

Abstract
Since the discovery of endothelin (ET)-1 in 1988, the main components of the signalling pathway have become established, comprising three structurally similar endogenous 21-amino acid peptides, ET-1, ET-2 and ET-3, that activate two GPCRs, ETA and ETB. Our aim in this review is to highlight the recent progress in ET research. The ET-like domain peptide, corresponding to prepro-ET-193–166, has been proposed to be co-synthesized and released with ET-1, to modulate the actions of the peptide. ET-1 remains the most potent vasoconstrictor in the human cardiovascular system with a particularly long-lasting action. To date, the major therapeutic strategy to block the unwanted actions of ET in disease, principally in pulmonary arterial hypertension, has been to use antagonists that are selective for the ETA receptor (ambrisentan) or that block both receptor subtypes (bosentan). Macitentan represents the next generation of antagonists, being more potent than bosentan, with longer receptor occupancy and it is converted to an active metabolite; properties contributing to greater pharmacodynamic and pharmacokinetic efficacy. A second strategy is now being more widely tested in clinical trials and uses combined inhibitors of ET-converting enzyme and neutral endopeptidase such as SLV306 (daglutril). A third strategy based on activating the ETB receptor, has led to the renaissance of the modified peptide agonist IRL1620 as a clinical candidate in delivering anti-tumour drugs and as a pharmacological tool to investigate experimental pathophysiological conditions. Finally, we discuss biased signalling, epigenetic regulation and targeting with monoclonal antibodies as prospective new areas for ET research.
Tables of Links
| TARGETS |
|---|
| GPCRsa |
| AT1 receptors |
| ETA receptors |
| ETB receptors |
| GPR37 |
| GPR37L |
| Hydroxycarboxylic acid receptors |
| μ opioid receptors |
| Enzymesb |
| Cathepsin A |
| Chymase |
| CYP3A4 |
| CYP2C19 |
| Endothelin-converting enzyme 1 |
| Endothelin-converting enzyme 2 |
| Neutral endopeptidase |
These Tables list protein targets and ligands that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b,).
| LIGANDS | |
|---|---|
| 5-Fluorouracil | Endothelin-3 |
| Ambrisentan | IRL1620 |
| Amyloid β-peptide | KC-12615 |
| Atrial natriuretic peptide (ANP) | Losartan |
| β-Catenin | Macitentan |
| Bosentan | NO |
| BQ123 | Prosaptide |
| BQ788 | PGI2 |
| BQ3020 | Sarafotoxin S6b |
| Captopril | Sarafotoxin S6c |
| Daglutril | Sitaxentan |
| Docetaxel | TGFβ1 |
| Doxorubicin | |
| Endothelin-1 | |
| Endothelin-2 |
This article, written by members of the International Union of Basic and Clinical Pharmacology Committee on Receptor Nomenclature and Drug Classification (NC-IUPHAR) subcommittee for the endothelin receptors, confirms the existing nomenclature for these receptors and reviews our current understanding of their structure, pharmacology and functions, and their likely physiological roles in health and disease. More information on these receptor families can be found in the Concise Guide to PHARMACOLOGY (http://onlinelibrary.wiley.com/doi/10.1111/bph.12445/abstract) and for each member of the family in the corresponding database http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=21&familyType=GPCR.
Introduction
Since the discovery of endothelin (ET)-1 in 1988 (Yanagisawa et al., 1988; Inoue et al., 1989) the components of the ET signalling pathway have become established, comprising three structurally similar endogenous 21-amino acid peptides, ET-1, ET-2 and ET-3, that activate two GPCRs, ETA (Arai et al., 1990) and ETB (Sakurai et al., 1990). In humans, ET-2 differs from ET-1 by only two amino acids, whereas ET-3 differs by six amino acids representing more substantial changes. ET-3 is the only isoform that can distinguish between the two receptor subtypes, having a similar potency at the ETA receptor as ET-1 and ET-2, but much lower affinity than these isoforms for the ETB receptor (Figure 1). Structurally, ETs are unusual among the mammalian peptides in possessing two disulphide bridges. This feature is shared by the sarafotoxins, a family of peptides that were isolated from snake venom in the same year as the discovery of ET-1 (Takasaki et al., 1988), and that provided the first selective agonist at the ETB receptor, sarafotoxin S6C (William et al., 1991).
Figure 1.
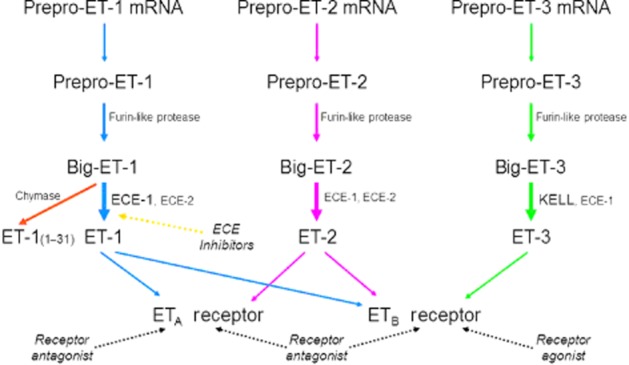
Scheme of the biosynthesis of ET peptides and their interaction with receptors. Based on information from the literature including Barton and Yanagisawa (2008), Turner and Tanzawa (1997) and Lee et al. (1999).
A number of features of the ET signalling pathway are unusual compared with other peptidergic systems and these continue to intrigue investigators, with over a thousand ET-related papers still published each year. ET-1 is the most abundant isoform in the human cardiovascular system, predominantly released from endothelial cells to cause potent and unusually long-lasting vasoconstriction that may persist for many hours. ET-1 is a key mediator in regulating vascular function in the majority of organs systems balanced by opposing vasodilators, particularly NO, prostacyclin and endothelium-derived hyperpolarizing factor. Endothelial cell dysfunction occurs in pathophysiological conditions such as pulmonary arterial hypertension (PAH) and is associated with loss of these dilators and increased synthesis of ET. The consequence of this is vasoconstriction, proliferation of many different cell types particularly vascular smooth muscle, fibrosis and inflammation; processes associated with vascular remodelling. In disease, the deleterious actions of ET in the vasculature are mainly mediated by the ETA receptor, whereas activation of ETB receptors results in many of the beneficial effects of the peptide that frequently act as a regulatory counterbalance (Davenport and Maguire, 2006). The formation of the disulphide bridge in the ET peptides blocks the N-terminal amino acid conferring resistance to enzymic degradation in plasma. Internalization by ETB scavenging receptors is therefore particularly important for termination of the ET signal in health and disease.
The major therapeutic strategy (Figure 1) to block the unwanted actions of ET in disease has been to use antagonists of ETA receptors or both receptor subtypes (Palmer, 2009) with the first clinical application being bosentan in PAH (Rubin et al., 2002). More recently, a second strategy has started to be more widely tested in clinical trials using inhibitors of ET-converting enzymes 1 (ECE-1; Xu et al., 1994) and 2 (ECE-2; Emoto and Yanagisawa, 1995), the major biosynthetic pathway of ET (Figure 1) at least in the human vasculature (Russell and Davenport, 1999a,b). A third emerging strategy based on biosimilar agonists at the ETB receptor (molecules similar, but not identical to the endogenous ligand) has led to the renaissance of IRL1620 as a clinical candidate in delivering anti-tumour drugs and in other pathophysiological conditions such as cerebral ischaemia.
The aim of this focused IUPHAR review is to highlight recent progress and some surprising new discoveries in the pharmacology of the ET system. The following references are recommended for more detailed information on specific ET research areas including the heart (Kelland and Webb, 2006; Kirkby et al., 2008; Kohan et al., 2012; Ohkita et al., 2012; Vignon-Zellweger et al., 2012; Drawnel et al., 2013), renal (Dhaun et al., 2011; 2012,; Kohan et al., 2011a,b,; Hyndman and Pollock, 2013), hypertension (Rautureau and Schiffrin, 2012; Speed and Pollock, 2013), PAH (Pernow et al., 2012; Rubin, 2012; Liu et al., 2013), cancer (Bagnato et al., 2011; Said and Theodorescu, 2012; Rosanò et al., 2013b), atherosclerosis and diabetes (Pernow et al., 2012).
ET peptides
Evidence for a new ET peptide: the ET-like domain peptide (ELDP)
The ELDP has been recently identified as a peptide corresponding to prepro-ET-193–166 (Yuzgulen et al., 2013) immediately adjacent to the gene sequence encoding big ET-1. The 74-amino acid peptide has been detected by HPLC and specific double recognition site immunoassays in conditioned media from two cell lines, endothelial (EA.hy 926) and epithelial (A549), as well as from primary cell cultures of human aortic endothelial cells that are known to secrete ET-1. In the aortic endothelial cells, the peptide was co-synthesized and co-released with ET-1. Plasma levels in untreated patients were 6.5 pmol·L−1, which compares with typical basal levels of immunoreactive ET-1 of 5 pmol·L−1 (Davenport et al., 1990). Levels of ELDP were significantly elevated in patients with heart failure suggesting a potential use as a bio-marker. While no effect was observed on BP in the anaesthetized rat, intriguingly ELDP significantly increased the duration of the pressor response of ET-1 (0.3 nmol·kg−1, likely to be a submaximal dose). Pretreatment of rat mesenteric arteries with 10 nM ELDP also potentiated a submaximal response of ET-1 by fivefold (Yuzgulen et al., 2013). It is not unexpected that a second peptide sharing a cleavage site with ET-1 would also be co-released, but it is intriguing that the peptide was able to potentiate ET-1 responses in vitro and in vivo. It is not yet reported, using saturation or competition-binding experiments, whether ELDP binds directly to ET receptors, binds to an allosteric site or whether the peptide modulates ET responses by other mechanisms. Intriguingly, ELDP encompasses the sequence of the putative ‘endothelin-like peptide’ corresponding to prepro-ET-1109–123 proposed in the original Nature paper by Yanagisawa et al. (1988). Eight out of 15 residues in the corresponding sequence in ET-1 are identical and the four Cys residues are perfectly conserved and flanked by dibasic pairs that are recognized by endopeptidase processing enzymes, to yield a cleaved peptide. However, a synthetic peptide corresponding to this sequence was devoid of agonist or antagonist activity against ET-1, in vascular preparations (Cade et al., 1990).
Global knockout of ET-2 gene reveals distinct phenotype compared with ET-1/ETA and ET-3/ETB gene deletions
ET–1-deficient homozygous mice die at birth of respiratory failure, which is secondary to severe craniofacial and cardiovascular abnormalities. ETA receptor and ECE-1 knockout mice have similar morphological abnormalities (Kurihara et al., 1994; Clouthier et al., 1998; Yanagisawa et al., 1998). The phenotype is similar to a spectrum of human conditions, CATCH 22 (cardiac anomaly, abnormal face, thymic hypoplasia, cleft palate, hypocalcaemia, chromosome 22 deletions) and established the importance of the ETA/ET-1 signalling system for cardiovascular and craniofacial development. Gene deletions for ET-3 and ETB receptors exhibit a different and non-overlapping phenotype to ET-1/ETA-deficient animals. They are viable at birth and survive for up to 8 weeks, but display aganglionic megacolon, as a result of absence of ganglion neurones, together with a disorder of the pigment in their coats (Baynash et al., 1994; Hosoda et al., 1994). In these mice, enteric nervous system precursors and neural crest-derived epidermal melanoblasts fail to colonize the intestine and skin. This phenotype resembles Hirschsprung's disease in man.
Deleting genes encoding all the key molecules, ET-1, ET-3, ETA, ETB, ECE-1 and ECE-2 has been accomplished in mice generating important information about their effect on phenotype. The deletion of the gene for ET-2 has now been reported. The physiological role of ET-2 has been puzzling. It had been assumed that the actions of ET-2 would be similar, if released, to the more widely distributed and abundant ET-1. Current antagonists block both ET-2 and ET-1 with the same potency and are not yet able to distinguish the actions of these peptides.
A key advance in the field was the generation by Chang et al. (2013) of a global ET-2 gene knockout mouse, which surprisingly exhibited a distinct phenotype to global ET-1 or ET-3 gene deletions. These mice showed severe growth retardation, internal starvation characterized by hypoglycaemia, ketonaemia and increased levels of starvation-induced genes. Mice were profoundly hypothermic and the median lifespan could be significantly extended by housing in a warm environment. The intestine was morphologically and functionally normal, which was unexpected as murine ET-2 (see Ling et al., 2013), also known as vasoactive intestinal contractor, is present throughout the gastrointestinal tract suggesting, in this tissue at least, in the absence of ET-2, ET-1 continues to mediate signalling. In agreement, intestinal epithelium-specific ET-2 knockout mice showed no abnormalities in growth and survival. In marked contrast, dramatic changes were observed in lung morphology and function. Mice had breathing difficulties after the first week exhibiting enlarged air spaces with substantial simplification of lung alveolar structure, larger lung capacities leading to abnormally elevated carbon dioxide (hypercapnia) and deficiency of oxygen (hypoxaemia) in the blood. Hypothermia and lung dysfunction might not be specific, but may be due to a secondary effect of internal starvation because of ET-2 deficiency. However, it is possible that these studies identify an important function for ET-2 in the pulmonary system. The authors showed that mRNA encoding ET-2 was only present in epithelial cells whereas receptor mRNA was mainly present in mesenchyme, consistent with a paracrine function for ET-2 in the lung.
Pharmacological significance: is ET-2 the inducible isoform?
