer, 2001). Motor proteins, such as the kinesin and dynein families, assist in cargo transport along microtubules to various sub-cellular locations, such as cilia, axons and importantly, the leading edge of migrating cells (Verhey and Gaertig, 2007). Disruptions in the levels of proteins involved in vesicular transport have been reported in cancer cells. For example, Yoon et al. (2005) observed that hypoxia increased MDA-MB-231 breast carcinoma invasion by modulating an important vesicular trafficking protein Rab11. Rab11-mediated trafficking contributes to invasion through transporting integrin α6β4 to the plasma membrane. Importantly, hypoxia-induced Rab11 trafficking is regulated by microtubule stability, supported by findings that hypoxia results in increased glutamylated (Glu) tubulin (stable microtubules) and that the microtubule polymerization inhibitor, colchicine, blocks Rab11 trafficking (Yoon et al., 2005).
An important microtubule-interacting protein, which regulates microtubule dynamics, is end-binding protein 1 (EB1). EB1, a microtubule plus-end binding protein, co-localizes and interacts with both cytoplasmic and spindle microtubules in interphase and mitotic cells respectively. Functionally, EB1 acts as a negative inhibitor of microtubule stability. EB1 promotes tumour cell migration by contributing to stable detyrosinated microtubules (Glu-MTs) (reviewed in Zhang et al., 2009). In addition to EB1, ATIP3 is involved in the regulation of microtubule dynamics. ATIP3 is a potent microtubule-stabilizing protein whose depletion increases microtubule dynamics (Molina et al., 2013). ATIP3 was reported to be a prognostic marker for overall survival in breast cancer, where low ATIP3 levels in metastatic tumours are associated with decreased patient survival (Rodrigues-Ferreira et al., 2009). Functionally, ATIP3 was found to mediate cell motility and directionality, and influence the number and size of metastases (Molina et al., 2013). By reducing microtubule dynamics, ATIP3 regulates the ability of microtubule tips to reach the cell cortex during migration, which may account for decreased cancer cell motility and metastasis.
Recent developments in imaging technology have begun to unearth how the cytoskeleton is asymmetrically distributed during migration. In addition to the critical roles of the Rho family GTPases in the regulation of the actin cytoskeleton during cell migration, recent studies have revealed that Rho family GTPases also control the dynamics of microtubules, and, in turn, microtubules influence the activities of Rho family GTPases (reviewed in Akhshi et al., 2014). The Rho family GTPase, Cdc42, is important for directional cell migration (Etienne-Manneville and Hall, 2001). Directional cell migration, key to the metastatic process, is initiated usually in response to extracellular cues, resulting in the formation of a highly polarized cell characterized by the asymmetrical distribution of signalling molecules and the cytoskeleton. Notably, during cell migration, Cdc42, most active towards the front of migrating cells, controls microtubule-organizing centre (MTOC) and Golgi apparatus reorientation towards the migrating direction where stabilization of microtubule plus ends occurs (reviewed in Vaughan and Dawe, 2011).
Cytoskeletal crosstalk
Recent observations highlight a role for FAs as key sites where the actin and microtubule cytoskeletons crosstalk. The molecular mechanisms by which microtubules and actin communicate to contribute to cell migration are complex. Initial studies utilizing microtubule-depolymerizing drugs, including colchicine, observed that an intact microtubule array is necessary for directional movement of fibroblasts and endothelial cells (reviewed in Kaverina and Straube, 2011). Intriguingly, cells exposed to microtubule inhibitors displayed delocalization of actin-based lamellipodia and membrane ruffles from the leading edge of migrating cells (Palazzo and Gundersen, 2002), leading to the belief that microtubules act to limit actin cytoskeletal activity to specified cellular locations. Very low concentrations of microtubule drugs, which inhibit microtubule dynamics without influencing microtubule organization or length, negatively affect lamellipodia formation, fibroblast migration and endothelial cell protrusion in 3D matrices (reviewed in Martins and Kolega, 2012). Altogether, these findings suggest that microtubule dynamics contribute to actin cytoskeletal regulation and potentially metastasis.
Further evidence of cross-communication and signalling between the microtubule and actin cytoskeleton involves the microtubule-interacting protein stathmin. Stathmin, a 19 kDa cytosolic phosphoprotein, is a microtubule destabilizing protein (Sobel and Tashjian, 1983). Growing evidence highlights a significant role for stathmin in tumour cell migration and metastasis. Stathmin overexpression correlates with metastatic disease progression and survival (Kuo et al., 2009). Additionally, stathmin plays a role in the migration of various healthy and cancer cells. Stathmin promotes cell movement by assisting in new leading edge microtubule growth in motile cells (Niethammer et al., 2004; Wittmann et al., 2004). Stathmin is highly expressed in a number of cancer types, including breast, leukaemia and neuroblastoma (Belletti and Baldassarre, 2011). It is associated with advanced and aggressive disease in neuroblastoma (Hailat et al., 1990) and suppression of this protein significantly reduced in vitro cell migration and invasion, and importantly, lung metastasis in an orthotopic xenograft model of neuroblastoma (Byrne et al., 2014). In this same study, stathmin suppression was found to modulate the phosphorylation of the actin-regulatory proteins, cofilin and MLC via ROCK signalling (Byrne et al., 2014). Moreover, stathmin has been linked to RhoA/ROCK signalling through its interactions with p27kip1, a member of the Cip/Kip family of cyclin-dependent kinase inhibitors (Baldassarre et al., 2005). Further studies are required to understand the mechanism of how stathmin influences tumour cell migration and metastasis. Our studies implicating stathmin in cell migration and metastasis clearly highlights crosstalk between the microtubule and actin cytoskeleton (Byrne et al., 2014). Stathmin's action on microtubule dynamics, while not fully elucidated, appears to influence tumour cell migration, invasion and metastasis via its effects on the actin cytoskeleton. Approaches of this nature, whereby the actin cytoskeleton can be targeted indirectly via the microtubule cytoskeleton, may allow pharmacological intervention that could potentially avoid many of the toxic effects of some actin-based therapies.
Metastasis: targeting the cytoskeleton
Before the cytoskeleton can be considered as a target in metastasis, the parts of the metastatic cascade that are suitable, and in particular most amenable to therapeutic intervention need to be determined. Steeg (2006) has demonstrated, by classifying patients as either exhibiting local, regional or distant disease, that the interruption of the metastatic process could be useful for the majority of individuals with cancer. Additionally, the authors argued that the last steps in the metastatic process (such as colonization and angiogenesis in distant secondary sites) may represent potential priority steps for therapeutic intervention and that this finding may allow more focused research (Steeg, 2006).
Gaining an understanding of how the cytoskeleton is involved in tumour cell migration, invasion and metastasis has lead to increased research and attention to exploit the cytoskeleton as a target to treat metastatic disease.
