

Abstract
Background and Purpose
Neurodegenerative diseases are now recognized to be multifunctional, whereby a heterogeneous set of reactions acts independently or cooperatively, leading eventually to the demise of neurons. This has led our group to design and synthesize the multifunctional, nontoxic, brain-permeable, iron chelator compound M30 with a range of pharmacological properties. Here, we have characterized the molecular targets of M30 in the brains of animal models of type 2 diabetes mellitus (T2DM).
Experimental Approach
Effects of M30 on molecular mechanisms associated with neuroprotection in the CNS were investigated-in the high-fat diet (HFD) and ob/ob transgenic mouse models of T2DM, using real-time PCR and Western blotting analyses. Brain monoamine oxidase (MAO) activity and catecholamine levels, and peripheral glucose tolerance were assayed after treatment in vivo.
Key Results
M30 increased cerebral levels of insulin and insulin receptor and phosphorylated-GSK-3β in HFD mice, compared with vehicle-treated HFD mice. In both T2DM mice models, M30 treatment significantly up-regulated cerebral hypoxia-inducible factor (HIF)-1α protein levels and induced the expression of several HIF-1 target genes involved in neuroprotection, glycolysis, neurogenesis, oxidative stress and anti-inflammation. Additionally, M30 inhibited MAO-A and -B activities in the cerebellum. Accordingly, M30 administration significantly reduced brain levels of dopamine metabolites and increased levels of 5-HT and noradrenaline. Glucose tolerance was also improved after M30 treatment in both models of T2DM.
Conclusions and Implications
In the brain of HFD and ob/ob transgenic mice, M30 exerted a variety of beneficial neuroprotective regulatory effects that may act synergistically to delay or prevent neurodegenerative processes associated with T2DM.
Tables of Links
| TARGETS | LIGANDS |
|---|---|
| GLUT-1, glucose transporter 1 | 5-HT |
| GSK-3β, glycogen synthase kinase 3β | Deprenyl (selegiline) |
| HO-1, haem oxygenase | Dopamine |
| InsR, insulin receptor | Noradrenaline |
| Insulin | Rasagiline |
| MAO-A, monoamine oxidase A | |
| MAO-B, monoamine oxidase B |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013a,b,c,,).
Introduction
Preclinical and clinical evidence supports a pathophysiological link between diabetes mellitus (DM) and Alzheimer's disease (AD). Defective insulin signalling, insulin deficiency, impaired glucose metabolism, mitochondrial dysfunction, oxidative stress (OS) and pro-inflammatory cytokine activation in the brain are major neuropathological features of both DM and AD (Correia et al., 2012; Ahmad, 2013). Data from animal models have shown that diet-induced insulin resistance and hyperinsulinaemia in AD mice significantly increased the accumulation of amyloid-β (Aβ) and reduced the levels/activity of insulin degrading enzyme (Ho et al., 2004). In addition, cross-breeding AD mice with diabetic mice has demonstrated that diabetic conditions induced Aβ deposition, associated with exacerbation of memory and cognitive dysfunction (Takeda et al., 2010). Insulin resistance in the brain of rats treated i.c.v. with streptozotocin generated cognitive impairments and neurodegeneration (Grunblatt et al., 2007).
Because neurons are not able to synthesize or store glucose, they are dependent on its transport across the blood–brain barrier (BBB), mediated by glucose transporters (GLUTs) levels (Scheepers et al., 2004). Levels of GLUT-1 and GLUT-3 were significantly reduced in AD brains and this was associated with decreased τ hyperphosphorylation and the density of neurofibrillary tangles (Liu et al., 2008). Previous reports in aged double transgenic (Tg) mouse model of AD, carrying mutations in the amyloid precursor protein (APP) and presenelin 1 (PS1) genes, suggested that Aβ deposition was closely related to hippocampal atrophy and GLUT-1 levels. These studies also showed that hippocampal Aβ must reach a sufficient level before there is a reduction in GLUT-1 levels, leading to impairment in glucose metabolism and hippocampal atrophy (Hooijmans et al., 2007).