The dramatic effects on the lung suggest a crucial role for ET-2 at birth, at least in mice. The lungs, and potentially the heart, remain major therapeutic targets for ET antagonists in humans in the treatment of PAH. In rodents, ET-2 was less widely distributed than ET-1, mainly found in heart, lung, ovary, stomach and all regions of the intestine (de la Monte et al., 1995; Takizawa et al., 2005). ET-2 expression in human tissue was similar, being present in the human heart (Plumpton et al., 1996b), lung (Marciniak et al., 1992), kidney (Karet and Davenport, 1996), vasculature, (Howard et al., 1992) intestine and ovaries (Palanisamy et al., 2006), but has not been investigated in pathophysiological tissue. In humans, alternatively spliced mRNA variants encoding ET-2 have been detected with a specific pattern of distribution in various tissues. Some of these variants contain sites for the post-transcriptional processing of preproET-2 into mature ET-2, which may be altered in a tissue-specific manner (O'Reilly et al., 1993). The best established model of spatial and temporal ET-2 signalling is in the ovary, a highly vascular tissue, which undergoes cyclic changes as follicles grow, rupture and transform into corpora lutea and eggs are periodically released (Ko et al., 2012). In rats, low levels of ET-1 are constitutively expressed throughout the ovulatory cycle, whereas ET-2 is induced transiently at much higher concentrations during the period of ovulation to luteal phases (Ko et al., 2006). ET-2 is expressed in the granulosa cells of periovulatory follicles, but not during other stages of follicular development. In mice, induced superovulation results in a dramatic increase in ET-2 mRNA expression (Palanisamy et al., 2006). ET-2 expression surged in response to gonadotropin and quickly declined by 13 h, which coincided with the time of follicular rupture. Crucially, both ET receptor subtypes are present and their ratio does not seem to change. Thus, the ET-2 gene appears to be switched on only when increased levels of ET are required, with ET–2-mediated contraction being the final signal facilitating ovulation (Ling et al., 2013).
ET-1 signalling is well established in neural crest migration. In the developing mouse retina, constitutive over-expression of ET-2 affects vascular development by inhibiting endothelial cell migration across the retinal surface and subsequent endothelial cell invasion into the retina, an action mediated by ETA receptors. Interestingly, over-expression is spatially localized as it has no obvious action on vascular structures in brain or skin (Rattner et al., 2013). Constitutive over-expression of ET-2 signalling also protected photoreceptors from light damage (Braunger et al., 2013). Similarly, Bramall et al. (2013) found expression of ET-2 mRNA was greatly increased in the photoreceptors of mouse models of inherited photoreceptor degeneration and, using the global ET-2 knockout mice, showed increased ET-2 expression was protective of the mutant photoreceptors.
Case for re-evaluating the role of ET-2
Targeting the ET-2 gene in mice provides compelling evidence that, while both ET-1 and ET-2 can coexist in the same tissue compartments, there is a critical, but distinct ET-2 pathway. A key role has now been established for ET-2 in ovarian physiology. This may be accomplished at the level of gene expression, but differences may also exist in peptide synthesis by ECEs and chymase, which may allow the two ET peptide pathways to be distinguished pharmacologically and become separate drug targets. Additionally, pharmacological differences have been identified, for example ET-2 dissociates from receptors much more rapidly than ET-1 and higher affinity has been reported, for example in the brain (Ling et al., 2013). Detailed studies comparing rat mesenteric resistance and basilar arteries demonstrated that ET-1 and ET-2 initiate and maintain vasoconstriction by different downstream mechanisms raising the prospect of ‘biased signalling’ mediated by two structurally different agonists activating the same receptor (Compeer et al., 2013).
Potential new therapeutic strategies exploiting ETB receptor agonists
The pharmacological rationale for this strategy is that ET-1, tonically released from endothelial cells, also interacts with endothelial cell ETB receptors. The importance of this counter-regulatory pathway has been underestimated to date. Endothelial cells line the vasculature of every organ and tissue in the body that receives blood supply. Although the cells represent ∼1% of the weight of the vessel wall, they have a combined mass comparable with some endocrine glands. Crucially, ET-1 feeding back onto endothelial receptors to release NO not only limits ETA-mediated vasoconstriction by stimulation of vascular cGMP, but also limits further ET-1 release. Thus in the vasculature, NO and other dilators are crucial in balancing the ET system, but these may be reduced or absent in pathophysiological conditions.
ET-1+/− heterozygous mice developed elevated BP and mild hypertension, rather than the fall in BP that might have been expected. Partial deletion of the gene allows survival and produced lower levels of ET-1 in plasma and lung tissue than wild type (Kurihara et al., 1994). These results suggest that ET-1 has an essential physiological role in cardiovascular homeostasis. Low levels promote vasodilatation whereas higher and pathophysiological concentrations of ET-1 increase BP and total peripheral vascular resistance. While ETA receptor selective antagonists such as BQ123 (Ihara et al., 1992) cause the expected vasodilatation in humans (Haynes and Webb, 1994), the ETB receptor selective antagonist BQ788 (Ishikawa et al., 1994) caused systemic vasoconstriction in healthy volunteers, showing that the main consequence of activation of endothelial ETB receptors by tonically secreted ET-1 was the physiological basal release of NO (Love et al., 2000). In agreement, initial vasodilatation can be detected in the human forearm vascular bed following infusion of low concentrations of ET-1 whereas higher doses caused sustained vasoconstriction (Kiowski et al., 1991). A contribution to vasoconstriction may also be the result of occupancy by ET-1 of the clearance ETB receptors causing an ETA-mediated vasoconstriction.
ETB agonists in chemotherapy: IRL1620
ET-1 acting on ETA receptors has been proposed to stimulate cell proliferation, migration, invasion, osteogenesis and angiogenesis in several cancers. New vessels forming in tumours are characterized by high densities of ETA receptors in smooth muscle, for example in glioblastoma multiforme in the brain (Harland et al., 1998). Conversely, ETB receptors may oppose tumour progression by promoting apoptosis and clearing ET-1 (Bagnato et al., 2011; Rosanò et al., 2013b). The strategy of stimulating ETB receptors to cause transient vasodilatation is being developed to increase the penetration of cytotoxic anti-tumour agents into tumours and to minimize the concentration in healthy tissue.
IRL1620 was originally developed as a tool compound (Takai et al., 1992). The N-terminus has an N-succinyl modification, which is likely to reduce metabolism by non-specific peptidases, but it is not orally active and requires injection. Despite these unpromising pharmacokinetic features, it is being used in vivo and has emerged as a possible clinical candidate in improving the delivery of drugs to tumours. IRL1620 infused into rats improved the efficacy of doxorubicin and 5-flurouracil by significantly increasing the amount of drug in tumours in rat models of prostrate and breast cancer. In addition, radiation-induced reduction in tumour volume was enhanced, suggesting IRL1620 can significantly increase the efficacy of radiotherapy in the treatment of solid tumours. The results suggest that for a given dose of drug, the efficacy in reducing the tumour could be improved (Gulati and Rai, 2004; Rajeshkumar et al., 2005a,b,; Lenaz et al., 2006; Rai et al., 2006; Gulati et al., 2012). A phase I trial to determine the safety, tolerability, pharmacokinetics and pharmacodynamics of IRL1620 (known as SPI-1620 licensed by Spectrum Pharmaceuticals, Technology Drive Irvine, CA, USA) in patients with recurrent progressive carcinoma has been successfully completed and shown to selectively and transiently increase tumour blood flow (Gulati et al., 2012; http://www.cancer.gov/clinicaltrials). A phase II trial was initiated in 2013 to determine the effectiveness of SPI-1620 in combination with docetaxel in patients with advanced biliary cancer (http://clinicaltrials.gov/ct2/show/NCT01773785) and in combination with docetaxel compared with docetaxel alone for patients with non small-cell lung cancer after failure of platinum-based chemotherapy (http://clinicaltrials.gov/show/NCT01741155).
ETB agonists in neuroprotection
The human brain contains the highest density of ET receptors, with the ETB receptor subtype comprising about 90%, in areas such as cerebral cortex (Harland et al., 1998). Binding and functional studies have demonstrated glia mainly express ETB receptors whereas ETA receptors are localized mainly on neurones (Morton and Davenport, 1992). Smooth muscle cells from large arteries and small intracerebral vessels only express ETA receptors (Adner et al., 1994; Harland et al., 1995; Pierre and Davenport, 1999) with endothelial cell ETB receptors mediating relaxation (Lucas et al., 1996). The small pial arteries and arterioles penetrating into the brain play a major role in the maintenance of cerebral blood flow (auto-regulation). These vessels are particularly sensitive to ET-1 compared with peripheral vessels and the peptide has been a long-standing candidate in the genesis or maintenance of cerebrovascular disorders such as delayed vasospasm associated with subarachnoid haemorrhage or stroke. ET-1 does not cross the blood–brain barrier from the plasma, but may do so when compromised by subarachnoid haemorrhage, stroke or head injury. Strategies for targeting cerebrovascular disease have focused previously on the use of ET receptor antagonists, firstly to block vascular receptors mediating cerebrovasospasm that may be responsible for delayed cerebral ischaemia seen after aneurysmal subarachnoid haemorrhage and could contribute to ischaemic core volume in stroke. Secondly, to block neural receptors that mediate increases in intracellular free calcium (Morton and Davenport, 1992) and initiate the pathophysiological processes leading to neuronal death.
A new emerging strategy is to use ETB receptor agonists such as IRL1620 to provide vasodilatation and neuroprotection. The peptide reduced neurological damage following permanent middle cerebral artery occlusion in rats, a model of focal ischaemic stroke. Animals received i.v. injections of IRL1620 after the occlusion, which dramatically reduced infarct volume (by more than 80% in the acute and 70% in the chronic study), prevented cerebral oedema, reduced oxidative stress markers and improved all neurological and motor function for up to 7 days (Leonard et al., 2011; 2012,; Leonard and Gulati, 2013). Rats treated with the amyloid peptide Aβ1–40 administered into the intracerebral vessels display increased markers of oxidative stress in the brain. IRL1620 significantly reduced oxidative stress and importantly the cognitive impairment (Briyal et al., 2014). As discussed later, a reduction in ECE activity is associated with accumulation of amyloid β-peptide and neurotoxicity early in progression of Alzheimer's disease (Eckman et al., 2001; 2003; 2006,,; Pacheco-Quinto and Eckman, 2013). These results are limited to disease models in a single species, and it is unclear whether the molecular mechanisms would translate to humans, but taken together, they suggest that an ETB receptor agonist might offer a new therapeutic strategy in Alzheimer's disease and provide neuroprotection following cerebral ischaemia in conditions such as stroke.
No evidence for further ETB receptor subtypes
Previous studies have suggested that ETB receptors could be further subdivided into ETB1 present on endothelial cells and ETB2 on smooth muscle cells. Studies continue to be published with this misleading nomenclature, but current evidence only supports the existence of two subtypes, ETA and ETB, according to NC-IUPHAR nomenclature (Davenport, 2002; Alexander et al., 2013a,b,). Firstly, Mizuguchi et al. (1997) demonstrated unequivocally that in ETB receptor knockout mice, both the direct constrictor and indirect vasodilator responses to the ETB agonist sarafotoxin S6C were abolished. Selective deletion of endothelial ETB receptors in mice (demonstrated by autoradiography to leave unaltered ETB receptors expressed by other cell types) impaired, as expected, the clearance of an i.v. bolus of labelled ET-1 compared with controls (Bagnall et al., 2006; Kelland et al., 2010). Secondly, Flynn et al. (1998) were unable to distinguish pharmacologically, in extensive competition-binding experiments, between ETB receptors expressed by human isolated endothelial and smooth muscle cells in culture. In concordance, saturation-binding assays in human tissue always found ETB radiolabelled ligands bound with a single affinity and Hill slopes close to unity with no suggestion of further subtypes (Molenaar et al., 1992; 1993,; Nambi et al., 1994) or in competition-binding versus radiolabelled ET-1 in human native (Peter and Davenport, 1995; 1996,; Russell and Davenport, 1996) or recombinant ETB receptors (Nambi et al., 1994; Reynolds et al., 1995). Clozel and Gray (1995) showed that endothelial and smooth muscle ETB receptors cannot be distinguished functionally.
Do ETs interact with any other GPCRs?
The virtually complete sequencing of the human genome has allowed the identification of all of the human gene sequences that could potentially encode GPCRs that are currently classified as ‘orphan’ to indicate that their endogenous ligand is not yet known (Foord et al., 2005; Davenport et al., 2013). In this catalogue, the most closely related to the ETA and ETB receptor subtypes are the orphan receptors GPR37 (also know as ET receptor type B-like or Parkin-associated ET receptor-like receptor) and its related receptor, GPR37L1. A recent high-throughput screen tested ∼10 000 biologically active compounds for binding to 82 remaining orphan GPCRs. None of the ∼20 ET peptides tested at high concentration (including all three mature isoform and their corresponding big ET precursors, C-terminal metabolites, BQ123 and the ETB receptor agonist BQ3020) activated any of the expressed receptors, including GPR37 or GPR37L, supporting the established concept of ETs binding to only two receptor subtypes. Two orphan neuropeptides, prosaptide and prosaposin, have recently been proposed as cognate ligands for GPR37 and GPR37L (Meyer et al., 2013).