Targeting cytoskeletal signalling pathways
One protein described in this review that is involved in cytoskeletal regulation and tumour cell migration and metastasis is ROCK. Recent studies have identified a remarkably diverse variety of functions of ROCK, such as regulation of cell morphology, motility, gene transcription, proliferation, differentiation and apoptosis. These findings may influence the manner in which ROCK inhibitors may be utilized in cancer therapy. It has been proposed that Rho-ROCK signalling, and its subsequent downstream cytoskeletal changes, promotes metastasis by supporting rounded blebbing-associated tumour cell movement. ROCK inhibition has been found to decrease the invasive and metastatic behaviour of tumour cells (Sahai and Marshall, 2003; Patel et al., 2014). Hence, therapies that target Rho or ROCK function may be effective in inhibiting the invasive behaviour of rounded tumour cells. Despite this, tumour cells have been observed to overcome inhibitors for a specific mode of motility by switching to a different mode (Sahai and Marshall, 2003). Combinations of inhibitors, targeting both modes of motility, may result in a synergistic decrease in tumour cell invasion and metastasis. While tumour cells have been observed to overcome past ROCK inhibitors, it is currently unclear whether tumour cells have the ability to overcome newer generations of ROCK inhibitors, such as those developed by Patel et al. (2014), where they presently warrant advanced preclinical studies.
Despite the early promise of pharmacological ROCK inhibition for the treatment of cancer, no compounds are clinically approved. Nevertheless, careful definition of key cancer types and expansion of the understanding of ROCK's role in cancer may one day lead to the application of ROCK inhibitors in cancer therapy. Notably, the observation that tumour cells have the ability to overcome ROCK inhibitors by switching from one mode of motility to a different mode, thus rendering the ROCK inhibitor ineffective has hampered development in this field (Rath and Olson, 2012). Targeting proteins involved in ROCK signalling is a potential alternative to targeting ROCK directly. Pharmacological inhibitors of the downstream effector of ROCK, LIMK, are being developed. A number of LIMK inhibitors have been reported to suppress cancer cell invasion (Ross-Macdonald et al., 2008; Prudent et al., 2012). For example, Prudent et al. (2012) identified a small-molecule LIMK inhibitor in a cell-based screen that induced a microtubule-stabilizing effect that was independent of any direct effects on the actin organization. The researchers concluded that LIMK functions as a signalling relay that regulates the dynamics of both actin and microtubules (Prudent et al., 2012). LIMK has also been implicated in resistance to tubulin-targeting agents in neuroblastoma (Po'uha et al., 2010), and the development of inhibitors of this pathway as a mode of chemosensitization of drug-resistant tumour cells is an attractive possibility. Given the role of LIMK in tumour metastasis and resistance, the development of targeted therapies against LIMK represents an attractive approach to treat cancer.
Targeting cytoskeletal and cytoskeletal-associated proteins
Given the fundamental nature of the actin cytoskeleton in multiple key aspects of tumour cell migration, invasion and metastasis, targeting the actin cytoskeleton may be an effective approach to develop novel therapeutics for metastatic disease. Pharmacological inhibitors of actin have failed clinical development due to non-specific targeting of actin in normal tissues leading to high levels of cardiotoxicity. Recently, Stehn et al. (2013) developed a first-in-class pharmacological inhibitor that targets Tm5NM1. Treatment of cancer cells with the lead compound TR100 disrupted Tm5NM1-containing actin filament populations and impaired tumour cell motility and viability. Importantly, TR100 exhibited no major adverse effects on cardiac structure and function, a common problem encountered with anti-actin compounds (Stehn et al., 2013). This provides an elegant proof of principle in how targeting specific components of the cell architecture can compromise the survival of tumour cells and lead to the development of novel treatment approaches for cancer.
In cancers resistant to tubulin-targeting agents, γ-actin has been identified as altered (Verrills et al., 2006a,b,) via, in part, effects on microtubule dynamics (Po'uha et al., 2013), and modulation of this protein in drug naïve cancer cells changed directional cell migration via alterations in ROCK signalling (Shum et al., 2011). These findings reveal opportunities to exploit the crosstalk and interactions between actin and microtubules in metastasis and drug resistance by dual targeting of specific components of the network.
Tubulin-targeting agents remain one of the most successful groups of agents in the clinic (Dumontet and Jordan, 2010). Although many studies focus on the anti-mitotic effects of these agents, it is becoming increasingly accepted that the efficacy of these agents is due to their ability to target both mitotic and interphase cells (Komlodi-Pasztor et al., 2011). Tubulin-targeting agents act not only as potent anti-cancer drugs but also as anti-angiogenic, and in some cases anti-vascular agents, and this has led to their use in metronomic chemotherapy to treat drug refractory and metastatic disease (reviewed in Pasquier et al., 2010). Epothilones are a new class of tubulin-targeting agents that have demonstrated clinical efficacy and are approved for the treatment of drug refractory metastatic breast cancer in the USA (Cristofanilli, 2012). A recently approved tubulin-targeting agent that has demonstrated clinical activity in drug refractory and metastatic cancer is eribulin (McBride and Butler, 2012). Although eribulin is known to suppress microtubule dynamics, its efficacy in drug refractory and metastatic disease is not well understood. Yoshida et al., (2014) found that eribulin exerted effects on EMT and mesenchymal-epithelial transition in human breast cancer cells in both in vitro and in vivo models. It is possible that other tubulin-targeting agents also exert broader effects on cell migration, invasion and metastasis and exploiting these in combination with actin pathway inhibitors is worthy of further study.
Conclusions
Metastasis is a complex process requiring dramatic reorganization of the cytoskeleton. Numerous proteins interacting with the actin and microtubule cytoskeletons, directly or indirectly, have been shown to significantly influence the migratory and metastatic phenotype of tumour cells. Emerging evidence on crosstalk between different components of the cytoskeleton in metastasis highlights the fact that the actin, IF and MT cytoskeletons do not work in isolation but are inextricably linked together in tumour cell migration and metastasis. Importantly, the strong relationship between cytoskeletal alterations and metastasis in the clinic (Table 1) provides important opportunities for potential therapeutic targets. The highly complex nature of cellular migration and invasion presents challenges in developing therapeutics where compensatory pathways may overcome the action of specific inhibitors. These challenges also provide opportunities to develop combination therapies to target multiple pathways associated with the cytoskeleton and treat drug refractory and metastatic cancer.
Table 1.