The concept of multifunctional and complex nature of neurodegenerative diseases led our group to develop a number of multifunctional, nontoxic, brain-permeable compounds with iron-chelation and anti-apoptotic properties. Among these compounds, M30 (5-[N-methyl-N-propargylaminomethyl]-8-hydroxyquinoline) was the most effective, exhibiting iron-chelating/radical scavenging potency and inhibition of iron-induced membrane lipid peroxidation. In addition, M30 was found to be a potent selective inhibitor of MAO-A and -B activities (Zheng et al., 2005a,b,). M30, was designed and synthesized from the prototype brain-permeable iron chelator, VK28, chemically attached to the propargyl moiety of the anti-Parkinsonian MAO-B inhibitor, rasagiline (Azilect®) (Gal et al., 2005; 2010,; Zheng et al., 2005a,b,; Avramovich-Tirosh et al., 2007; 2010,; Weinreb et al., 2010).
Several reports have suggested that the neuroprotective effects of iron-chelating agents can be attributed to their ability to prevent the iron from redox cycling and to inhibit hydroxyl radical formation via the Fenton reaction (Sayre et al., 2001). An additional level of neuroprotection by iron chelators indicates their involvement in inhibiting the iron-dependent hypoxia-inducible factor (HIF)-1α-prolyl-4-hydroxylase (PHD), resulting in stabilization and activation of HIF-1α and induction of HIF-1 target genes that may contribute to cell survival, increase glycolysis and inhibit mitochondrial production of reactive oxygen species in neurons (Zaman et al., 1999). In agreement, it was reported that M30 regulated a number of neuroprotective/adaptive and pro-survival signalling pathways in the brain that might function as therapeutic targets in neurodegenerative diseases (Gal et al., 2005; 2010,; Kupershmidt et al., 2009; 2012a,b,,).
The present study provides further insight into the molecular targets associated with neuroprotection, that were affected by the iron-chelating and propargyl moieties of M30, in the brains of mice with high-fat diet (HFD)-induced obesity and insulin resistance. These effects included regulation of insulin, insulin receptor (InsR)/phosphorylated glycogen synthase kinase (p-GSK)-3β levels; HIF-1α signalling pathway and specific HIF-1α-related target genes; MAO-A and -B activities, amine metabolism and expression levels of APP. The regulatory effects of the drug were also assessed in the brains of genetically induced diabetes ob/ob mice (leptin−/−).
Methods
Animals and treatment
All animal care and experimental procedures were carried out in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals, and were approved by the Animal Ethics Committee of the Technion, Haifa, Israel. A total of 45 animals were used in the experiments described here. The dose of M30 used in these experiments was based on our previous animal studies (Gal et al., 2005; Kupershmidt et al., 2012b).
HFD mice
Male C57BL/6J mice at 8 months old were obtained from Harlan Laboratories, Inc. (Israel). C57BL/6J mice were randomly assigned into three groups: mice fed with normal diet (ND); vehicle-treated HFD mice; and M30-treated HFD mice, n = 7 in each experimental group. Animals in the ND group were fed with standard laboratory chow. The dietary formulation for the ND group was 11% calories from fat, 65% calories from carbohydrate and 24% calories from protein. The dietary formulation for the HFD group (D12492; Research Diet Inc, New Brunswick, NJ, USA) was 60% calories from fat, 20% calories from carbohydrate and 20% calories from protein. All animals were given free access to the diet and drinking water. M30 (5 mg⋅kg−1) and vehicle were administered by oral gavage, three times a week, starting at 8 months of age and continuing for a further 5 months. HFD was given 3 months after the start of M30 treatment.
At the end of the experiment, the mice that were fed with HFD and treated with vehicle were significantly heavier, compared with ND mice (47.6 ± 2.43 g vs. 31.7 ± 1.13 g, respectively; P < 0.05), while M30-treated mice had a slightly lower weight than the vehicle-treated HFD mice (44.1 ± 2.46 g vs. 47.6 ± 2.43 g respectively). Ingestion of a HFD over a long period leads to insulin resistance and obesity, which are core findings of type 2 diabetes mellitus (T2DM; Kahn et al., 2001; Goldstein, 2002). As expected, glucose tolerance tests (GTT) revealed that HFD mice had impaired tolerance, compared with ND mice [AUC values calculated over 2 h after the glucose load, (g⋅L−1 × min): 559 ± 18 vs. 297 ± 6.8; P < 0.05 respectively]. However, after treatment with M30, the HFD mice showed a lower response to the GTT, compared with vehicle-treated HFD mice [AUC levels (g⋅L−1 × min): 432 ± 20 vs. 559 ± 18; P < 0.05 respectively]. We further investigated the effect of M30 on glucose metabolism in C57BL/6 mice fed with ND. M30-treated mice showed a lower increase in glucose values, compared with vehicle-treated control mice, as corroborated by the significant decrease in AUC levels (g⋅L−1 × min) (339 ± 30 vs. 561 ± 37; P < 0.05). At the end of the experiment, mice were killed by cervical dislocation and the brains were dissected and frozen at −80°C for further analysis.