Clinical application of ET antagonists
Bosentan, ambrisentan and withdrawal of sitaxentan
PAH is a progressive condition with no cure and has a major impact on the ability to lead a normal life. It is an orphan disease (∼100 000 patients in US and Europe). PAH involves constriction of pulmonary arteries and is characterized by high BP in the lungs, ultimately leading to right heart failure and death. A number of pathways have been implicated in the development of PAH including bone morphogenetic proteins, prostacyclin and ET-1. Restoring the imbalance between constriction and vasodilatation of blood vessels is the basis for current medical therapies, although the cause of death is right heart failure. Although ETA receptors are significantly increased in the right ventricle of patients with PAH (Kuc et al., 2014) and in the left ventricle of patients with heart failure (Zolk et al., 1999), surprisingly, ET receptor antagonists have clinical efficacy in the former, but not the latter group (Kohan et al., 2012).
Bosentan (Tracleer, Ro47-0203) was the first ET receptor antagonist to be introduced into the clinic for the treatment of PAH (Rubin et al., 2002) and, as an orally acting agent, at the time represented a major advance over existing therapies such as prostacyclin analogues. Bosentan is classified as a mixed ETA/ETB receptor antagonist blocking both receptors (Figures 2 and 3). The second antagonist to enter the clinic in 2007 was ambrisentan (Letairis, Volibris, LU208075, Figures 2 and 3), which was reported to display some ETA receptor selectivity (Vatter and Seifert, 2006) followed by the most highly selective ETA receptor antagonist sitaxentan (Thelin, TBC11251) (Barst et al., 2004). While hepatotoxicity is a known side effect of ET antagonists, it is usually reversible and related to dose. Unfortunately, cases of idiosyncratic hepatitis resulting in acute liver failure leading to death have been reported with sitaxentan and the compound was withdrawn in 2010 (Don et al., 2012).
Figure 2.
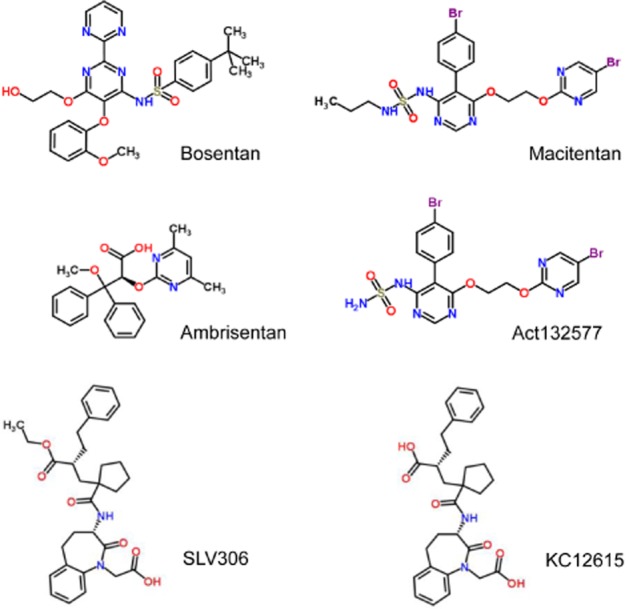
Structures of ET receptor antagonists in clinical use bosentan, ambrisentan and macitentan. The structures of the NEP/ECE inhibitor pro-drug SLV306 and its active metabolite are also shown.
Figure 3.
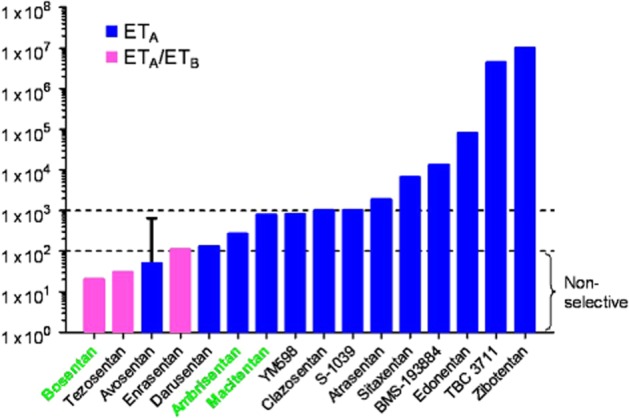
Selectivity of ET receptor antagonists for ETA versus ETB receptors shown on the vertical axis as reported by the companies that discovered the compounds. Selectivity was mainly determined by measuring affinity constants in separate competition assays against [125I]-ET-1 using human recombinant ETA versus ETB receptors and may not reflect selectivity measured in clinically relevant native tissues. Bosentan, ambrisentan and macitentan are currently approved for clinical use and are highlighted.
Next generation of ET antagonists: macitentan
Despite the current use of ET receptor antagonists and drugs targeting the two other principal pathways- that of NO with PDE5 inhibitors and that involving prostacyclin (PGI2) – meta-analysis of PAH trials shows existing therapies only moderately increased the most widely used objective evaluation of functional exercise capacity (6 min walk distance) by 11%. The prognosis for patients with PAH remains poor with ∼15% mortality within 1 year. There remains an urgent need for new efficacious treatments that has lead to the development of macitentan.
Macitentan (Opsumit, ACT-064992, Figures 2 and 3) represents the next generation of orally active ET receptor antagonists and was developed by modifying the structure of bosentan to improve efficacy and tolerability (Bolli et al., 2012). Macitentan is described as a dual antagonist that blocks both ETA and ETB receptors and it inhibited [125I]-ET-1 binding to human recombinant ETA receptors with an IC50 of 0.2nM and to ETB receptors with an IC50 of 391 nM. On the basis of these results, macitentan displays about 800-fold selectivity. A phase III clinical trial was successfully completed in 2012 (Pulido et al., 2013), and the compound gained approval from the US FDA in 2013 for the treatment of PAH. Macitentan is metabolized by the cytochrome P450 system, predominantly CYP3A4 and to a lesser extent the CYP2C19 iso-enzyme. Unlike other antagonists currently in use, one of the metabolites of macitentan, ACT-132577 (Figure 2), is pharmacologically active. Although it has a lower potency than the parent compound ACT-132577 reaches higher plasma concentrations, with a longer half-life of about 48 h (Iglarz et al., 2008; Sidharta et al., 2011; 2013a, b). These factors are likely to contribute to improved activity of macitentan compared with bosentan. While in vitro studies suggested macitentan was likely to interact with other drugs (Weiss et al., 2013), other observed pharmacokinetic benefits included fewer interactions with other drugs at clinically used concentrations, no requirement to alter doses in patients with renal or hepatic impairment, improved hepatic safety and reduced oedema/fluid retention compared with bosentan. Key differences were also identified in the pharmacodynamic parameters. For example, in calcium release assays macitentan was more potent (KB = 0.1 nM) than bosentan (KB = 1.1 nM) and had a significantly longer receptor occupancy (17 min compared with 70 s) (Iglarz et al., 2008; Bruderer et al., 2012a,b; 2013; Gatfield et al., 2012). The authors suggested that the macitentan binding site differed slightly from the bosentan binding site and that this difference in interaction with amino acids in the receptor contributed to the slow dissociation of macitentan from the receptor, particularly leading to insurmountable antagonism. A number of clinical trials are actively recruiting (Patel and McKeage, 2014) including the use of macitentan for the treatment of digital ulcers in patients with systemic sclerosis, Eisenmenger's syndrome and, perhaps the most challenging, in patients with brain tumours (glioblastoma).
Compounds interacting with ET-1 synthesis and metabolism
Members of the neprilysin (NEP)-like family of zinc metalloendopeptidases play key roles in the ET pathway (Turner and Murphy, 1996; Turner et al., 2001). Neutral endopeptidase (NEP) is a membrane-bound thermolysin-like zinc metalloendopeptidase, which is particularly abundant in human kidneys. The enzyme metabolizes a number of peptides including enkephalins, tachykinins, natriuretic peptides as well as the ETs (Turner and Tanzawa, 1997). Inactivation of ET-1 is via a two-stage process, opening of the Ser5–Leu6 bond, followed by cleavage at the amino side of Ile19 resulting in an inactive peptide, which is inhibited by phosphoramidon (Skolovsky et al., 1990). Pharmacological intervention in the pathway is challenging because NEP-like enzymes also include the synthetic enzymes ECE-1, ECE-2 and KELL. The ECEs are also inhibited by phosphoramidon and ECE inhibitors currently in clinical trials have significant NEP inhibitory activity and it seems counter-intuitive to inhibit the degradative pathway. However, in practice, inactivation of ET-1 is thought to be mainly via binding and internalization of the ETB receptor and ET-1 is essentially stable in plasma. Binding to ETB receptors, particularly in those organs such as the lung expressing high densities of the subtype, are critical for inactivation of the peptide. After internalization of the ligand–receptor complex to the lysosome, ET-1 is thought to be degraded, like other peptides, by cathepsin A. In support, cathepsin A knockout mice showed reduced ET-1 degradation and significantly increased arterial BP. In humans, genetic defects of cathepsin A include hypertension and cardiomyopathies (Seyrantepe et al., 2008).
ECE-1
It is now well established that ET is synthesized in a three-step process, with pre-pro-ET-1 initially cleaved by a signal peptidase to proET-1, which is in turn cleaved by a furin enzyme to an inactive precursor big ET-1 (Figure 1). Although low MW inhibitors of furins have been reported, furins cleave a number of other proteins to mature or active forms and therefore are not an easy tractable drug target for selectively reducing ET-1, without altering other pathways. Targeting the ECE enzymes responsible for transformation of big ET-1 to the mature, biologically active ET-1 has been more promising (Xu et al., 1994; Turner and Murphy, 1996). In humans there are four isoforms, ECE-1a–d, derived from a single gene by the action of different promoters. Structurally, they differ only in the amino acid sequence of the extreme N-terminus. ECE-1 localizes to the small secretory vesicles of the constitutive pathway from where ET-1 is continuously released to maintain normal vascular tone. Unusually for vasoactive peptides, ET-1 is also synthesized by ECE-1 and stored in specialized Weibel–Palade bodies within endothelial cells until its release following an external physiological or pathophysiological stimulus (the regulated pathway) to produce further vasoconstriction (Russell et al., 1998a,b; Russell and Davenport, 1999b).
In addition to intracellular endothelial cell ECE, the enzyme is also present on vascular smooth muscle, efficiently converts big ET-1 in human vessels in vitro and is up-regulated in atherosclerosis (Maguire et al., 1997; Maguire and Davenport, 1998). Given the larger volume of the smooth muscle compared with the single layer of endothelium, smooth muscle ECE may be a more important source of ET-1 in pathophysiological conditions.
ECE-2
ET-1 is also synthesized by a second membrane-bound metalloprotease, ECE-2 (Emoto and Yanagisawa, 1995; Yanagisawa et al., 2000; Lorenzo et al., 2001) with ∼60% sequence similarity to ECE-1. It is distinguishable from ECE-1 by having an optimum pH of 5.5 for activity. In human endothelial cells, ECE-2 was localized to the acidified environment of vesicles of the secretory pathway, but unlike ECE-1 it is not found in storage granules (Russell and Davenport, 1999b). Four isoforms exist, differing in their N-terminus: ECE-2a-1 and ECE-2a-2 are expressed predominantly in peripheral tissues and ECE-2b-1 and ECE-2b-2 in the brain, possibly representing the neuronal isoforms (Ikeda et al., 2002). The physiological importance of this pathway for ET-1 synthesis remains to be determined, as ECE-2 also metabolizes other peptides such as bradykinin. However, the requirement for an acidic pH suggests a role in pathophysiological conditions associated with low pH such as ischaemia. ECE-1/ECE-2 knockout mice display increased developmental defects compared with deletion of ECE-1 or ECE-2.
Alternative, non-ECE synthetic pathway: chymase
ET-1 can also be synthesized indirectly by chymase, a serine protease present in mast cells. Big ET-1 is converted to ET-11–31 by cleaving the Tyr31–Gly32 bond (Figure 1), which in turn is converted to the mature peptide via Trp21–Val22 bond (Fecteau et al., 2005; D'Orleans-Juste et al., 2008). The existence of an alternative pathway was originally predicted when ET-1 and ET-2 were detected in embryos of the ECE-1/ECE-2 double-knockout mouse (Yanagisawa et al., 2000).
The importance of this alternative pathway remains unclear, but importantly ET-11–31 was equipotent with big ET-1 in causing vasoconstriction in human isolated vessels, including coronary arteries, and this was associated with the appearance of measurable levels of ET-1 in the bathing medium. ET-11–31 displayed no selectivity between ETA and ETB receptors in human heart and vasoconstriction was fully blocked by ETA receptor selective antagonists, reflecting the predominance of the ETA receptor on vascular smooth muscle (Maguire et al., 2001; Maguire and Davenport, 2004). The precise physiological role of mast cells within human blood vessels is unclear, but following degranulation, which may occur under pathophysiological conditions, the mast cell chymase is associated with interstitial spaces with the potential to convert circulating big ET-1 and provide a further source of ET-1. Mast cell expression is increased in cardiovascular disease, for example in atherosclerotic lesions. It is therefore possible that the contribution of this pathway within the vasculature, leading to over-expression of ET-1, may be underestimated particularly in conditions of endothelial malfunction where opposing levels of endogenous vasodilators may be reduced. It is unclear whether under conditions of NEP/ECE inhibition the rising levels of big ET-1 would favour increased conversion by the serine protease pathway, thus increasing the pressor effect via ETA receptors or whether excretion of unmetabolized big ET-1 by the kidney would be sufficient to remove the elevated levels of precursors (Johnström et al., 2010).