Relationship between cytoskeletal-associated and regulatory proteins with metastatic disease
| Group | Gene | Cancer type | Alteration | Additional information | Refs. |
|---|---|---|---|---|---|
| Cofilin/ADF | CFL1 | Breast | Down-regulation (RNA) | Decreased expression in lymph node metastases | Hao et al., 2004 |
| ATIP3 | MTUS1 | Breast | Down-regulation (RNA) | Decreased expression among metastatic tumours associated with DFS | Rodrigues-Ferreira et al., 2009 |
| Arp2/3 | ACTR2 | Breast | Up-regulation (protein) | Increased lymph node metastases and reduced DFS and OS | Iwaya et al., 2007a |
| Colorectal | Up-regulation (mRNA and protein) | Increased hepatic metastases | Iwaya et al., 2007b | ||
| Formins | DIAPH3 | Prostatic | Increased chromosomal deletion containing DIAPH3 gene | Di Vizio et al., 2009 | |
| FMNL2 | Colorectal | Up-regulation (mRNA and protein) | Increased lymph node metastases | Zhu et al., 2008 | |
| WASP | WASL | Breast | Down-regulation (mRNA and protein) | Increased lymph node metastases and reduced DFS and OS | Martin et al., 2008 |
| ESCC | Up-regulation (protein) | Increased lymph node metastases | Wang et al., 2010a | ||
| Colorectal | Up-regulation (mRNA) | Increased expression in liver metastases | Yanagawa et al., 2001 | ||
| WAVE | WASF2 | Colorectal | Up-regulation (mRNA and protein) | Increased hepatic metastases | Iwaya et al., 2007b |
| Lung | Up-regulation (protein) | Increased lymph node metastasis and reduced DFS and OS | Semba et al., 2006 | ||
| Cortactin | CTTN | Colorectal | Up-regulation (protein) | Increased distal metastasis and reduced OS | Lee et al., 2009; Cai et al., 2010 |
| ESCC | Up-regulation (mRNA and protein) | Increased lymph node metastasis | Luo et al., 2006 | ||
| Gastric | Up-regulation (protein) | Increased lymph node metastasis and reduced DFS and OS | Li et al., 2008; Wang et al., 2010b | ||
| Hepatocellular | Up-regulation (protein) | Increased distal metastasis | Chuma et al., 2004 | ||
| HNSCC | Up-regulation (mRNA and protein) | Increased lymph node and distal metastasis and reduced DFS and OS | Takes et al., 1997; Gibcus et al., 2008; Hsu et al., 2009; Rodrigo et al., 2009 | ||
| Melanoma | Up-regulation (protein) | Increased distal metastasis and reduced DFS | Xu et al., 2010 | ||
| OSCC | Up-regulation (protein) | Increased lymph node metastasis | Yamada et al., 2010 | ||
| Ena/VASP | ENAH | Breast | Up-regulation (protein) | Increased expression in metastases | Di Modugno et al., 2006 |
| Colorectal | Up-regulation (protein) | Increased lymph node metastasis | Toyoda et al., 2009 | ||
| ERM | VIL2 | Osteosarcoma | Up-regulation (protein) | Increased expression in metastases | Khanna et al., 2001; 2004, |
| Rhabdomyosarcoma | Up-regulation (protein) | Increased expression in metastases | Yu et al., 2004 | ||
| RAS | RhoC | Breast | Up-regulation (protein) | Expression strongly correlates with advanced stage disease | Kleer et al., 2002 |
| Bladder | Up-regulation (protein) | Increased expression in lymph node metastases and reduced DFS and OS | Kamai et al., 2003 | ||
| HNSCC | Up-regulation (mRNA) | Increased expression in metastatic stage III and IV disease | Schmalbach et al., 2004 | ||
| Ovarian | Up-regulation (mRNA) | Increased expression in metastatic tumours and increased expression in metastatic stage III and IV disease | Horiuchi et al., 2003 | ||
| RhoA | Bladder | Up-regulation (mRNA) | Increased expression in lymph node metastases and reduced DFS and OS | Kamai et al., 2003 | |
| Ovarian | Up-regulation (mRNA) | Increased expression in metastatic tumours and increased expression in metastatic stage III and IV disease | Horiuchi et al., 2003 | ||
| Testicular | Up-regulation (protein) | Increased expression in higher stage disease | Kamai et al., 2004 | ||
| AGC | ROCK | Bladder | Up-regulation (protein) | Increased expression in lymph node metastases and reduced DFS and OS | Kamai et al., 2003 |
| Testicular | Up-regulation (protein) | Increased expression in higher stage disease | Kamai et al., 2004 | ||
| STMN | Stathmin | Ovarian | Up-regulation (mRNA) | Increased expression in metastases | Wei et al., 2008 |
| Endometrial | Up-regulation (protein) | Increased expression in lymph node metastases | Trovik et al., 2011 |
DFS, disease-free survival; ESCC, oesophageal squamous cell carcinoma; HNSCC, head and neck squamous cell carcinoma; OS, overall survival; OSCC, oral squamous cell carcinoma.
Acknowledgments
The authors are supported by the Children's Cancer Institute Australia for Medical Research, which is affiliated with the University of New South Wales (UNSW) and Sydney Children's Hospital and by grants from the National Health and Medical Research Council (NHMRC) (M. K. & J. A. M.), Cancer Council New South Wales (M. K.), Kids Cancer Project (M. K.), Cancer Institute New South Wales Career Development Fellowship (J. A. M.) and NHMRC Senior Research Fellowship (M. K.). C. M. F. is supported by a Postgraduate Scholarship from the Australian Centre for NanoMedicine, UNSW.
Glossary
- ABP
actin-binding protein
- EB1
end-binding protein 1
- ECM
extracellular matrix
- FA
focal adhesion
- F-actin
filamentous actin
- FC
focal contact
- GEF
guanine nucleotide exchange factor
- G-actin
monomeric actin
- IF
intermediate filament
- LIMK
LIM kinase
- MLC
myosin light chain
- MT
microtubule
- MTOC
microtubule-organizing centre
- ROCK
Rho-associated coiled-coil forming protein kinase
- Tm
tropomyosin
- Tm5NM1
tropomyosin 5 non-muscle isoform 1
Author contributions
C. M. F., J. A. M. and M. K. planned and prepared the review outline. C. M. F. produced the first draft and all three authors were involved in writing or revising sections of the review.