ob/ob Tg mice
Male, leptin-deficient, ob/ob transgenic (Tg) mice bred on the C57BL/6J genetic background were obtained from Harlan Laboratories, Inc. (Israel). This experiment included wild-type, vehicle-treated ob/ob Tg mice and M30-treated ob/ob Tg mice (n = 8 in each experimental group). M30 (1 mg⋅kg−1) and vehicle were given by oral gavage three times a week, starting at 6 weeks of age and continuing for 5 months. At the beginning of the experiment, the body weights of ob/ob Tg mice were significantly higher than those of the non-Tg mice (22.8 ± 0.55 g vs. 16.7 ± 0.31 g; P < 0.05 respectively). At the end of the experiment, the body weight of M30-treated ob/ob Tg mice was similar to that of vehicle-treated ob/ob mice (51.1 ± 0.64 g vs. 50.8 ± 0.88 g respectively). Impaired GTT was demonstrated in ob/ob Tg mice, compared with non-Tg mice [AUC levels (g⋅L−1 × min): 685 ± 22 vs. 245 ± 27; P < 0.05 respectively]. However, following treatment with M30, the ob/ob Tg mice showed a lower increase in glucose AUC values, compared with vehicle-treated ob/ob Tg mice [AUC (g⋅L−1 × min): 480 ± 32 vs. 685 ± 22; P < 0.05]. At the end of the experiment, the brains removed and frozen, as described above.
Western immunoblotting analysis
For Western blot analysis, brain sections were homogenized in Tris-sucrose buffer, pH = 7.4 [containing a mixture of protease inhibitors (Roche, Inc., Basel, Switzerland) and phosphatase inhibitors], and centrifuged at 1000× g for 10 min at 4°C. The supernatant was collected and stored at −80°C. The protein concentration was determined using the Bradford method. Equal amounts of proteins per sample were loaded and subjected to SDS-PAGE (4–12% Bis-Tris gels) and blotted on Protran nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany). Membranes were treated with blocking buffer and incubated with primary antibodies overnight at 4°C, followed by incubation with HRP-conjugated secondary antibodies diluted in the same buffers for 1 h at 25°C. Detection was achieved using Western blotting detection reagent, ECL system (Amersham Pharmacia, Amersham, UK). Band intensities were quantified using the computerized imaging program Bio-1D (VilberLourmat Biotechnology, Collegien, France) and values were normalized to β-actin or β-tubulin protein intensity.
Total RNA extraction, reverse transcription (RT) and quantitative real-time RT-PCR
Isolation of total RNA was performed using PerfectPure RNA Cultured Cell Kit (5PRIME Inc., Gaithersburg, MD, USA), as recommended by the manufacturer's protocol. Total RNA was denatured and reverse transcribed using random hexanucleotides as described (Avramovich-Tirosh et al., 2010). Quantitative real-time RT-PCR was performed by LightCycler and FastStart DNA Master SYBR Green I ready-to-use PCR mix according to the manufacturer's protocol (Roche Applied Science, Penzberg, Germany). cDNA was amplified in 10 μL of total volume. The sequence of HIF-1α, enolase-1, VEGF, heme oxygenase-1 (HO-1), catalase, 18S-rRNA and Erk-2 used for quantitative real-time RT-PCR is shown in Table 1. The primers superoxide dismutase (SOD)-1, aldolase A, GLUT-1 and -3, insulin, and γ-tubulin were purchased from Qiagen (Hilden, Germany). The results were analysed on the provided program of the LightCycler. The relative expression level of a given mRNA was assessed by normalizing to the housekeeping genes Erk-2, 18S-rRNA and/or γ-tubulin.
Table 1.