KELL and ET-3 synthesis
Although big ET-3 is converted by ECE-1 to ET-3, owing to difference in the C-terminus the efficiency is much less than for ET-1. In contrast, big ET-3 is reported to be efficiently converted by KELL (Lee et al., 1999). KELL is a membrane-bound glycoprotein expressed in human erythrocytes and one of the major antigens; it is also related to mammalian NEP-like enzymes including ECE-1 and ECE-2 (Turner and Tanzawa, 1997). If KELL is the main synthetic pathway for ET-3, a possible benefit of inhibiting ECE would be to increase the ratio of ET-3 to ET-1, which could then differentially produce beneficial vasodilatation via the ETB receptor, but this speculative hypothesis has not been tested.
Pharmacological inhibition of ECE by research compounds
A combination of phosphoramidon and thiorphan has been widely used to identify ECE activity. This is based on the finding that the conversion of big ET-1 to ET-1 is inhibited by phosphoramidon, but not by thiorphan, and has been shown both in vitro and in vivo. Importantly for evaluating the significance of animal models, both compounds have also been used in clinical studies to characterize big ET-1 conversion (see Webb, 1995; Plumpton et al., 1996a; Hand et al., 1999). Low MW, non-peptide ECE inhibitors have been developed and one that has been widely used in vitro and in vivo animal models and is commercially available is CGS26303 (De Lombaert et al., 1994). CGS26303 inhibited conversion of all three big ETs in human isolated blood vessels but, importantly, did not interfere with the interaction of mature peptides with ET receptors (Yap et al., 2000). Although primarily an NEP inhibitor, SOL1, a more recent combined NEP/ECE non-peptide inhibitor with modest inhibition of ECE-1 in vitro, was remarkably potent in vivo, fully blocking the big ET–1-induced rise in BP at a dose of 10 μmol·kg−1 (Nelissen et al., 2012).
A disadvantage of using phosphoramidon is that it is not selective for ECE. An alternative tool compound is PD159790, which inhibits ECE-1 with an IC50 value of 3 μM; at this concentration the compound is selective for ECE-1 over NEP (Ahn et al., 1998). PD159790 has been shown experimentally in HUVECs to inhibit conversion of big ET-1 at pH 6.9, optimum for ECE-1, but did not affect big ET-1 conversion to the mature peptide at pH 5.4, optimum for ECE-2 (Russell and Davenport, 1999a). The compound did not inhibit the further metabolism of ET-11–31, the chymase product of big ET-1 (Maguire et al., 2001) and can be used to distinguish between the three different pathways for ET synthesis. While the mature peptide is located in intracellular Weibel–Palade bodies or secretory vesicles within endothelial cells and a proportion of big ET-1 is converted to ET-1 intracellularly, it is not reported whether ECE inhibitors can cross the plasma membrane to access these intracellular sites. The main effects of these inhibitors may be on external ECE. In agreement with this proposal, the SLV306 metabolite KC-12615 (see later) effectively prevented conversion of exogenous big ET-1 in human vasculature (Seed et al., 2012).
Emerging NEP/ECE inhibitors
Selective inhibitors of ECE have not progressed into clinical applications. SLV306 (daglutril, Figure 2) is an orally active, mixed enzyme inhibitor of both ECE and NEP. It is a pro-drug being converted in vivo to the active metabolite, KC-12615. This latter molecule has a pharmacological profile similar to phosphoramidon, inhibiting NEP in the nanomolar range, but with more modest inhibition in the micromolar range for ECE (Meil et al., 1998; Jeng et al., 2002). The therapeutic basis is that while inhibition of NEP alone increased plasma concentrations of atrial natriuretic factor (ANP) to cause vasodilatation, NEP inhibitors are ineffective as anti-hypertensives, probably because NEP also degrades vasoconstrictor peptides including ET. A combined ECE/NEP inhibitor would be predicted to reduce the systemic conversion of big ET-1 to the mature peptide and increase dilator peptides such as ANP. SLV306 is well tolerated with few or none of the side effects such as increases in liver function, oedema, observed with ET receptor antagonists (Dickstein et al., 2004; Parvanova et al., 2013). A potential disadvantage is big ET-1 might still be converted to ET-1 by an alternative pathway such as chymase. However, in animal models with normal renal function, this did not occur and big ET-1 labelled with the positron emitter 18F was rapidly accumulated unchanged in the kidney following inhibition of NEP/ECE, with no evidence of conversion by another pathway (Johnström et al., 2010).
The effect of a combined NEP/ECE inhibitor has been tested in volunteers in a randomized, double-blind trial. Following oral administration of three increasing doses of SLV306 (to reach an average target concentration of 75, 300, 1200 ng·mL−1 of the active metabolite KC-12615), big ET-1 was infused into 13 male volunteers at a rate of 8 and 12 pmol·kg−1·min−1 (20 min each). At the two highest concentrations tested, SLV306 dose-dependently attenuated the rise in BP after big ET-1 infusion. There was a significant increase in circulating big ET-1 levels compared with placebo, indicating that SLV306 was inhibiting an increasing proportion of endogenous ECE activity. Importantly, plasma ANP concentrations also significantly increased, consistent with systemic NEP inhibition (Seed et al., 2012).
SLV306 in animal models and patients with type 2 diabetes and nephropathy
Diabetes causes activation of the renal ET system, which leads to progressive renal damage by cell proliferation and interstitial inflammation. Inhibitors of the renin–angiotensin system are widely used in treatment for hypertensive patients with type 2 diabetes, but are less effective in the advanced stages of diabetic renal disease. Studies in an animal model suggested that SLV306 had a similar efficacy to the angiotensin converting enzyme (ACE) inhibitor captopril in reducing proteinuria and preventing nephrosclerosis (Thone-Reinke et al., 2004). In this study, rats were treated with streptozotocin for twenty weeks and the effects of SLV306 (30 mg·kg−1 per day) compared with those of captopril (10 mg·kg−1 per day). SLV306 significantly decreased renal interstitial matrix content as well as protein and albumin excretion in diabetic rats, independent of BP and was as effective as captopril. These results suggested SLV306 treatment on top of blocking the renin–angiotensin system might have an additional benefit in reducing BP and improving renal function.
Parvanova et al. (2013) tested the efficacy of SLV306 in 45 patients with type 2 diabetes mellitus who had albuminuria and were already receiving the angiotensin receptor antagonist losartan, together with up to two additional anti-hypertensive drugs, in a randomized, crossover, double-blind, placebo-controlled trial. Although 8 weeks of treatment with SLV306 together with losartan did not significantly alter urinary albumin excretion or renal haemodynamic measures, the authors showed for the first time that the combination decreased ambulatory BP (particularly for systolic hypertension) in this patient group that are often resistant to treatment. There was a small, but significant increase in plasma big ET-1, consistent with ECE inhibition, but surprisingly not in pro-ANP. Increases in the natriuretic peptides was measured in healthy volunteers by Seed et al. (2012). Interestingly, the effect of SLV306 in this study on BP was higher at night (10 versus 12 mmHg). This is of potential importance as increased hypertension at night is a strong cardiovascular risk factor in this patient population. The molecular mechanism is not yet known, as plasma levels of big ET-1 were not reported separately for daytime versus night. The study was comparatively short and did not reveal significant changes in albumin excretion as predicted from animal studies. Long-term trials are required to determine whether the observed lowering of BP by SLV306 will translate into longer-term renal and cardio-protection.
SLV306 and congestive heart failure
The effect of three single oral doses of SLV306 was tested in patients with congestive heart failure who underwent right-sided heart catheterization in a randomized, double-blind, placebo-controlled design (Dickstein et al., 2004). Pulmonary pressures and right atrial pressure decreased significantly in all SLV306 dose groups with the maximum decrease occurring at 6–8 h. Despite plasma levels of the drug increasing with dose, there was no clear dose–response relationship, which may have been the result of the comparatively small numbers (18–20) in the study.
Insight in NEP/ECE inhibition from animal models
The efficacy of inhibiting NEP/ECE in animal models associated with increases in the ET signalling pathway has provided clues to future clinical applications. The development of nephropathy in diabetes is associated with a poor outcome, eventually leading to end-stage renal disease. In patients with diabetes, urinary excretion of protein and albumin rises and is associated with increased risk of cardiovascular disease. In diabetic rats, SLV306 decreased renal matrix protein content, protein and albumin excretion. The magnitude of these effects was comparable to those of ACE inhibition and independent of BP (Thone-Reinke et al., 2004). Currently, there are few drugs for the treatment of chronic renal failure. SLV338, a NEP/ECE inhibitor, abolished renal tissue damage (interstitial fibrosis, glomerulosclerosis, renal arterial remodelling) in rat models of both acute kidney failure as well as chronic renal failure. The compound preserved kidney function and reduced mortality (Sharkovska et al., 2011). In spontaneously hypertensive stroke-prone rats, SLV338 significantly improved survival in comparison with the vehicle-treated group in a BP-independent manner and could offer a new therapeutic approach for primary stroke prevention and improvement of mortality (Wengenmayer et al., 2011). SLV338 was also tested for cardiac protection in rat model of experimental renovascular hypertension (two-kidney, one-clip). SLV338 prevented cardiac remodelling to the same extent as losartan, but in a BP-independent manner. This effect was at least partly mediated via suppression of cardiac TGF-β1 expression (Kalk et al., 2011).
ET has been proposed to be a mediator in toxic liver injury. However, while SLV338 largely prevented the activation of the ET system it did not prevent D-galactosamine-induced acute liver injury in rats. The authors speculated that SLV388 should be tested in a less severe model of liver injury, as very severe intoxication might not be relevantly amenable to pharmacological interventions (Hocher et al., 2011).
ECE-1 and amyloid deposition
The strategy in the cardiovascular and renal systems has been to inhibit ECE-1 activity. However, evidence is emerging that ECE-1 may function in the brain as a novel enzyme degrading amyloid β-peptides at several sites. Deposition of amyloid in the brain in Alzheimer's disease is determined not only by its production, but also by its catabolism. ECE-1 inhibition produces, in addition to extracellular accumulation, accumulation of intracellular amyloid β-peptides within endosomal/lysosomal and autophagic vesicles and an intracellular pool, is partly regulated by ECE activity at the sites of production. Reduction in ECE activity leads to accumulation of amyloid β-peptide, which is associated with neurotoxicity early in progression of Alzheimer's disease (Eckman et al., 2001; 2003; 2006,,; Pacheco-Quinto and Eckman, 2013). The clearance of Aβ1–40 in mice was almost completely inhibited by phosphoramidon as well as insulin indicating that human Aβ1–40 was degraded, at least in part, by a phosphoramidon-sensitive pathway, implicating both ECE and NEP (Ito et al., 2013).
To date, these investigations have comprised in vitro or in vivo rodent studies It is not yet clear whether enhancing ECE-1 activity is a potential drug target in Alzheimer's disease rather than inhibiting ECE-1, as in the periphery. ECE-like immunoreactivity has been localized to afferent and efferent fibres of neurones and neuronal cell bodies of mixed morphology in human brain (Giaid et al., 1991). Drugs increasing ECE activity such as enzyme enhancers or recombinant ECE would have to cross the blood–brain barrier, and it is not clear what effect this would have on ET signalling in the periphery.
What are the new ET drug targets in the future?
Epigentics
Epigenetics can be defined as heritable changes in phenotype through mechanisms other than changes in DNA sequence. Epigenetic changes will therefore be preserved when cells divide and affect normal development and disease progression. Processes mediating epigenetic regulation include DNA methylation and histone modification, which involves post-translational covalent modification of histone proteins by a range of writers, erasers and readers. This in turn modulates the ability of associated DNA to be transcribed. The histone code is read by specific families of proteins such as the bromodomains. These are of pharmacological significance because of the recent discovery of low MW inhibitors, which selectively modulate gene expression (Prinjha et al., 2012).
Epigenetic regulation is of particular importance in the ET pathway with transcription being the primary level of ET-1 regulation of the gene EDN1 by histone modifications and DNA methylation (Welch et al., 2013). Silencing of the EDNRB gene by DNA methylation during development of tumours results in the down-regulation of the receptor. As a result, promotion of apoptosis via the ETB receptor is reduced or lost, suggesting the ETB receptor could be a target for epigenetic drugs or ETB agonists where ET may be the cause of some tumour types, including melanomas and oligodendrogliomas (Bagnato et al., 2011). Intriguingly, epigenetic inactivation of ET-2 and ET-3 mRNA and protein was found in rat and human colon tumours and cancer cell lines, as a result of hypermethylation of EDN2 and EDN3 genes. Restoring expression of ET-2 and ET-3 in human cells significantly attenuated the migration and invasion of human colon cancer cells (Wang et al., 2013). As ET-3 displays high affinity for the ETB receptor, forced expression of ET-3 might antagonize the actions of ET-1 mediated through ETA receptors. Such a mechanism would be consistent with proposed beneficial effects of the ETB receptor agonist, IRL-1620, in cancer.
Life before birth – is ET a critical pathway?
Maternal malnutrition and uteroplacental vascular insufficiency causes foetal growth restriction or intrauterine growth retardation. Low birthweight is linked to the later development of cardiovascular disease and hypertension. Maternal treatment with dexamethasone increased ET-1 constrictor responses and ETA receptor expression in placental arteries from the foetus (Docherty et al., 2001; Kutzler et al., 2003). Maternal nutrient restriction increased the histone acetylation and hypoxia inducible factor-1α (HIF-1α) binding levels in the ET-1 gene promoter of pulmonary vein endothelial cells (PVECs) in intrauterine growth restriction (IUGR) newborn rats, and continued up to 6 weeks after birth. These epigenetic changes could result in an IUGR rat being highly sensitive to hypoxia later in life, causing more significant PAH or pulmonary vascular remodelling. Recently, Xu et al. (2013) have shown that restricting the diets of pregnant rats so that they were undernourished increased the histone acetylation and HIF-1α binding levels in the proximal promoter region of ET-1, up-regulating the expression of ET-1, and this continued for 6 weeks after birth of the offspring. The authors speculate that that this intrauterine growth retardation could cause varying degrees of PAH later in life.