Conflict of interest
None.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhshi TK, Wernike D, Piekny A. Microtubules and actin crosstalk in cell migration and division. Cytoskeleton (Hoboken) 2014;71:1–23. doi: 10.1002/cm.21150. [DOI] [PubMed] [Google Scholar]
- Amin E, Dubey BN, Zhang SC, Gremer L, Dvorsky R, Moll JM, et al. Rho-kinase: regulation, (dys)function, and inhibition. Biol Chem. 2013;394:1399–1410. doi: 10.1515/hsz-2013-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ara T, DeClerck YA. Mechanisms of invasion and metastasis in human neuroblastoma. Cancer Metastasis Rev. 2006;25:645–657. doi: 10.1007/s10555-006-9028-9. [DOI] [PubMed] [Google Scholar]
- Bach CT, Creed S, Zhong J, Mahmassani M, Schevzov G, Stehn J, et al. Tropomyosin isoform expression regulates the transition of adhesions to determine cell speed and direction. Mol Cell Biol. 2009;29:1506–1514. doi: 10.1128/MCB.00857-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach CT, Schevzov G, Bryce NS, Gunning PW, O'Neill GM. Tropomyosin isoform modulation of focal adhesion structure and cell migration. Cell Adh Migr. 2010;4:226–234. doi: 10.4161/cam.4.2.10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri-Yarmand R, Mazumdar A, Sahin AA, Kumar R. LIM kinase 1 increases tumor metastasis of human breast cancer cells via regulation of the urokinase-type plasminogen activator system. Int J Cancer. 2006;118:2703–2710. doi: 10.1002/ijc.21650. [DOI] [PubMed] [Google Scholar]
- Baldassarre G, Belletti B, Nicoloso MS, Schiappacassi M, Vecchione A, Spessotto P, et al. p27(Kip1)-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell. 2005;7:51–63. doi: 10.1016/j.ccr.2004.11.025. [DOI] [PubMed] [Google Scholar]
- Belletti B, Baldassarre G. Stathmin: a protein with many tasks. New biomarker and potential target in cancer. Expert Opin Ther Targets. 2011;15:1249–1266. doi: 10.1517/14728222.2011.620951. [DOI] [PubMed] [Google Scholar]
- Bernard O. Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol. 2007;39:1071–1076. doi: 10.1016/j.biocel.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Bharadwaj S, Thanawala R, Bon G, Falcioni R, Prasad GL. Resensitization of breast cancer cells to anoikis by tropomyosin-1: role of Rho kinase-dependent cytoskeleton and adhesion. Oncogene. 2005;24:8291–8303. doi: 10.1038/sj.onc.1208993. [DOI] [PubMed] [Google Scholar]
- Broussard JA, Webb DJ, Kaverina I. Asymmetric focal adhesion disassembly in motile cells. Curr Opin Cell Biol. 2008;20:85–90. doi: 10.1016/j.ceb.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Byrne FA, Yang Y, Phillips AA, Hansford LM, Fletcher JI, Ormandy CJ, et al. RNAi-mediated stathmin suppression reduces lung metastasis in an orthotopic neuroblastoma mouse model. Oncogene. 2014;33:882–890. doi: 10.1038/onc.2013.11. [DOI] [PubMed] [Google Scholar]
- Cai JH, Zhao R, Zhu JW, Jin XL, Wan FJ, Liu K, et al. Expression of cortactin correlates with a poor prognosis in patients with stages II-III colorectal adenocarcinoma. J Gastrointest Surg. 2010;14:1248–1257. doi: 10.1007/s11605-010-1247-2. [DOI] [PubMed] [Google Scholar]
- Capetanaki Y, Bloch RJ, Kouloumenta A, Mavroidis M, Psarras S. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp Cell Res. 2007;313:2063–2076. doi: 10.1016/j.yexcr.2007.03.033. [DOI] [PubMed] [Google Scholar]
- Chang L, Goldman RD. Intermediate filaments mediate cytoskeletal crosstalk. Nat Rev Mol Cell Biol. 2004;5:601–613. doi: 10.1038/nrm1438. [DOI] [PubMed] [Google Scholar]
- Charras G, Paluch E. Blebs lead the way: how to migrate without lamellipodia. Nat Rev Mol Cell Biol. 2008;9:730–736. doi: 10.1038/nrm2453. [DOI] [PubMed] [Google Scholar]
- Chaudary N, Hill RP. Hypoxia and metastasis. Clin Cancer Res. 2007;13:1947–1949. doi: 10.1158/1078-0432.CCR-06-2971. [DOI] [PubMed] [Google Scholar]
- Chuma M, Sakamoto M, Yasuda J, Fujii G, Nakanishi K, Tsuchiya A, et al. Overexpression of cortactin is involved in motility and metastasis of hepatocellular carcinoma. J Hepatol. 2004;41:629–636. doi: 10.1016/j.jhep.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Cordes N, van Beuningen D. Cell adhesion to the extracellular matrix protein fibronectin modulates radiation-dependent G2 phase arrest involving integrin-linked kinase (ILK) and glycogen synthase kinase-3beta (GSK-3beta) in vitro. Br J Cancer. 2003;88:1470–1479. doi: 10.1038/sj.bjc.6600912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofanilli M. Advancements in the treatment of metastatic breast cancer (MBC): the role of ixabepilone. J Oncol. 2012;2012:703858. doi: 10.1155/2012/703858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Modugno F, Mottolese M, Di Benedetto A, Conidi A, Novelli F, Perracchio L, et al. The cytoskeleton regulatory protein hMena (ENAH) is overexpressed in human benign breast lesions with high risk of transformation and human epidermal growth factor receptor-2-positive/hormonal receptor-negative tumors. Clin Cancer Res. 2006;12:1470–1478. doi: 10.1158/1078-0432.CCR-05-2027. [DOI] [PubMed] [Google Scholar]
- Di Vizio D, Kim J, Hager MH, Morello M, Yang W, Lafargue CJ, et al. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009;69:5601–5609. doi: 10.1158/0008-5472.CAN-08-3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling P, Meleady P, Dowd A, Henry M, Glynn S, Clynes M. Proteomic analysis of isolated membrane fractions from superinvasive cancer cells. Biochim Biophys Acta. 2007;1774:93–101. doi: 10.1016/j.bbapap.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- Eitaki M, Yamamori T, Meike S, Yasui H, Inanami O. Vincristine enhances amoeboid-like motility via GEF-H1/RhoA/ROCK/Myosin light chain signaling in MKN45 cells. BMC Cancer. 2012;12:469. doi: 10.1186/1471-2407-12-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001;106:489–498. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- Fackler OT, Grosse R. Cell motility through plasma membrane blebbing. J Cell Biol. 2008;181:879–884. doi: 10.1083/jcb.200802081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev. 2010;29:285–293. doi: 10.1007/s10555-010-9224-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin Ensign SP, Mathews IT, Symons MH, Berens ME, Tran NL. Implications of Rho GTPase signaling in glioma cell invasion and tumor progression. Front Oncol. 2013;3:241. doi: 10.3389/fonc.2013.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan PP, Kavallaris M. Tubulin-targeted drug action: functional significance of class ii and class IVb beta-tubulin in vinca alkaloid sensitivity. Cancer Res. 2008;68:9817–9824. doi: 10.1158/0008-5472.CAN-08-1501. [DOI] [PubMed] [Google Scholar]