Sequences of primers used in quantitative real-time RT-PCR
| mRNA | Oligonucleotide sequence (5′-3′) forward | Oligonucleotide sequence (5′-3′) reverse |
|---|---|---|
| HIF-1α | CCTTCATCGGAAACTCCAAA | TGGGGCATGGTAAAAGAAAG |
| Enolase-1 | GGTTCTCATGCTGGCAACAAGT | TAAACCTCTGCTCCAATGCGC |
| VEGF | CAGGACAAGCAGAGCACGTC | CAAGCCAGCACTCAGAGCACAGCC |
| HO-1 | ACCCGCTTCCATAAGGCTTTA | CAGCCTTGTGCCGAAAGAC |
| Catalase | CAGGACAAGCAGAGCACGTC | CAAGCCAGCACTCAGAGCACAGCC |
| 18S-rRNA | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG |
| Erk-2 | TGAAGTTGAACAGGCTCTGG | GCTGGAATCGAGCAGTCTCT |
MAO activity assay
The cerebellum was homogenized using a glass Teflon homogenizer in ice-cold sucrose buffer (10 mM Tris-HCl buffer, pH 7.4, containing 0.32 M sucrose) followed by the addition of sucrose buffer to a final concentration of 1 mL homogenate. The homogenates were centrifuged at 300× g for 2 min at 4°C. The supernatant fraction was removed and used to determine MAO activity. MAO-A and -B activities were measured according to Gal et al. (2005). Briefly, protein homogenate was added to a suitable dilution of the enzyme preparation. The mixture was incubated with 0.15 μM L-deprenyl or clorgyline (for determination of MAO-A or -B respectively). Incubation was carried for 1 h at 37°C prior to the addition of [14C]5-HT binoxalate (400 μM) for determination of MAO-A, or [14C]phenylethylamine (400 μM) for determination of MAO-B, and further incubation for 30 or 20 min respectively. The reaction was stopped with ice-cold citric acid (2 M) and radioactivity was determined by liquid scintillation.
Catecholamine analysis
The striatum was dissected, weighed and homogenized in 400 μL 0.1 M perchloric acid using a glass Teflon homogenizer. The levels of catecholamines and their metabolites – dopamine (DA), 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), noradrenaline (NA), 5-HT and 5-hydroxyindoleacetic acid (5-HIAA) – were determined with reverse-phase HPLC using a Hypersil column H30DS-2409, 12.5 cm × 4.6 mm i.d. (Hichrom, Theale, UK). The dual electrode analytical cell operated in a redox mode with 0.3 V oxidation and 0.35 V reduction potential. The mobile phase consisted of 0.1 M phosphate buffer, pH 2.75, containing 0.2 mM EDTA, 0.2 mM octane sulfonic acid, 2.5% methanol and 4.5% acetonitrile, and was run at a flow rate of 0.9 mL min−1. Results were analysed using a Coularray for Windows 32 software. The HPLC system consists of an ESA Coulochem Model 5600 electrochemical detector (ESA analytical with a Model 6210 8-channel sensor, Thermo Scientific, Sunnyvale, CA, USA).
Data analysis
All values are expressed as mean ± SEM. For statistical analysis, one-way anova followed by Student's t-test was performed and a value of P < 0.05 was considered significant.
Materials
The following antibodies were used for Western blotting analysis: rabbit polyclonal antibodies against phospho-GSK-3α/β (Ser21/9) and GSK-3β from Cell Signaling (Beverly, MA, USA); InsRβ (c19) antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA); GLUT-1 antibody (Millipore, Billerica, MA, USA); monoclonal rabbit antibody against HIF-1α (Epitomics Inc., Burlingame, CA, USA); β-actin and APP C-terminal antibodies purchased from Sigma (St Louis, MO, USA). The multifunctional iron chelator M30 (5-[N-methyl-N-propargylaminomethyl]-8-hydroxyquinoline; MW 299.3; Zheng et al., 2005a,b,) was synthesized and kindly provided by Varinel Inc. (Philadelphia, PA, USA). M30 exhibts good aqueous solubility and selectively chelates iron, compared with zinc and copper. The drug is not cytotoxic, as shown by genotoxicity assays performed in several cell lines, cytochrome p450 isoenzymes inhibition and voltage-dependent potassium channel blocking tests (Zheng et al., 2005a,b,). [14C]5-HT binoxalate (57.5 mCi mmol−1) and [14C]phenylethylamine (43.8 mCi mmol−1) were purchased from PerkinElmer (Massachusetts, USA). Other chemicals and reagents were of the highest analytical grade and purchased from local commercial sources.
Results
The regulatory effect of M30 on insulin, InsR and p-GSK-3β in the hippocampus and frontal cortex of HFD mice
One of the main links between DM and neurodegenerative diseases is impaired insulin signalling (De la Monte and Wands, 2008). Here, we tested the effects of chronic administration of M30 on the levels of insulin protein, its receptor InsR and the phosphorylation of GSK-3β at Ser9, in the hippocampus and frontal cortex of HFD mice.