Generally, increased levels of histone acetylation are associated with increased transcriptional activity, whereas decreased levels of acetylation are correlated with suppressed gene expression. These data show that the open chromatin domains marked by histone H3 and H3K9/18 acetylation at the proximal promoter of ET-1 in IUGR rats are essential for transcription. Up-regulated ET-1 protein expression in PVEC from IUGR hypoxia rats is closely associated with the presence of increased acetylated H3 histones.
Biased signalling in the ET pathway
Pharmacology is undergoing a revolution in understanding the mechanism of ‘biased signalling’ via GPCRs. It was originally thought that ligands binding to a receptor would equally activate the G-protein pathway to produce a physiological response such as vasoconstriction (such as ET-1 acting on an ETA receptor) as well as activating the β-arrestin pathway, which eventually leads to desensitization, receptor internalization and ‘silencing’ of the pathway. It is now clear that some ligands are biased to one pathway over the other and secondly, rather than silencing, β-arrestin can activate alternative signalling pathways, some of which may be pathophysiological leading to longer-term signalling responses such as migration and proliferation.
Both ET receptor subtypes follow an β-arrestin and dynamin/clathrin-dependent mechanism of internalization, but it has been established that ETA receptors are recycled to the plasma membrane for further signalling while ETB receptors are targeted to lysosomes and degraded (Bremnes et al., 2000). In epithelial ovarian cancer, activation of ET-1/ETA receptor signalling is linked to many tumour-promoting processes including proliferation, angiogenesis, invasion and metastasis. NF-κB is an important signalling molecule in immunity, inflammation and cancer and β-arrestin is required for ET–1-induced NF-κB activation (Cianfrocca et al., 2014). ET-1 promoted podocyte migration via ETA receptors and increased β-arrestin-1, sustaining renal injury, a pathogenetic pathway that can affect podocyte phenotype in proliferative glomerular disorders (Buelli et al., 2014). β-Arrestin-1 has also been found to be a nuclear transcriptional regulator of ET–1-induced β-catenin signalling, an important mechanism for controlling cell division and progression of epithelial ovarian cancer and necessary for epigenetic modification, such as histone acetylation, and gene expression (Rosanò et al., 2009; 2013a,). In addition, these effects are blocked by ET receptor antagonists and support a role for ETA-mediated/β-arrestin-1 facilitating inter-protein interaction in invasive and metastatic behaviour of ovarian cancer.
Biased ET ligands?
Agonists that are biased towards β-arrestin signalling for parathyroid hormone and angiotensin AT1 receptors have been identified. G-protein pathway-selective agonists have been identified for nicotinic acid (nomenclature revised by NC-IUPHAR to hydroxycarboxylic acid) and μ-opioid receptors (Luttrell, 2014). The race is now to determine whether such strategies can be exploited therapeutically.
Do biased ligands (ligands that binding to the same receptor but activate different signalling pathways) exist for ET receptors? The study by Compeer et al. (2013) already mentioned suggested ET-1 and ET-2 initiate and maintain vasoconstriction by different downstream mechanisms. Biased signalling can be identified by comparing the affinities of ligands in β-arrestin recruitment assays with a G–protein-mediated response such as vasoconstriction (Maguire et al., 2012). In this study the rank order of potency for β-arrestin recruitment at the ETA (ET-1 ≥ ET-2 > > ET-3) and ETB (ET-1 = ET-2 = ET-3) receptors was as expected and there was no obvious major differences in potency of ETs when comparing with G–protein-mediated constrictor assays in human vessels. However, at the ETA receptor sarafotoxin S6b was a partial agonist in β-arrestin recruitment, but a full agonist in causing constriction, suggesting the possibility of biased ligands. Such a bias could have been selected for during evolution by prolonging the effects of envenomation of the mammalian prey. While bosentan displays no selectivity for ETA over ETB receptors in radioligand binding and G-protein functional assays, unexpectedly, it was significantly more effective an inhibitor of β-arrestin recruitment mediated by ETA, compared with the ETB receptors (Maguire et al., 2012). The result for bosentan is intriguing as many of the detrimental actions of ET-1, particularly in cancer, may use the β-arrestin pathway, and this suggests the potential to block a deleterious pathway while preserving activation of a beneficial pathway.
The next 25 years – quo vadis?
Since the discovery of ET, intense medicinal chemistry programmes have identified receptor antagonists, ETB receptor agonists and inhibitors of the key synthetic enzyme, ECE-1. Both mixed ETA/ETB and ETA selective receptor antagonists have become established in the treatment of PAH, while NEP/ECE inhibitors such as SLV306 have promise as an alternative to receptor blockade and IRL1620 and other ETB receptor ligands have potential in improving cancer therapy.
All of these are approaches that have exploited low MW compounds. Over 50 therapeutic monoclonal antibodies have been approved for clinical use, but none yet against a GPCR target, emphasizing the technical challenge. Endomab-B1, a monoclonal antibody has recently been reported to bind with subnanomolar affinity for the ETB receptor, competed with ET-1 binding with greater efficacy than BQ788, and functions as an antagonist to block the ET–1-induced IP3-calcium signalling pathway (Allard et al., 2013). Whether this antibody has clinical applications remains to be discovered.
Acknowledgments
We thank the British Heart Foundation (PS/02/001, PG/05/127/19872, FS/12/64/130001), the Wellcome Trust Programme in Metabolic and Cardiovascular Disease (096822/Z/11/Z), the National Institute for Health Research Cambridge Biomedical Research Centre and the Pulmonary Hypertension Association UK.
Glossary
- ECE-1
endothelin-converting enzyme-1
- ECE-2
endothelin-converting enzyme-2
- ELDP
endothelin-like domain peptide
- ET
endothelin
- HIF-1α
hypoxia inducible factor-1α
- NEP
neutral endopeptidase
- PAH
pulmonary arterial hypertension
Conflict of interest
None.
References
- Adner M, Jansen I, Edvinsson L. Endothelin-A receptors mediate contraction in human cerebral, meningeal and temporal arteries. J Auton Nerv Syst. 1994;49(Suppl):S117–S121. doi: 10.1016/0165-1838(94)90098-1. [DOI] [PubMed] [Google Scholar]
- Ahn K, Sisneros AM, Herman SB, Pan SM, Hupe D, Lee C, et al. Novel selective quinazoline inhibitors of endothelin converting enzyme-1. Biochem Biophys Res Commun. 1998;243:184–190. doi: 10.1006/bbrc.1998.8081. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard B, Wijkhuisen A, Borrull A, Deshayes F, Priam F, Lamourette P, et al. Generation and characterization of rendomab-B1, a monoclonal antibody displaying potent and specific antagonism of the human endothelin B receptor. MAbs. 2013;5:56–69. doi: 10.4161/mabs.22696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai H, Hori S, Aramori I, Ohkubo H, Nakanishi S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature. 1990;348:730–732. doi: 10.1038/348730a0. [DOI] [PubMed] [Google Scholar]
- Bagnall AJ, Kelland NF, Gulliver-Sloan F, Davenport AP, Gray GA, Yanagisawa M, et al. Deletion of endothelial cell endothelin B receptors does not affect blood pressure or sensitivity to salt. Hypertension. 2006;48:286–293. doi: 10.1161/01.HYP.0000229907.58470.4c. [DOI] [PubMed] [Google Scholar]
- Bagnato A, Loizidou M, Pflug BR, Curwen J, Growcott J. Role of the endothelin axis and its antagonists in the treatment of cancer. Br J Pharmacol. 2011;163:220–233. doi: 10.1111/j.1476-5381.2011.01217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barst RJ, Langleben D, Frost A, Horn EM, Oudiz R, Shapiro S, et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;169:441–447. doi: 10.1164/rccm.200307-957OC. [DOI] [PubMed] [Google Scholar]
- Barton M, Yanagisawa M. Endothelin: 20 years from discovery to therapy. Can J Physiol Pharmacol. 2008;86:485–498. doi: 10.1139/Y08-059. [DOI] [PubMed] [Google Scholar]
- Baynash AG, Hosoda K, Giaid A, Richardson JA, Emoto N, Hammer RE, et al. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell. 1994;79:1277–1285. doi: 10.1016/0092-8674(94)90018-3. [DOI] [PubMed] [Google Scholar]
- Bolli MH, Boss C, Binkert C, Buchmann S, Bur D, Hess P, et al. The discovery of N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-p ropylsulfamide (Macitentan), an orally active, potent dual endothelin receptor antagonist. J Med Chem. 2012;55:7849–7861. doi: 10.1021/jm3009103. [DOI] [PubMed] [Google Scholar]
- Bramall AN, Szego MJ, Pacione LR, Chang I, Diez E, D'Orleans-Juste P, et al. Endothelin-2-mediated protection of mutant photoreceptors in inherited photoreceptor degeneration. PLoS ONE. 2013;8:e58023. doi: 10.1371/journal.pone.0058023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunger BM, Ohlmann A, Koch M, Tanimoto N, Volz C, Yang Y, et al. Constitutive overexpression of Norrin activates Wnt/beta-catenin and endothelin-2 signaling to protect photoreceptors from light damage. Neurobiol Dis. 2013;50:1–12. doi: 10.1016/j.nbd.2012.09.008. [DOI] [PubMed] [Google Scholar]
- Bremnes T, Paasche JD, Mehlum A, Sandberg C, Bremnes B, Attramadal H. Regulation and intracellular trafficking pathways of the endothelin receptors. J Biol Chem. 2000;275:17596–17604. doi: 10.1074/jbc.M000142200. [DOI] [PubMed] [Google Scholar]
- Briyal S, Shepard C, Gulati A. Endothelin receptor type B agonist, IRL-1620, prevents beta amyloid (Abeta) induced oxidative stress and cognitive impairment in normal and diabetic rats. Pharmacol Biochem Behav. 2014;120C:65–72. doi: 10.1016/j.pbb.2014.02.008. [DOI] [PubMed] [Google Scholar]
- Bruderer S, Aanismaa P, Homery MC, Hausler S, Landskroner K, Sidharta PN, et al. Effect of cyclosporine and rifampin on the pharmacokinetics of macitentan, a tissue-targeting dual endothelin receptor antagonist. AAPS J. 2012a;14:68–78. doi: 10.1208/s12248-011-9316-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruderer S, Hopfgartner G, Seiberling M, Wank J, Sidharta PN, Treiber A, et al. Absorption, distribution, metabolism, and excretion of macitentan, a dual endothelin receptor antagonist, in humans. Xenobiotica. 2012b;42:901–910. doi: 10.3109/00498254.2012.664665. [DOI] [PubMed] [Google Scholar]
- Bruderer S, Marjason J, Sidharta PN, Dingemanse J. Pharmacokinetics of macitentan in Caucasian and Japanese subjects: the influence of ethnicity and sex. Pharmacology. 2013;91:331–338. doi: 10.1159/000351704. [DOI] [PubMed] [Google Scholar]
- Buelli S, Rosano L, Gagliardini E, Corna D, Longaretti L, Pezzotta A, et al. Beta-arrestin-1 drives endothelin-1-mediated podocyte activation and sustains renal injury. J Am Soc Nephrol. 2014;25:523–533. doi: 10.1681/ASN.2013040362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cade C, Lumma WC, Jr, Mohan R, Rubanyi GM, Parker-Botelho LH. Lack of biological activity of preproendothelin [110–130] in several endothelin assays. Life Sci. 1990;47:2097–2103. doi: 10.1016/0024-3205(90)90308-e. [DOI] [PubMed] [Google Scholar]
- Chang I, Bramall AN, Baynash AG, Rattner A, Rakheja D, Post M, et al. Endothelin-2 deficiency causes growth retardation, hypothermia, and emphysema in mice. J Clin Invest. 2013;123:2643–2653. doi: 10.1172/JCI66735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianfrocca R, Tocci P, Semprucci E, Spinella F, Di Castro V, Bagnato A, et al. Beta-arrestin 1 is required for endothelin-1-induced NF-kappaB activation in ovarian cancer cells. Life Sci. 2014 doi: 10.1016/j.lfs.2014.01.078. doi: 10.1016/j.lfs.2014.01.078. [DOI] [PubMed] [Google Scholar]