- Gan PP, Pasquier E, Kavallaris M. Class III beta-tubulin mediates sensitivity to chemotherapeutic drugs in non small cell lung cancer. Cancer Res. 2007;67:9356–9363. doi: 10.1158/0008-5472.CAN-07-0509. [DOI] [PubMed] [Google Scholar]
- Geiger TR, Peeper DS. Metastasis mechanisms. Biochim Biophys Acta. 2009;1796:293–308. doi: 10.1016/j.bbcan.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Gibcus JH, Mastik MF, Menkema L, de Bock GH, Kluin PM, Schuuring E, et al. Cortactin expression predicts poor survival in laryngeal carcinoma. Br J Cancer. 2008;98:950–955. doi: 10.1038/sj.bjc.6604246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KJ, Jones JC. Desmosomes and hemidesmosomes: structure and function of molecular components. FASEB J. 1996;10:871–881. doi: 10.1096/fasebj.10.8.8666164. [DOI] [PubMed] [Google Scholar]
- Gunnersen JM, Spirkoska V, Smith PE, Danks RA, Tan SS. Growth and migration markers of rat C6 glioma cells identified by serial analysis of gene expression. Glia. 2000;32:146–154. [PubMed] [Google Scholar]
- Gunning P, O'Neill G, Hardeman E. Tropomyosin-based regulation of the actin cytoskeleton in time and space. Physiol Rev. 2008;88:1–35. doi: 10.1152/physrev.00001.2007. [DOI] [PubMed] [Google Scholar]
- Gupton SL, Waterman-Storer CM. Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration. Cell. 2006;125:1361–1374. doi: 10.1016/j.cell.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Hailat N, Strahler J, Melhem R, Zhu XX, Brodeur G, Seeger RC, et al. N-myc gene amplification in neuroblastoma is associated with altered phosphorylation of a proliferation related polypeptide (Op18) Oncogene. 1990;5:1615–1618. [PubMed] [Google Scholar]
- Hall A. Rho family GTPases. Biochem Soc Trans. 2012;40:1378–1382. doi: 10.1042/BST20120103. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hao X, Sun B, Hu L, Lahdesmaki H, Dunmire V, Feng Y, et al. Differential gene and protein expression in primary breast malignancies and their lymph node metastases as revealed by combined cDNA microarray and tissue microarray analysis. Cancer. 2004;100:1110–1122. doi: 10.1002/cncr.20095. [DOI] [PubMed] [Google Scholar]
- Horita Y, Ohashi K, Mukai M, Inoue M, Mizuno K. Suppression of the invasive capacity of rat ascites hepatoma cells by knockdown of Slingshot or LIM kinase. J Biol Chem. 2008;283:6013–6021. doi: 10.1074/jbc.M706538200. [DOI] [PubMed] [Google Scholar]
- Horiuchi A, Imai T, Wang C, Ohira S, Feng Y, Nikaido T, et al. Up-regulation of small GTPases, RhoA and RhoC, is associated with tumor progression in ovarian carcinoma. Lab Invest. 2003;83:861–870. doi: 10.1097/01.lab.0000073128.16098.31. [DOI] [PubMed] [Google Scholar]
- Hsu KF, Lin CK, Yu CP, Tzao C, Lee SC, Lee YY, et al. Cortactin, fascin, and survivin expression associated with clinicopathological parameters in esophageal squamous cell carcinoma. Dis Esophagus. 2009;22:402–408. doi: 10.1111/j.1442-2050.2008.00921.x. [DOI] [PubMed] [Google Scholar]
- Huang S, Ingber DE. The structural and mechanical complexity of cell-growth control. Nat Cell Biol. 1999;1:E131–E138. doi: 10.1038/13043. [DOI] [PubMed] [Google Scholar]
- Ishiwata T, Matsuda Y, Naito Z. Nestin in gastrointestinal and other cancers: effects on cells and tumor angiogenesis. World J Gastroenterol. 2011;17:409–418. doi: 10.3748/wjg.v17.i4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaya K, Norio K, Mukai K. Coexpression of Arp2 and WAVE2 predicts poor outcome in invasive breast carcinoma. Mod Pathol. 2007a;20:339–343. doi: 10.1038/modpathol.3800741. [DOI] [PubMed] [Google Scholar]
- Iwaya K, Oikawa K, Semba S, Tsuchiya B, Mukai Y, Otsubo T, et al. Correlation between liver metastasis of the colocalization of actin-related protein 2 and 3 complex and WAVE2 in colorectal carcinoma. Cancer Sci. 2007b;98:992–999. doi: 10.1111/j.1349-7006.2007.00488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Jiang H, Yu XM, Zhou XM, Wang XH, Su D. Correlation between microtubule-associated gene expression and chemosensitivity of patients with stage II non-small cell lung cancer. Exp Ther Med. 2013;5:1506–1510. doi: 10.3892/etm.2013.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Kamai T, Tsujii T, Arai K, Takagi K, Asami H, Ito Y, et al. Significant association of Rho/ROCK pathway with invasion and metastasis of bladder cancer. Clin Cancer Res. 2003;9:2632–2641. [PubMed] [Google Scholar]
- Kamai T, Yamanishi T, Shirataki H, Takagi K, Asami H, Ito Y, et al. Overexpression of RhoA, Rac1, and Cdc42 GTPases is associated with progression in testicular cancer. Clin Cancer Res. 2004;10:4799–4805. doi: 10.1158/1078-0432.CCR-0436-03. [DOI] [PubMed] [Google Scholar]
- Karlsson R, Pedersen ED, Wang Z, Brakebusch C. Rho GTPase function in tumorigenesis. Biochim Biophys Acta. 2009;1796:91–98. doi: 10.1016/j.bbcan.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Katsumi A, Orr AW, Tzima E, Schwartz MA. Integrins in mechanotransduction. J Biol Chem. 2004;279:12001–12004. doi: 10.1074/jbc.R300038200. [DOI] [PubMed] [Google Scholar]
- Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10:194–204. doi: 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- Kaverina I, Straube A. Regulation of cell migration by dynamic microtubules. Semin Cell Dev Biol. 2011;22:968–974. doi: 10.1016/j.semcdb.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshamouni VG, Michailidis G, Grasso CS, Anthwal S, Strahler JR, Walker A, et al. Differential protein expression profiling by iTRAQ-2DLC-MS/MS of lung cancer cells undergoing epithelial-mesenchymal transition reveals a migratory/invasive phenotype. J Proteome Res. 2006;5:1143–1154. doi: 10.1021/pr050455t. [DOI] [PubMed] [Google Scholar]
- Khanna C, Khan J, Nguyen P, Prehn J, Caylor J, Yeung C, et al. Metastasis-associated differences in gene expression in a murine model of osteosarcoma. Cancer Res. 2001;61:3750–3759. [PubMed] [Google Scholar]
- Khanna C, Wan X, Bose S, Cassaday R, Olomu O, Mendoza A, et al. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat Med. 2004;10:182–186. doi: 10.1038/nm982. [DOI] [PubMed] [Google Scholar]
- Kim H, Nakamura F, Lee W, Hong C, Perez-Sala D, McCulloch CA. Regulation of cell adhesion to collagen via beta1 integrins is dependent on interactions of filamin A with vimentin and protein kinase C epsilon. Exp Cell Res. 2010a;316:1829–1844. doi: 10.1016/j.yexcr.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Kim H, Nakamura F, Lee W, Shifrin Y, Arora P, McCulloch CA. Filamin A is required for vimentin-mediated cell adhesion and spreading. Am J Physiol Cell Physiol. 2010b;298:C221–C236. doi: 10.1152/ajpcell.00323.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleer CG, van Golen KL, Zhang Y, Wu ZF, Rubin MA, Merajver SD. Characterization of RhoC expression in benign and malignant breast disease: a potential new marker for small breast carcinomas with metastatic ability. Am J Pathol. 2002;160:579–584. doi: 10.1016/S0002-9440(10)64877-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo T. Mitosis is not a key target of microtubule agents in patient tumors. Nat Rev Clin Oncol. 2011;8:244–250. doi: 10.1038/nrclinonc.2010.228. [DOI] [PubMed] [Google Scholar]