- Clouthier DE, Hosoda K, Richardson JA, Williams SC, Yanagisawa H, Kuwaki T, et al. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development. 1998;125:813–824. doi: 10.1242/dev.125.5.813. [DOI] [PubMed] [Google Scholar]
- Clozel M, Gray GA. Are there different ETB receptors mediating constriction and relaxation? J Cardiovasc Pharmacol. 1995;26(Suppl. 3):S262–S264. [PubMed] [Google Scholar]
- Compeer MG, Janssen GM, De Mey JG. Endothelin-1 and endothelin-2 initiate and maintain contractile responses by different mechanisms in rat mesenteric and cerebral arteries. Br J Pharmacol. 2013;170:1199–1209. doi: 10.1111/bph.12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport AP. International Union of Pharmacology. XXIX. Update on endothelin receptor nomenclature. Pharmacol Rev. 2002;54:219–226. doi: 10.1124/pr.54.2.219. [DOI] [PubMed] [Google Scholar]
- Davenport AP, Maguire JJ. Endothelin. Handb Exp Pharmacol. 2006;152:295–329. doi: 10.1007/3-540-32967-6_9. [DOI] [PubMed] [Google Scholar]
- Davenport AP, Ashby MJ, Easton P, Ella S, Bedford J, Dickerson C, et al. A sensitive radioimmunoassay measuring endothelin-like immunoreactivity in human plasma: comparison of levels in patients with essential hypertension and normotensive control subjects. Clin Sci (Lond) 1990;78:261–264. doi: 10.1042/cs0780261. [DOI] [PubMed] [Google Scholar]
- Davenport AP, Alexander SP, Sharman JL, Pawson AJ, Benson HE, Monaghan AE, et al. International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacol Rev. 2013;65:967–986. doi: 10.1124/pr.112.007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lombaert S, Ghai RD, Jeng AY, Trapani AJ, Webb RL. Pharmacological profile of a non-peptidic dual inhibitor of neutral endopeptidase 24.11 and endothelin-converting enzyme. Biochem Biophys Res Commun. 1994;204:407–412. doi: 10.1006/bbrc.1994.2473. [DOI] [PubMed] [Google Scholar]
- Dhaun N, Goddard J, Webb DJ. Endothelin antagonism in patients with nondiabetic chronic kidney disease. Contrib Nephrol. 2011;172:243–254. doi: 10.1159/000328704. [DOI] [PubMed] [Google Scholar]
- Dhaun N, Webb DJ, Kluth DC. Endothelin-1 and the kidney-beyond BP. Br J Pharmacol. 2012;167:720–731. doi: 10.1111/j.1476-5381.2012.02070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickstein K, De Voogd HJ, Miric MP, Willenbrock R, Mitrovic V, Pacher R, et al. Effect of single doses of SLV306, an inhibitor of both neutral endopeptidase and endothelin-converting enzyme, on pulmonary pressures in congestive heart failure. Am J Cardiol. 2004;94:237–239. doi: 10.1016/j.amjcard.2004.03.074. [DOI] [PubMed] [Google Scholar]
- Docherty CC, Kalmar-Nagy J, Engelen M, Koenen SV, Nijland M, Kuc RE, et al. Effect of in vivo fetal infusion of dexamethasone at 0.75 GA on fetal ovine resistance artery responses to ET-1. Am J Physiol Regul Integr Comp Physiol. 2001;281:R261–R268. doi: 10.1152/ajpregu.2001.281.1.R261. [DOI] [PubMed] [Google Scholar]
- Don GW, Joseph F, Celermajer DS, Corte TJ. Ironic case of hepatic dysfunction following the global withdrawal of sitaxentan. Intern Med J. 2012;42:1351–1354. doi: 10.1111/imj.12007. [DOI] [PubMed] [Google Scholar]
- D'Orleans-Juste P, Houde M, Rae GA, Bkaily G, Carrier E, Simard E. Endothelin-1 (1–31): from chymase-dependent synthesis to cardiovascular pathologies. Vascul Pharmacol. 2008;49:51–62. doi: 10.1016/j.vph.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Drawnel FM, Archer CR, Roderick HL. The role of the paracrine/autocrine mediator endothelin-1 in regulation of cardiac contractility and growth. Br J Pharmacol. 2013;168:296–317. doi: 10.1111/j.1476-5381.2012.02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckman EA, Reed DK, Eckman CB. Degradation of the Alzheimer's amyloid beta peptide by endothelin-converting enzyme. J Biol Chem. 2001;276:24540–24548. doi: 10.1074/jbc.M007579200. [DOI] [PubMed] [Google Scholar]
- Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB. Alzheimer's disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J Biol Chem. 2003;278:2081–2084. doi: 10.1074/jbc.C200642200. [DOI] [PubMed] [Google Scholar]
- Eckman EA, Adams SK, Troendle FJ, Stodola BA, Kahn MA, Fauq AH, et al. Regulation of steady-state beta-amyloid levels in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme. J Biol Chem. 2006;281:30471–30478. doi: 10.1074/jbc.M605827200. [DOI] [PubMed] [Google Scholar]
- Emoto N, Yanagisawa M. Endothelin-converting enzyme-2 is a membrane-bound,phosphoramidon-sensitive metalloprotease with acidic pH optimum. J Biol Chem. 1995;270:15262–15268. doi: 10.1074/jbc.270.25.15262. [DOI] [PubMed] [Google Scholar]
- Fecteau MH, Honore JC, Plante M, Labonte J, Rae GA, D'Orleans-Juste P. Endothelin-1 (1–31) is an intermediate in the production of endothelin-1 after big endothelin-1 administration in vivo. Hypertension. 2005;46:87–92. doi: 10.1161/01.HYP.0000170460.24604.23. [DOI] [PubMed] [Google Scholar]
- Flynn MA, Haleen SJ, Welch KM, Cheng XM, Reynolds EE. Endothelin B receptors on human endothelial and smooth-muscle cells show equivalent binding pharmacology. J Cardiovasc Pharmacol. 1998;32:106–116. doi: 10.1097/00005344-199807000-00017. [DOI] [PubMed] [Google Scholar]
- Foord SM, Bonner TI, Neubig RR, Rosser EM, Pin JP, Davenport AP, et al. International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol Rev. 2005;57:279–288. doi: 10.1124/pr.57.2.5. [DOI] [PubMed] [Google Scholar]
- Gatfield J, Mueller Grandjean C, Sasse T, Clozel M, Nayler O. Slow receptor dissociation kinetics differentiate macitentan from other endothelin receptor antagonists in pulmonary arterial smooth muscle cells. PLoS ONE. 2012;7:e47662. doi: 10.1371/journal.pone.0047662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaid A, Gibson SJ, Herrero MT, Gentleman S, Legon S, Yanagisawa M, et al. Topographical localisation of endothelin mRNA and peptide immunoreactivity in neurones of the human brain. Histochemistry. 1991;95:303–314. doi: 10.1007/BF00266781. [DOI] [PubMed] [Google Scholar]
- Gulati A, Rai A. Endothelin-1-induced vasodilatation in rat Breast tumor is mediated through endothelin-B receptors. J Cardiovasc Pharmacol. 2004;44:S483–S486. doi: 10.1097/01.fjc.0000166308.63808.16. [DOI] [PubMed] [Google Scholar]
- Gulati A, Sunila ES, Kuttan G. IRL-1620, an endothelin-B receptor agonist, enhanced radiation induced reduction in tumor volume in Dalton's Lymphoma Ascites tumor model. Arzneimittelforschung. 2012;62:14–17. doi: 10.1055/s-0031-1295430. [DOI] [PubMed] [Google Scholar]
- Hand MF, Haynes WG, Webb DJ. Reduced endogenous endothelin-1-mediated vascular tone in chronic renal failure. Kidney Int. 1999;55:613–620. doi: 10.1046/j.1523-1755.1999.00291.x. [DOI] [PubMed] [Google Scholar]
- Harland SP, Kuc RE, Pickard JD, Davenport AP. Characterization of endothelin receptors in human brain cortex, gliomas, and meningiomas. J Cardiovasc Pharmacol. 1995;26(Suppl. 3):S408–S411. [PubMed] [Google Scholar]
- Harland SP, Kuc RE, Pickard JD, Davenport AP. Expression of endothelin(A) receptors in human gliomas and meningiomas, with high affinity for the selective antagonist PD156707. Neurosurgery. 1998;43:890–898. doi: 10.1097/00006123-199810000-00097. [DOI] [PubMed] [Google Scholar]
- Haynes WG, Webb DJ. Contribution of endogenous generation of endothelin-1 to basal vascular tone. Lancet. 1994;344:852–854. doi: 10.1016/s0140-6736(94)92827-4. [DOI] [PubMed] [Google Scholar]
- Hocher B, Heiden S, von Websky K, Rahnenfuhre J, Kalk P, Pfab T. Dual endothelin-converting enzyme/neutral endopeptidase blockade in rats with D-galactosamine-induced liver failure. Eur J Med Res. 2011;16:275–279. doi: 10.1186/2047-783X-16-6-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda K, Hammer RE, Richardson JA, Baynash AG, Cheung JC, Giaid A, et al. Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell. 1994;79:1267–1276. doi: 10.1016/0092-8674(94)90017-5. [DOI] [PubMed] [Google Scholar]
- Howard PG, Plumpton C, Davenport AP. Anatomical localization and pharmacological activity of mature endothelins and their precursors in human vascular tissue. J Hypertens. 1992;10:1379–1386. doi: 10.1097/00004872-199211000-00010. [DOI] [PubMed] [Google Scholar]
- Hyndman KA, Pollock JS. Nitric oxide and the A and B of endothelin of sodium homeostasis. Curr Opin Nephrol Hypertens. 2013;22:26–31. doi: 10.1097/MNH.0b013e32835b4edc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglarz M, Binkert C, Morrison K, Fischli W, Gatfield J, Treiber A, et al. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther. 2008;327:736–745. doi: 10.1124/jpet.108.142976. [DOI] [PubMed] [Google Scholar]
- Ihara M, Noguchi K, Saeki T, Fukuroda T, Tsuchida S, Kimura S, et al. Biological profiles of highly potent novel endothelin antagonists selective for the ET(A) receptor. Life Sci. 1992;50:247–255. doi: 10.1016/0024-3205(92)90331-i. [DOI] [PubMed] [Google Scholar]
- Ikeda S, Emto N, Alimsardjono H, Yokoyama M, Matsuo M. Molecular isolation and characterization of novel four subisoforms of ECE-2. Biochem Biophys Res Commun. 2002;293:421–426. doi: 10.1016/S0006-291X(02)00252-8. [DOI] [PubMed] [Google Scholar]
- Inoue A, Yanagisawa M, Kimura S, Kasuya Y, Miyauchi T, Goto K, et al. The human endothelin family: three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc Natl Acad Sci U S A. 1989;86:2863–2867. doi: 10.1073/pnas.86.8.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa K, Ihara M, Noguchi K, Mase T, Mino N, Saeki T, et al. Biochemical and pharmacological profile of a potent and selective endothelinB-receptor antagonist, BQ-788. Proc Natl Acad Sci U S A. 1994;91:4892–4896. doi: 10.1073/pnas.91.11.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Matsumiya K, Ohtsuki S, Kamiie J, Terasaki T. Contributions of degradation and brain-to-blood elimination across the blood–brain barrier to cerebral clearance of human amyloid-beta peptide(1–40) in mouse brain. J Cereb Blood Flow Metab. 2013;33:1770–1777. doi: 10.1038/jcbfm.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeng AY, Mulder P, Kwan AL, Battistini B. Nonpeptidic endothelin-converting enzyme inhibitors and their potential therapeutic applications. Can J Physiol Pharmacol. 2002;80:440–449. doi: 10.1139/y02-025. [DOI] [PubMed] [Google Scholar]
- Johnström P, Fryer TD, Richards HK, Maguire JJ, Clark JC, Pickard JD, et al. Positron emission tomography of [18F]-big endothelin-1 reveals renal excretion but tissue-specific conversion to [18F]-endothelin-1 in lung and liver. Br J Pharmacol. 2010;159:812–819. doi: 10.1111/j.1476-5381.2010.00641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalk P, Sharkovska Y, Kashina E, von Websky K, Relle K, Pfab T, et al. Endothelin-converting enzyme/neutral endopeptidase inhibitor SLV338 prevents hypertensive cardiac remodeling in a blood pressure-independent manner. Hypertension. 2011;57:755–763. doi: 10.1161/HYPERTENSIONAHA.110.163972. [DOI] [PubMed] [Google Scholar]
- Karet FE, Davenport AP. Localization of endothelin peptides in human kidney. Kidney Int. 1996;49:382–387. doi: 10.1038/ki.1996.56. [DOI] [PubMed] [Google Scholar]
- Kelland NF, Kuc RE, McLean DL, Azfer A, Bagnall AJ, Gray GA, et al. Endothelial cell-specific ETB receptor knockout: autoradiographic and histological characterisation and crucial role in the clearance of endothelin-1. Can J Physiol Pharmacol. 2010;88:644–651. doi: 10.1139/Y10-041. [DOI] [PubMed] [Google Scholar]
- Kelland NF, Webb DJ. Clinical trials of endothelin antagonists in heart failure: a question of dose? Exp Biol Med (Maywood) 2006;231:696–699. [PubMed] [Google Scholar]
- Kiowski W, Luscher TF, Linder L, Buhler FR. Endothelin-1-induced vasoconstriction in humans. Reversal by calcium channel blockade but not by nitrovasodilators or endothelium-derived relaxing factor. Circulation. 1991;83:469–475. doi: 10.1161/01.cir.83.2.469. [DOI] [PubMed] [Google Scholar]
- Kirkby NS, Hadoke PW, Bagnall AJ, Webb DJ. The endothelin system as a therapeutic target in cardiovascular disease: great expectations or bleak house? J Pharmacol. 2008;153:1105–1119. doi: 10.1038/sj.bjp.0707516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko C, Gieske MC, Al-Alem L, Hahn Y, Su W, Gong MC, et al. Endothelin-2 in ovarian follicle rupture. Endocrinology. 2006;147:1770–1779. doi: 10.1210/en.2005-1228. [DOI] [PubMed] [Google Scholar]
- Ko C, Meidan R, Bridges PJ. Why two endothelins and two receptors for ovulation and luteal regulation? Life Sci. 2012;91:501–506. doi: 10.1016/j.lfs.2012.05.010. [DOI] [PubMed] [Google Scholar]
- Kohan DE, Inscho EW, Wesson D, Pollock DM. Physiology of endothelin and the kidney. Compr Physiol. 2011a;1:883–919. doi: 10.1002/cphy.c100039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev. 2011b;91:1–77. doi: 10.1152/physrev.00060.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan DE, Cleland JG, Rubin LJ, Theodorescu D, Barton M. Clinical trials with endothelin receptor antagonists: what went wrong and where can we improve? Life Sci. 2012;91:528–539. doi: 10.1016/j.lfs.2012.07.034. [DOI] [PubMed] [Google Scholar]
- Kuc RE, Carlebur M, Maguire JJ, Yang P, Long L, Toshner M, et al. Modulation of endothelin receptors in the failing right ventricle of the heart and vasculature of the lung in human pulmonary arterial hypertension. Life Sci. 2014 doi: 10.1016/j.lfs.2014.02.020. doi: 10.1016/j.lfs.2014.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara Y, Kurihara H, Suzuki H, Kodama T, Maemura K, Nagai R, et al. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature. 1994;368:703–710. doi: 10.1038/368703a0. [DOI] [PubMed] [Google Scholar]
- Kutzler MA, Molnar J, Schlafer DH, Kuc RE, Davenport AP, Nathanielsz PW. Maternal dexamethasone increases endothelin-1 sensitivity and endothelin a receptor expression in ovine foetal placental arteries. Placenta. 2003;24:392–402. doi: 10.1053/plac.2002.0920. [DOI] [PubMed] [Google Scholar]
- Lee S, Lin M, Mele A, Cao Y, Farmar J, Russo D, et al. Proteolytic processing of big endothelin-3 by the kell blood group protein. Blood. 1999;94:1440–1450. [PubMed] [Google Scholar]
- Lenaz L, Gulati A, Sunila E. IRL-1620 increases the efficacy of radiation treatment in mice bearing lymphoma cell induced tumors. Blood. 2006;108:269B. [Google Scholar]
- Leonard MG, Gulati A. Endothelin B receptor agonist, IRL-1620, enhances angiogenesis and neurogenesis following cerebral ischemia in rats. Brain Res. 2013;1528:28–41. doi: 10.1016/j.brainres.2013.07.002. [DOI] [PubMed] [Google Scholar]
- Leonard MG, Briyal S, Gulati A. Endothelin B receptor agonist, IRL-1620, reduces neurological damage following permanent middle cerebral artery occlusion in rats. Brain Res. 2011;1420:48–58. doi: 10.1016/j.brainres.2011.08.075. [DOI] [PubMed] [Google Scholar]
- Leonard MG, Briyal S, Gulati A. Endothelin B receptor agonist, IRL-1620, provides long-term neuroprotection in cerebral ischemia in rats. Brain Res. 2012;1464:14–23. doi: 10.1016/j.brainres.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Ling L, Maguire JJ, Davenport AP. Endothelin-2, the forgotten isoform: emerging role in the cardiovascular system, ovarian development, immunology and cancer. Br J Pharmacol. 2013;168:283–295. doi: 10.1111/j.1476-5381.2011.01786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Chen J, Gao Y, Deng B, Liu K. Endothelin receptor antagonists for pulmonary arterial hypertension. Cochrane Database Syst Rev. 2013;(2) doi: 10.1002/14651858.CD004434.pub5. CD004434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo MN, Khan RY, Wang Y, Tai SC, Chan GC, Cheung AH, et al. Human endothelin converting enzyme-2 (ECE2): characterization of mRNA species and chromosomal localization. Biochim Biophys Acta. 2001;1522:46–52. doi: 10.1016/s0167-4781(01)00283-4. [DOI] [PubMed] [Google Scholar]
- Love MP, Ferro CJ, Haynes WG, Plumpton C, Davenport AP, Webb DJ, et al. Endothelin receptor antagonism in patients with chronic heart failure. Cardiovasc Res. 2000;47:166–172. doi: 10.1016/s0008-6363(00)00081-x. [DOI] [PubMed] [Google Scholar]
- Lucas GA, White LR, Juul R, Cappelen J, Aasly J, Edvinsson L. Relaxation of human temporal artery by endothelin ETB receptors. Peptides. 1996;17:1139–1144. doi: 10.1016/s0196-9781(96)00177-5. [DOI] [PubMed] [Google Scholar]
- Luttrell LM. Minireview: more than just a hammer: ligand ‘bias’ and pharmaceutical discovery. Mol Endocrinol. 2014;28:281–294. doi: 10.1210/me.2013-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire JJ, Davenport AP. Increased response to big endothelin-1 in atherosclerotic human coronary artery: functional evidence for up-regulation of endothelin-converting enzyme activity in disease. Br J Pharmacol. 1998;125:238–240. doi: 10.1038/sj.bjp.0702102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire JJ, Davenport AP. Alternative pathway to endothelin-converting enzyme for the synthesis of endothelin in human blood vessels. J Cardiovasc Pharmacol. 2004;44(Suppl. 1):S27–S29. doi: 10.1097/01.fjc.0000166219.65593.af. [DOI] [PubMed] [Google Scholar]
- Maguire JJ, Johnson CM, Mockridge JW, Davenport AP. Endothelin converting enzyme (ECE). Activity in human vascular smooth muscle. Br J Pharmacol. 1997;122:1647–1654. doi: 10.1038/sj.bjp.0701564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire JJ, Kuc RE, Davenport AP. Vasoconstrictor activity of novel endothelin peptide, ET-1(1–31), in human mammary and coronary arteries in vitro. Br J Pharmacol. 2001;134:1360–1366. doi: 10.1038/sj.bjp.0704384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire JJ, Kuc RE, Pell VR, Green A, Brown M, Kumar S, et al. Comparison of human ETA and ETB receptor signalling via G-protein and beta-arrestin pathways. Life Sci. 2012;91:544–549. doi: 10.1016/j.lfs.2012.03.021. [DOI] [PubMed] [Google Scholar]
- Marciniak SJ, Plumpton C, Barker PJ, Huskisson NS, Davenport AP. Localization of immunoreactive endothelin and proendothelin in the human lung. Pulm Pharmacol. 1992;5:175–182. doi: 10.1016/0952-0600(92)90038-i. [DOI] [PubMed] [Google Scholar]
- Meil J, Wurl M, Thormahlen D, Rose H. Pharmacology of the active metabolite of SLV 306 – a mixed inhibitor of NEP and ECE. Naunyn Schmiedebergs Arch Pharmacol. 1998;358(Suppl. 1):R513. [Google Scholar]
- Meyer RC, Giddens MM, Schaefer SA, Hall RA. GPR37 and GPR37L1 are receptors for the neuroprotective and glioprotective factors prosaptide and prosaposin. Proc Natl Acad Sci U S A. 2013;110:9529–9534. doi: 10.1073/pnas.1219004110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi T, Nishiyama M, Moroi K, Tanaka H, Saito T, Masuda Y, et al. Analysis of two pharmacologically predicted endothelin B receptor subtypes by using the endothelin B receptor gene knockout mouse. Br J Pharmacol. 1997;120:1427–1430. doi: 10.1038/sj.bjp.0701054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar P, Kuc RE, Davenport AP. Characterization of two new ETB selective radioligands, [125I]-BQ3020 and [125I]-[Ala1,3,11,15]ET-1 in human heart. Br J Pharmacol. 1992;107:637–639. doi: 10.1111/j.1476-5381.1992.tb14498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar P, O'Reilly G, Sharkey A, Kuc RE, Harding DP, Plumpton C, et al. Characterization and localization of endothelin receptor subtypes in the human atrioventricular conducting system and myocardium. Circ Res. 1993;72:526–538. doi: 10.1161/01.res.72.3.526. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Quertermous T, Hong CC, Bloch KD. Regional and maturation-associated expression of endothelin 2 in rat gastrointestinal tract. J Histochem Cytochem. 1995;43:203–209. doi: 10.1177/43.2.7822776. [DOI] [PubMed] [Google Scholar]
- Morton AJ, Davenport AP. Cerebellar neurons and glia respond differentially to endothelins and sarafotoxin S6b. Brain Res. 1992;581:299–306. doi: 10.1016/0006-8993(92)90721-k. [DOI] [PubMed] [Google Scholar]
- Nambi P, Pullen M, Spielman W. Species differences in the binding characteristics of [125I]IRL-1620, a potent agonist specific for endothelin-B receptors. J Pharmacol Exp Ther. 1994;268:202–207. [PubMed] [Google Scholar]
- Nelissen J, Lemkens P, Sann H, Bindl M, Bassissi F, Jasserand D, et al. Pharmacokinetic and pharmacodynamic properties of SOL1: a novel dual inhibitor of neutral endopeptidase and endothelin converting enzyme. Life Sci. 2012;91:587–592. doi: 10.1016/j.lfs.2012.01.015. [DOI] [PubMed] [Google Scholar]
- Ohkita M, Tawa M, Kitada K, Matsumura Y. Pathophysiological roles of endothelin receptors in cardiovascular diseases. J Pharmacol Sci. 2012;119:302–313. doi: 10.1254/jphs.12r01cr. [DOI] [PubMed] [Google Scholar]
- O'Reilly G, Charnock-Jones DS, Morrison JJ, Cameron IT, Davenport AP, Smith SK. Alternatively spliced mRNAs for human endothelin-2 and their tissue distribution. Biochem Biophys Res Commun. 1993;193:834–840. doi: 10.1006/bbrc.1993.1701. [DOI] [PubMed] [Google Scholar]
- Pacheco-Quinto J, Eckman EA. Endothelin-converting enzymes degrade intracellular beta-amyloid produced within the endosomal/lysosomal pathway and autophagosomes. J Biol Chem. 2013;288:5606–5615. doi: 10.1074/jbc.M112.422964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanisamy GS, Cheon YP, Kim J, Kannan A, Li Q, Sato M, et al. A novel pathway involving progesterone receptor, endothelin-2, and endothelin receptor B controls ovulation in mice. Mol Endocrinol. 2006;20:2784–2795. doi: 10.1210/me.2006-0093. [DOI] [PubMed] [Google Scholar]
- Palmer MJ. Endothelin receptor antagonists: status and learning 20 years on. Prog Med Chem. 2009;47:203–237. doi: 10.1016/S0079-6468(08)00205-1. [DOI] [PubMed] [Google Scholar]
- Parvanova A, van der Meer IM, Iliev I, Perna A, Gaspari F, Trevisan R, et al. Effect on blood pressure of combined inhibition of endothelin-converting enzyme and neutral endopeptidase with daglutril in patients with type 2 diabetes who have albuminuria: a randomised, crossover, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2013;1:19–27. doi: 10.1016/S2213-8587(13)70029-9. [DOI] [PubMed] [Google Scholar]
- Patel T, McKeage K. Macitentan: first global approval. Drugs. 2014;74:127–133. doi: 10.1007/s40265-013-0156-6. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernow J, Shemyakin A, Böhm F. New perspectives on endothelin-1 in atherosclerosis and diabetes mellitus. Life Sci. 2012;91:507–516. doi: 10.1016/j.lfs.2012.03.029. [DOI] [PubMed] [Google Scholar]
- Peter MG, Davenport AP. Delineation of endothelin receptors in human left ventricular smooth-muscle cells. J Cardiovasc Pharmacol. 1995;26(Suppl. 3):S355–S357. [PubMed] [Google Scholar]
- Peter MG, Davenport AP. Characterization of the endothelin receptor selective agonist, BQ3020 and antagonists BQ123, FR139317, BQ788, 50235, Ro462005 and bosentan in the heart. Br J Pharmacol. 1996;117:455–462. doi: 10.1111/j.1476-5381.1996.tb15212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre LN, Davenport AP. Blockade and reversal of endothelin-induced constriction in pial arteries from human brain. Stroke. 1999;30:638–643. doi: 10.1161/01.str.30.3.638. [DOI] [PubMed] [Google Scholar]
- Plumpton C, Ferro CJ, Haynes WG, Webb DJ, Davenport AP. The increase in human plasma immunoreactive endothelin but not big endothelin-1 or its C-terminal fragment induced by systemic administration of the endothelin antagonist TAK-044. Br J Pharmacol. 1996a;119:311–314. doi: 10.1111/j.1476-5381.1996.tb15987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumpton C, Ashby MJ, Kuc RE, O'Reilly G, Davenport AP. Expression of endothelin peptides and mRNA in the human heart. Clin Sci (Lond) 1996b;90:37–46. doi: 10.1042/cs0900037. [DOI] [PubMed] [Google Scholar]
- Prinjha RK, Witherington J, Lee K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol Sci. 2012;33:146–153. doi: 10.1016/j.tips.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Pulido T, Adzerikho I, Channick RN, Delcroix M, Galie N, Ghofrani HA, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–818. doi: 10.1056/NEJMoa1213917. [DOI] [PubMed] [Google Scholar]
- Rai A, Rajeshkumar NV, Gulati A. Effect of the ETB receptor agonist, IRL-1620, on paclitaxel plasma pharmacokinetics of breast tumor rats. Exp Biol Med. 2006;231:1120–1122. [PubMed] [Google Scholar]