- Kuo MF, Wang HS, Kuo QT, Shun CT, Hsu HC, Yang SH, et al. High expression of stathmin protein predicts a fulminant course in medulloblastoma. J Neurosurg Pediatr. 2009;4:74–80. doi: 10.3171/2009.2.PEDS08287. [DOI] [PubMed] [Google Scholar]
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley RR, Fidler IJ. The seed and soil hypothesis revisited–the role of tumor-stroma interactions in metastasis to different organs. Int J Cancer. 2011;128:2527–2535. doi: 10.1002/ijc.26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YY, Yu CP, Lin CK, Nieh S, Hsu KF, Chiang H, et al. Expression of survivin and cortactin in colorectal adenocarcinoma: association with clinicopathological parameters. Dis Markers. 2009;26:9–18. doi: 10.3233/DMA-2009-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees JG, Bach CT, Bradbury P, Paul A, Gunning PW, O'Neill GM. The actin-associating protein Tm5NM1 blocks mesenchymal motility without transition to amoeboid motility. Oncogene. 2011;30:1241–1251. doi: 10.1038/onc.2010.516. [DOI] [PubMed] [Google Scholar]
- Li X, Zheng H, Hara T, Takahashi H, Masuda S, Wang Z, et al. Aberrant expression of cortactin and fascin are effective markers for pathogenesis, invasion, metastasis and prognosis of gastric carcinomas. Int J Oncol. 2008;33:69–79. [PubMed] [Google Scholar]
- Lim Y, Lim ST, Tomar A, Gardel M, Bernard-Trifilo JA, Chen XL, et al. PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J Cell Biol. 2008;180:187–203. doi: 10.1083/jcb.200708194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luduena RF. A hypothesis on the origin and evolution of tubulin. Int Rev Cell Mol Biol. 2013;302:41–185. doi: 10.1016/B978-0-12-407699-0.00002-9. [DOI] [PubMed] [Google Scholar]
- Luo ML, Shen XM, Zhang Y, Wei F, Xu X, Cai Y, et al. Amplification and overexpression of CTTN (EMS1) contribute to the metastasis of esophageal squamous cell carcinoma by promoting cell migration and anoikis resistance. Cancer Res. 2006;66:11690–11699. doi: 10.1158/0008-5472.CAN-06-1484. [DOI] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- Martin TA, Pereira G, Watkins G, Mansel RE, Jiang WG. N-WASP is a putative tumour suppressor in breast cancer cells, in vitro and in vivo, and is associated with clinical outcome in patients with breast cancer. Clin Exp Metastasis. 2008;25:97–108. doi: 10.1007/s10585-007-9120-8. [DOI] [PubMed] [Google Scholar]
- Martins GG, Kolega J. A role for microtubules in endothelial cell protrusion in three-dimensional matrices. Biol Cell. 2012;104:271–286. doi: 10.1111/boc.201100088. [DOI] [PubMed] [Google Scholar]
- Martoglio AM, Tom BD, Starkey M, Corps AN, Charnock-Jones DS, Smith SK. Changes in tumorigenesis- and angiogenesis-related gene transcript abundance profiles in ovarian cancer detected by tailored high density cDNA arrays. Mol Med. 2000;6:750–765. [PMC free article] [PubMed] [Google Scholar]
- McBride A, Butler SK. Eribulin mesylate: a novel halichondrin B analogue for the treatment of metastatic breast cancer. Am J Health Syst Pharm. 2012;69:745–755. doi: 10.2146/ajhp110237. [DOI] [PubMed] [Google Scholar]
- McCarroll JA, Gan PP, Liu M, Kavallaris M. betaIII-tubulin is a multifunctional protein involved in drug sensitivity and tumorigenesis in non-small cell lung cancer. Cancer Res. 2010;70:4995–5003. doi: 10.1158/0008-5472.CAN-09-4487. [DOI] [PubMed] [Google Scholar]
- Mierke CT, Rosel D, Fabry B, Brabek J. Contractile forces in tumor cell migration. Eur J Cell Biol. 2008;87:669–676. doi: 10.1016/j.ejcb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina A, Velot L, Ghouinem L, Abdelkarim M, Bouchet BP, Luissint AC, et al. ATIP3, a novel prognostic marker of breast cancer patient survival, limits cancer cell migration and slows metastatic progression by regulating microtubule dynamics. Cancer Res. 2013;73:2905–2915. doi: 10.1158/0008-5472.CAN-12-3565. [DOI] [PubMed] [Google Scholar]
- Mouneimne G, Soon L, DesMarais V, Sidani M, Song X, Yip SC, et al. Phospholipase C and cofilin are required for carcinoma cell directionality in response to EGF stimulation. J Cell Biol. 2004;166:697–708. doi: 10.1083/jcb.200405156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouneimne G, DesMarais V, Sidani M, Scemes E, Wang W, Song X, et al. Spatial and temporal control of cofilin activity is required for directional sensing during chemotaxis. Curr Biol. 2006;16:2193–2205. doi: 10.1016/j.cub.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Niethammer P, Bastiaens P, Karsenti E. Stathmin-tubulin interaction gradients in motile and mitotic cells. Science. 2004;303:1862–1866. doi: 10.1126/science.1094108. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Palazzo AF, Gundersen GG. Microtubule-actin cross-talk at focal adhesions. Sci STKE. 2002;2002:pe31. doi: 10.1126/stke.2002.139.pe31. [DOI] [PubMed] [Google Scholar]
- Pan Y, Jing R, Pitre A, Williams BJ, Skalli O. Intermediate filament protein synemin contributes to the migratory properties of astrocytoma cells by influencing the dynamics of the actin cytoskeleton. FASEB J. 2008;22:3196–3206. doi: 10.1096/fj.08-106187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier E, Kavallaris M, Andre N. Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol. 2010;7:455–465. doi: 10.1038/nrclinonc.2010.82. [DOI] [PubMed] [Google Scholar]
- Patel RA, Liu Y, Wang B, Li R, Sebti SM. Identification of novel ROCK inhibitors with anti-migratory and anti-invasive activities. Oncogene. 2014;33:550–555. doi: 10.1038/onc.2012.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock JG, Miller AL, Bradley WD, Rodriguez OC, Webb DJ, Koleske AJ. The Abl-related gene tyrosine kinase acts through p190RhoGAP to inhibit actomyosin contractility and regulate focal adhesion dynamics upon adhesion to fibronectin. Mol Biol Cell. 2007;18:3860–3872. doi: 10.1091/mbc.E07-01-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrin S, Mellor H. Actin stress fibres. J Cell Sci. 2007;120(Pt 20):3491–3499. doi: 10.1242/jcs.018473. [DOI] [PubMed] [Google Scholar]