- Rajeshkumar NV, Rai A, Gulati A. Endothelin B receptor agonist, IRL 1620, a novel adjuvant to enhance the delivery and efficacy of paclitaxel in a rat mammary tumor model. Clin Cancer Res. 2005a;11:9138S–9239S. doi: 10.1007/s10549-005-9000-3. [DOI] [PubMed] [Google Scholar]
- Rajeshkumar NV, Rai A, Gulati A. Endothelin B receptor agonist, IRL 1620, enhances the anti-tumor efficacy of paclitaxel in breast tumor rats. Breast Cancer Res Treat. 2005b;94:237–247. doi: 10.1007/s10549-005-9000-3. [DOI] [PubMed] [Google Scholar]
- Rattner A, Yu H, Williams J, Smallwood PM, Nathans J. Endothelin-2 signaling in the neural retina promotes the endothelial tip cell state and inhibits angiogenesis. Proc Natl Acad Sci U S A. 2013;110:E3830–E3839. doi: 10.1073/pnas.1315509110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautureau Y, Schiffrin E. Endothelin in hypertension: an update. Curr Opin Nephrol Hypertens. 2012;21:128–136. doi: 10.1097/MNH.0b013e32834f0092. [DOI] [PubMed] [Google Scholar]
- Reynolds EE, Keiser JA, Haleen SJ, Walker DM, Olszewski B, Schroeder RL, et al. Pharmacological characterization of PD 156707, an orally active ETA receptor antagonist. J Pharmacol Exp Ther. 1995;273:1410–1417. [PubMed] [Google Scholar]
- Rosanò L, Cianfrocca R, Masi S, Spinella F, Di Castro V, Biroccio A, et al. β-Arrestin links endothelin A receptor to beta-catenin signaling to induce ovarian cancer cell invasion and metastasis. Proc Natl Acad Sci U S A. 2009;106:2806–2811. doi: 10.1073/pnas.0807158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosanò L, Cianfrocca R, Tocci P, Spinella F, Di Castro V, Spadaro F, et al. β-Arrestin-1 is a nuclear transcriptional regulator of endothelin-1-induced beta-catenin signaling. Oncogene. 2013a;32:5066–5077. doi: 10.1038/onc.2012.527. [DOI] [PubMed] [Google Scholar]
- Rosanò L, Spinella F, Bagnato A. Endothelin 1 in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2013b;13:637–651. doi: 10.1038/nrc3546. [DOI] [PubMed] [Google Scholar]
- Rubin LJ. Endothelin receptor antagonists for the treatment of pulmonary artery hypertension. Life Sci. 2012;91:517–521. doi: 10.1016/j.lfs.2012.07.033. [DOI] [PubMed] [Google Scholar]
- Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]
- Russell FD, Davenport AP. Characterization of the binding of endothelin ETB selective ligands in human and rat heart. Br J Pharmacol. 1996;119:631–636. doi: 10.1111/j.1476-5381.1996.tb15720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell FD, Davenport AP. Evidence for intracellular endothelin-converting enzyme-2 expression in cultured human vascular endothelial cells. Circ Res. 1999a;84:891–896. doi: 10.1161/01.res.84.8.891. [DOI] [PubMed] [Google Scholar]
- Russell FD, Davenport AP. Secretory pathways in endothelin synthesis. Br J Pharmacol. 1999b;26:391–398. doi: 10.1038/sj.bjp.0702315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell FD, Skepper JN, Davenport AP. Human endothelial cell storage granules: a novel intracellular site for isoforms of the endothelin-converting enzyme. Circ Res. 1998a;83:314–321. doi: 10.1161/01.res.83.3.314. [DOI] [PubMed] [Google Scholar]
- Russell FD, Skepper JN, Davenport AP. Evidence using immunoelectron microscopy for regulated and constitutive pathways in the transport and release of endothelin. J Cardiovasc Pharmacol. 1998b;31:424–430. doi: 10.1097/00005344-199803000-00014. [DOI] [PubMed] [Google Scholar]
- Said N, Theodorescu D. Permissive role of endothelin receptors in tumor metastasis. Life Sci. 2012;91:522–527. doi: 10.1016/j.lfs.2012.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Yanagisawa M, Takuwa Y, Miyazaki H, Kimura S, Goto K, et al. Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature. 1990;348:732–735. doi: 10.1038/348732a0. [DOI] [PubMed] [Google Scholar]
- Seed A, Kuc RE, Maguire JJ, Hillier C, Johnston F, Essers H, et al. The dual endothelin converting enzyme/neutral endopeptidase inhibitor SLV-306 (daglutril), inhibits systemic conversion of big endothelin-1 in humans. Life Sci. 2012;91:743–748. doi: 10.1016/j.lfs.2012.03.022. [DOI] [PubMed] [Google Scholar]
- Seyrantepe V, Hinek A, Peng J, Fedjaev M, Ernest S, Kadota Y, et al. Enzymatic activity of lysosomal carboxypeptidase (cathepsin) A is required for proper elastic fiber formation and inactivation of endothelin-1. Circulation. 2008;117:1973–1981. doi: 10.1161/CIRCULATIONAHA.107.733212. [DOI] [PubMed] [Google Scholar]
- Sharkovska Y, Kalk P, von Websky K, Relle K, Pfab T, Alter M, et al. Renoprotective effects of combined endothelin-converting enzyme/neutral endopeptidase inhibitor SLV338 in acute and chronic experimental renal damage. Clin Lab. 2011;57:507–515. [PubMed] [Google Scholar]
- Sidharta PN, van Giersbergen PL, Halabi A, Dingemanse J. Macitentan: entry-into-humans study with a new endothelin receptor antagonist. Eur J Clin Pharmacol. 2011;67:977–984. doi: 10.1007/s00228-011-1043-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidharta PN, Lindegger N, Ulc I, Dingemanse J. Pharmacokinetics of the novel dual endothelin receptor antagonist macitentan in subjects with hepatic or renal impairment. J Clin Pharmacol. 2013a;55:291–300. doi: 10.1002/jcph.193. [DOI] [PubMed] [Google Scholar]
- Sidharta PN, van Giersbergen PL, Dingemanse J. Safety, tolerability, pharmacokinetics, and pharmacodynamics of macitentan, an endothelin receptor antagonist, in an ascending multiple-dose study in healthy subjects. J Clin Pharmacol. 2013b;54:291–300. doi: 10.1002/jcph.152. [DOI] [PubMed] [Google Scholar]
- Skolovsky M, Galron R, Kloog Y, Bdolah A, Indig FE, Blumberg S, et al. Endothelins are more sensitive than sarafotoxins to neutral endopeptidase: possible physiological significance. Proc Natl Acad Sci U S A. 1990;87:4702–4706. doi: 10.1073/pnas.87.12.4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speed JS, Pollock DM. Endothelin, kidney disease, and hypertension. Hypertension. 2013;61:1142–1145. doi: 10.1161/HYPERTENSIONAHA.113.00595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasaki C, Tamiya N, Bdolah A, Wollberg Z, Kochva E. Sarafotoxins S6: several isotoxins from Atractaspis engaddensis (burrowing asp) venom that affect the heart. Toxicon. 1988;26:543–548. doi: 10.1016/0041-0101(88)90234-6. [DOI] [PubMed] [Google Scholar]
- Takai M, Umemura I, Yamasaki K, Watakabe T, Fujitani Y, Oda K, et al. A potent and specific agonist, Suc-[Glu9,Ala11,15]-endothelin-1(8–21), IRL 1620, for the ETB receptor. Biochem Biophys Res Commun. 1992;184:953–959. doi: 10.1016/0006-291x(92)90683-c. [DOI] [PubMed] [Google Scholar]
- Takizawa S, Uchide T, Adur J, Kozakai T, Kotake-Nara E, Quan J, et al. Differential expression of endothelin-2 along the mouse intestinal tract. J Mol Endocrinol. 2005;35:201–209. doi: 10.1677/jme.1.01787. [DOI] [PubMed] [Google Scholar]
- Thone-Reinke C, Simon K, Richter CM, Godes M, Neumayer HH, Thormahlen D, et al. Inhibition of both neutral endopeptidase and endothelin-converting enzyme by SLV306 reduces proteinuria and urinary albumin excretion in diabetic rats. J Cardiovasc Pharmacol. 2004;44:S76–S79. doi: 10.1097/01.fjc.0000166208.12297.8d. [DOI] [PubMed] [Google Scholar]
- Turner AJ, Murphy LJ. Molecular pharmacology of endothelin converting enzymes. Biochem Pharmacol. 1996;26:91–102. doi: 10.1016/0006-2952(95)02036-5. [DOI] [PubMed] [Google Scholar]
- Turner AJ, Tanzawa K. Mammalian membrane metallopeptidases: NEP, ECE, KELL, and PEX. FASEB J. 1997;11:355–364. doi: 10.1096/fasebj.11.5.9141502. [DOI] [PubMed] [Google Scholar]
- Turner AJ, Isaac RE, Coates D. The neprilysin (NEP). family of zinc metalloendopeptidases: genomics and function. Bioessays. 2001;23:261–269. doi: 10.1002/1521-1878(200103)23:3<261::AID-BIES1036>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Vatter H, Seifert V. Ambrisentan, a non-peptide endothelin receptor antagonist. Cardiovasc Drug Rev. 2006;24:63–76. doi: 10.1111/j.1527-3466.2006.00063.x. [DOI] [PubMed] [Google Scholar]
- Vignon-Zellweger N, Heiden S, Miyauchi T, Emoto N. Endothelin and endothelin receptors in the renal and cardiovascular systems. Life Sci. 2012;91:490–500. doi: 10.1016/j.lfs.2012.03.026. [DOI] [PubMed] [Google Scholar]
- Wang R, Lohr CV, Fischer K, Dashwood WM, Greenwood JA, Ho E, et al. Epigenetic inactivation of endothelin-2 and endothelin-3 in colon cancer. Int J Cancer. 2013;132:1004–1012. doi: 10.1002/ijc.27762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb DJ. Endogenous endothelin generation maintains vascular tone in humans. J Hum Hypertens. 1995;9:459–463. [PubMed] [Google Scholar]
- Weiss J, Theile D, Ruppell MA, Speck T, Spalwisz A, Haefeli WE. Interaction profile of macitentan, a new non-selective endothelin-1 receptor antagonist, in vitro. Eur J Pharmacol. 2013;701:168–175. doi: 10.1016/j.ejphar.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Welch AK, Jacobs ME, Wingo CS, Cain BD. Early progress in epigenetic regulation of endothelin pathway genes. Br J Pharmacol. 2013;168:327–334. doi: 10.1111/j.1476-5381.2012.01826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengenmayer C, Krikov M, Mueller S, Lucht K, Villringer A, Hocher B, et al. Novel therapy approach in primary stroke prevention: simultaneous inhibition of endothelin converting enzyme and neutral endopeptidase in spontaneously hypertensive, stroke-prone rats improves survival. Neurol Res. 2011;33:201–207. doi: 10.1179/016164111X12881719352534. [DOI] [PubMed] [Google Scholar]
- William DL, Jones KL, Pettibone DJ, Lis EV, Clineschmidt BV. Sarafotoxin S6c, an agonist which distinguishes between endothelin receptor subtypes. Biochem Biophys Res Commun. 1991;175:556–561. doi: 10.1016/0006-291x(91)91601-8. [DOI] [PubMed] [Google Scholar]
- Xu D, Emoto N, Giaid A, Slaughter C, Kaw S, deWit D, et al. ECE-1: a membrane-bound metalloprotease that catalyzes the proteolytic activation of big endothelin-1. Cell. 1994;78:473–485. doi: 10.1016/0092-8674(94)90425-1. [DOI] [PubMed] [Google Scholar]
- Xu XF, Lv Y, Gu WZ, Tang LL, Wei JK, Zhang LY, et al. Epigenetics of hypoxic pulmonary arterial hypertension following intrauterine growth retardation rat: epigenetics in PAH following IUGR. Respir Res. 2013;14:20. doi: 10.1186/1465-9921-14-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa H, Yanagisawa M, Kapur RP, Richardson JA, Williams SC, Clouthier DE, et al. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development. 1998;125:825–836. doi: 10.1242/dev.125.5.825. [DOI] [PubMed] [Google Scholar]
- Yanagisawa H, Hammer RE, Richardson JA, Emoto N, Williams SC, Takeda SI, et al. Disruption of ECE-1 and ECE-2 reveals a role for endothelin-converting enzyme-2 in murine cardiac development. J Clin Invest. 2000;105:1373–1382. doi: 10.1172/JCI7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa M, Inoue A, Ishikawa T, Kasuya Y, Kimura S, Kumagaye S, et al. Primary structure, synthesis, and biological activity of rat endothelin, an endothelium-derived vasoconstrictor peptide. Proc Natl Acad Sci U S A. 1988;85:6964–6967. doi: 10.1073/pnas.85.18.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap EY, Battistini B, McKay KO. Contraction to big endothelin-1, big endothelin-2 and big endothelin-3, and endothelin-converting enzyme inhibition in human isolated bronchi. Br J Pharmacol. 2000;129:170–176. doi: 10.1038/sj.bjp.0703006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzgulen J, Wood EG, Villar IC, Douthwaite JA, Patel NSA, Jegard J, et al. Characterisation of the ‘endothelin-like domain peptide’ (ELDP) co-synthesised with endothelin-1 from the EDN1 gene. Life Sciences. 2013;93 DOI: 10.1016/j.lfs.2013.12.037. [Google Scholar]
- Zolk O, Quattek J, Sitzler G, Schrader T, Nickenig G, Schnabel P, et al. Expression of endothelin-1, endothelin-converting enzyme, and endothelin receptors in chronic heart failure. Circulation. 1999;99:2118–2123. doi: 10.1161/01.cir.99.16.2118. [DOI] [PubMed] [Google Scholar]