- Perry SV. Vertebrate tropomyosin: distribution, properties and function. J Muscle Res Cell Motil. 2001;22:5–49. doi: 10.1023/a:1010303732441. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- Po'uha ST, Shum MS, Goebel A, Bernard O, Kavallaris M. LIM-kinase 2, a regulator of actin dynamics, is involved in mitotic spindle integrity and sensitivity to microtubule-destabilizing drugs. Oncogene. 2010;29:597–607. doi: 10.1038/onc.2009.367. [DOI] [PubMed] [Google Scholar]
- Po'uha ST, Honore S, Braguer D, Kavallaris M. Partial depletion of gamma-actin suppresses microtubule dynamics. Cytoskeleton (Hoboken) 2013;70:148–160. doi: 10.1002/cm.21096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudent R, Vassal-Stermann E, Nguyen CH, Pillet C, Martinez A, Prunier C, et al. Pharmacological inhibition of LIM kinase stabilizes microtubules and inhibits neoplastic growth. Cancer Res. 2012;72:4429–4439. doi: 10.1158/0008-5472.CAN-11-3342. [DOI] [PubMed] [Google Scholar]
- Rath N, Olson MF. Rho-associated kinases in tumorigenesis: re-considering ROCK inhibition for cancer therapy. EMBO Rep. 2012;13:900–908. doi: 10.1038/embor.2012.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico B, Beggs HE, Schahin-Reed D, Kimes N, Schmidt A, Reichardt LF. Control of axonal branching and synapse formation by focal adhesion kinase. Nat Neurosci. 2004;7:1059–1069. doi: 10.1038/nn1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Rodrigo JP, Garcia-Carracedo D, Garcia LA, Menendez S, Allonca E, Gonzalez MV, et al. Distinctive clinicopathological associations of amplification of the cortactin gene at 11q13 in head and neck squamous cell carcinomas. J Pathol. 2009;217:516–523. doi: 10.1002/path.2462. [DOI] [PubMed] [Google Scholar]
- Rodrigues-Ferreira S, Di Tommaso A, Dimitrov A, Cazaubon S, Gruel N, Colasson H, et al. 8p22 MTUS1 gene product ATIP3 is a novel anti-mitotic protein underexpressed in invasive breast carcinoma of poor prognosis. PLoS ONE. 2009;4:e7239. doi: 10.1371/journal.pone.0007239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Macdonald P, de Silva H, Guo Q, Xiao H, Hung CY, Penhallow B, et al. Identification of a nonkinase target mediating cytotoxicity of novel kinase inhibitors. Mol Cancer Ther. 2008;7:3490–3498. doi: 10.1158/1535-7163.MCT-08-0826. [DOI] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Rotty JD, Wu C, Bear JE. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat Rev Mol Cell Biol. 2013;14:7–12. doi: 10.1038/nrm3492. [DOI] [PubMed] [Google Scholar]
- Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2:133–142. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol. 2003;5:711–719. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68:3033–3046. doi: 10.1007/s00018-011-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Mitra SK. Multiple connections link FAK to cell motility and invasion. Curr Opin Genet Dev. 2004;14:92–101. doi: 10.1016/j.gde.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Schmalbach CE, Chepeha DB, Giordano TJ, Rubin MA, Teknos TN, Bradford CR, et al. Molecular profiling and the identification of genes associated with metastatic oral cavity/pharynx squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2004;130:295–302. doi: 10.1001/archotol.130.3.295. [DOI] [PubMed] [Google Scholar]
- Schroeder A, Heller DA, Winslow MM, Dahlman JE, Pratt GW, Langer R, et al. Treating metastatic cancer with nanotechnology. Nat Rev Cancer. 2012;12:39–50. doi: 10.1038/nrc3180. [DOI] [PubMed] [Google Scholar]
- Scott RW, Hooper S, Crighton D, Li A, Konig I, Munro J, et al. LIM kinases are required for invasive path generation by tumor and tumor-associated stromal cells. J Cell Biol. 2010;191:169–185. doi: 10.1083/jcb.201002041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba S, Iwaya K, Matsubayashi J, Serizawa H, Kataba H, Hirano T, et al. Coexpression of actin-related protein 2 and Wiskott-Aldrich syndrome family verproline-homologous protein 2 in adenocarcinoma of the lung. Clin Cancer Res. 2006;12:2449–2454. doi: 10.1158/1078-0432.CCR-05-2566. [DOI] [PubMed] [Google Scholar]
- Shum MS, Pasquier E, Po'uha ST, O'Neill GM, Chaponnier C, Gunning PW, et al. γ-Actin regulates cell migration and modulates the ROCK signaling pathway. FASEB J. 2011;25:4423–4433. doi: 10.1096/fj.11-185447. [DOI] [PubMed] [Google Scholar]
- Sihag RK, Inagaki M, Yamaguchi T, Shea TB, Pant HC. Role of phosphorylation on the structural dynamics and function of types III and IV intermediate filaments. Exp Cell Res. 2007;313:2098–2109. doi: 10.1016/j.yexcr.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha P, Hutter G, Kottgen E, Dietel M, Schadendorf D, Lage H. Increased expression of epidermal fatty acid binding protein, cofilin, and 14-3-3-sigma (stratifin) detected by two-dimensional gel electrophoresis, mass spectrometry and microsequencing of drug-resistant human adenocarcinoma of the pancreas. Electrophoresis. 1999;20:2952–2960. doi: 10.1002/(SICI)1522-2683(19991001)20:14<2952::AID-ELPS2952>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Small JV, Stradal T, Vignal E, Rottner K. The lamellipodium: where motility begins. Trends Cell Biol. 2002;12:112–120. doi: 10.1016/s0962-8924(01)02237-1. [DOI] [PubMed] [Google Scholar]
- Sobel A, Tashjian AH., Jr Distinct patterns of cytoplasmic protein phosphorylation related to regulation of synthesis and release of prolactin by GH cells. J Biol Chem. 1983;258:10312–10324. [PubMed] [Google Scholar]
- Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12:895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- Stehn JR, Haass NK, Bonello T, Desouza M, Kottyan G, Treutlein H, et al. A novel class of anticancer compounds targets the actin cytoskeleton in tumor cells. Cancer Res. 2013;73:5169–5182. doi: 10.1158/0008-5472.CAN-12-4501. [DOI] [PubMed] [Google Scholar]
- Takahashi K. WAVE2 protein complex coupled to membrane and microtubules. J Oncol. 2012;2012:590531. doi: 10.1155/2012/590531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takes RP, Baatenburg de Jong RJ, Schuuring E, Hermans J, Vis AA, Litvinov SV, et al. Markers for assessment of nodal metastasis in laryngeal carcinoma. Arch Otolaryngol Head Neck Surg. 1997;123:412–419. doi: 10.1001/archotol.1997.01900040048008. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Toyoda A, Kawana H, Azuhata K, Yu J, Omata A, Kishi H, et al. Aberrant expression of human ortholog of mammalian enabled (hMena) in human colorectal carcinomas: implications for its role in tumor progression. Int J Oncol. 2009;34:53–60. [PubMed] [Google Scholar]
- Trovik J, Wik E, Stefansson IM, Marcickiewicz J, Tingulstad S, Staff AC, et al. Stathmin overexpression identifies high-risk patients and lymph node metastasis in endometrial cancer. Clin Cancer Res. 2011;17:3368–3377. doi: 10.1158/1078-0432.CCR-10-2412. [DOI] [PubMed] [Google Scholar]
- Tsuruta D, Jones JC. The vimentin cytoskeleton regulates focal contact size and adhesion of endothelial cells subjected to shear stress. J Cell Sci. 2003;116(Pt 24):4977–4984. doi: 10.1242/jcs.00823. [DOI] [PubMed] [Google Scholar]
- Turhani D, Krapfenbauer K, Thurnher D, Langen H, Fountoulakis M. Identification of differentially expressed, tumor-associated proteins in oral squamous cell carcinoma by proteomic analysis. Electrophoresis. 2006;27:1417–1423. doi: 10.1002/elps.200500510. [DOI] [PubMed] [Google Scholar]
- Tybulewicz VL, Henderson RB. Rho family GTPases and their regulators in lymphocytes. Nat Rev Immunol. 2009;9:630–644. doi: 10.1038/nri2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan S, Dawe HR. Common themes in centriole and centrosome movements. Trends Cell Biol. 2011;21:57–66. doi: 10.1016/j.tcb.2010.09.004. [DOI] [PubMed] [Google Scholar]
- Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582:2093–2101. doi: 10.1016/j.febslet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Gaertig J. The tubulin code. Cell Cycle. 2007;6:2152–2160. doi: 10.4161/cc.6.17.4633. [DOI] [PubMed] [Google Scholar]
- Verrills NM, Liem NL, Liaw TY, Hood BD, Lock RB, Kavallaris M. Proteomic analysis reveals a novel role for the actin cytoskeleton in vincristine resistant childhood leukemia – an in vivo study. Proteomics. 2006a;6:1681–1694. doi: 10.1002/pmic.200500417. [DOI] [PubMed] [Google Scholar]
- Verrills NM, Po'uha ST, Liu ML, Liaw TY, Larsen MR, Ivery MT, et al. Alterations in gamma-actin and tubulin-targeted drug resistance in childhood leukemia. J Natl Cancer Inst. 2006b;98:1363–1374. doi: 10.1093/jnci/djj372. [DOI] [PubMed] [Google Scholar]
- de Vicente JC, Rosado P, Lequerica-Fernandez P, Allonca E, Villallain L, Hernandez-Vallejo G. Focal adhesion kinase overexpression: correlation with lymph node metastasis and shorter survival in oral squamous cell carcinoma. Head Neck. 2013;35:826–830. doi: 10.1002/hed.23038. [DOI] [PubMed] [Google Scholar]
- Wang W, Goswami S, Lapidus K, Wells AL, Wyckoff JB, Sahai E, et al. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004;64:8585–8594. doi: 10.1158/0008-5472.CAN-04-1136. [DOI] [PubMed] [Google Scholar]
- Wang W, Mouneimne G, Sidani M, Wyckoff J, Chen X, Makris A, et al. The activity status of cofilin is directly related to invasion, intravasation, and metastasis of mammary tumors. J Cell Biol. 2006;173:395–404. doi: 10.1083/jcb.200510115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Eddy R, Condeelis J. The cofilin pathway in breast cancer invasion and metastasis. Nat Rev Cancer. 2007;7:429–440. doi: 10.1038/nrc2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WS, Zhong HJ, Xiao DW, Huang X, Liao LD, Xie ZF, et al. The expression of CFL1 and N-WASP in esophageal squamous cell carcinoma and its correlation with clinicopathological features. Dis Esophagus. 2010a;23:512–521. doi: 10.1111/j.1442-2050.2009.01035.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Cao W, Mo M, Wang W, Wu H, Wang J. VEGF and cortactin expression are independent predictors of tumor recurrence following curative resection of gastric cancer. J Surg Oncol. 2010b;102:325–330. doi: 10.1002/jso.21644. [DOI] [PubMed] [Google Scholar]
- Wei SH, Lin F, Wang X, Gao P, Zhang HZ. Prognostic significance of stathmin expression in correlation with metastasis and clinicopathological characteristics in human ovarian carcinoma. Acta Histochem. 2008;110:59–65. doi: 10.1016/j.acthis.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Wittmann T, Waterman-Storer CM. Cell motility: can Rho GTPases and microtubules point the way? J Cell Sci. 2001;114(Pt 21):3795–3803. doi: 10.1242/jcs.114.21.3795. [DOI] [PubMed] [Google Scholar]
- Wittmann T, Bokoch GM, Waterman-Storer CM. Regulation of microtubule destabilizing activity of Op18/stathmin downstream of Rac1. J Biol Chem. 2004;279:6196–6203. doi: 10.1074/jbc.M307261200. [DOI] [PubMed] [Google Scholar]
- Wyckoff JB, Pinner SE, Gschmeissner S, Condeelis JS, Sahai E. ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr Biol. 2006;16:1515–1523. doi: 10.1016/j.cub.2006.05.065. [DOI] [PubMed] [Google Scholar]
- Xu XZ, Garcia MV, Li TY, Khor LY, Gajapathy RS, Spittle C, et al. Cytoskeleton alterations in melanoma: aberrant expression of cortactin, an actin-binding adapter protein, correlates with melanocytic tumor progression. Mod Pathol. 2010;23:187–196. doi: 10.1038/modpathol.2009.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, Yanamoto S, Kawasaki G, Mizuno A, Nemoto TK. Overexpression of cortactin increases invasion potential in oral squamous cell carcinoma. Pathol Oncol Res. 2010;16:523–531. doi: 10.1007/s12253-009-9245-y. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 2007;1773:642–652. doi: 10.1016/j.bbamcr.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagawa R, Furukawa Y, Tsunoda T, Kitahara O, Kameyama M, Murata K, et al. Genome-wide screening of genes showing altered expression in liver metastases of human colorectal cancers by cDNA microarray. Neoplasia. 2001;3:395–401. doi: 10.1038/sj.neo.7900185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee HF, Jr, Melton AC, Tran BN. RhoA/rho-associated kinase mediates fibroblast contractile force generation. Biochem Biophys Res Commun. 2001;280:1340–1345. doi: 10.1006/bbrc.2001.4291. [DOI] [PubMed] [Google Scholar]
- Yoon SO, Shin S, Mercurio AM. Hypoxia stimulates carcinoma invasion by stabilizing microtubules and promoting the Rab11 trafficking of the alpha6beta4 integrin. Cancer Res. 2005;65:2761–2769. doi: 10.1158/0008-5472.CAN-04-4122. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Ozawa Y, Kimura T, Sato Y, Kuznetsov G, Xu S, et al. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. Br J Cancer. 2014;101:1497–1505. doi: 10.1038/bjc.2014.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Khan J, Khanna C, Helman L, Meltzer PS, Merlino G. Expression profiling identifies the cytoskeletal organizer ezrin and the developmental homeoprotein Six-1 as key metastatic regulators. Nat Med. 2004;10:175–181. doi: 10.1038/nm966. [DOI] [PubMed] [Google Scholar]
- Zhang T, Zaal KJ, Sheridan J, Mehta A, Gundersen GG, Ralston E. Microtubule plus-end binding protein EB1 is necessary for muscle cell differentiation, elongation and fusion. J Cell Sci. 2009;122(Pt 9):1401–1409. doi: 10.1242/jcs.039255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XL, Liang L, Ding YQ. Overexpression of FMNL2 is closely related to metastasis of colorectal cancer. Int J Colorectal Dis. 2008;23:1041–1047. doi: 10.1007/s00384-008-0520-2. [DOI] [PubMed] [Google Scholar]