As shown in Figure 1, M30 (5 mg⋅kg−1) significantly increased the transcripts of insulin in the hippocampus and frontal cortex of HFD mice, compared with vehicle-treated HFD mice. Figure 1A demonstrates that hippocampal InsR (β subunit) protein levels were decreased in vehicle-treated HFD mice, compared with ND mice, while M30 (5 mg⋅kg−1) significantly up-regulated hippocampal InsR, compared with vehicle-treated HFD mice. Moreover, M30 increased InsR protein levels in the frontal cortex of HFD mice (Figure 1B). As GSK-3β is a central player in the insulin signalling, activated by insulin via InsR (Gabbay et al., 1996), we also examined the effect of M30 on the levels of p-GSK-3β-Ser9 (the inactivated form) in the hippocampus and frontal cortex of HFD mice. Figure 1 shows that the hippocampal and cortical p-GSK-3β levels were lower in vehicle-treated HFD mice, compared with ND mice. M30 treatment significantly increased p-GSK-3β levels, compared with the vehicle-treated HFD mice, both in the hippocampus and frontal cortex.
Figure 1.
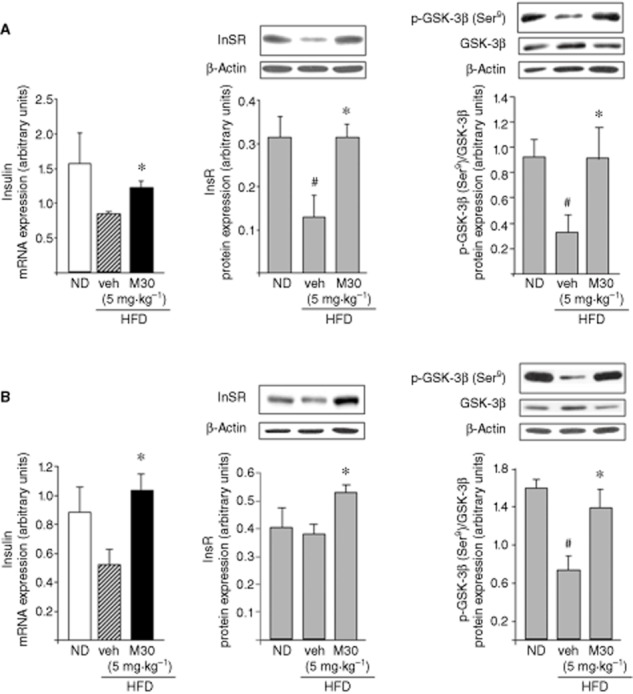
Effect of M30 on mRNA expression levels of insulin and protein levels of InsR and p-GSK-3β in the hippocampus and frontal cortex of HFD mice. mRNA expression levels of insulin and representative Western blots of InsR and p-GSK-3β (Ser9) in (A) hippocampus and (B) frontal cortex of mice fed with ND or HFD treated with vehicle or M30 (5 mg⋅kg−1). Western blot graphs represent densitometry quantification of the lanes normalized to β-actin or total GSK-3β. The amount of mRNA product was measured by quantitative real-time RT-PCR and normalized to the housekeeping gene 18S-rRNA and γ-tubulin. Data are expressed as arbitrary units and represent means ± SEM (n = 7). #P < 0.05 versus ND mice; *P < 0.05 versus vehicle-treated HFD mice.
The effect of M30 on cerebral HIF-1α and HIF-1α-related neuroprotective genes in HFD mice
It is well known that neurons are not able to synthesize or store glucose; they are dependent on glucose transport across the BBB, mediated by GLUTs (Scheepers et al., 2004). Moreover, several studies have shown that insulin resistance and hyperglycaemia were associated with reduced expression levels of GLUT-1 and GLUT-3 in the brain (Hou et al., 2007). In the current study, hippocampal GLUT-1 protein levels were decreased in vehicle-treated HFD mice, compared with ND mice (Figure 2A). However, M30 (5 mg⋅kg−1) significantly up-regulated hippocampal GLUT-1 protein levels, compared with vehicle-treated HFD mice (Figure 2A). Additionally, M30 increased protein expression of GLUT-1 in the frontal cortex of HFD mice, compared with vehicle-treated HFD mice (Figure 2B).
Figure 2.
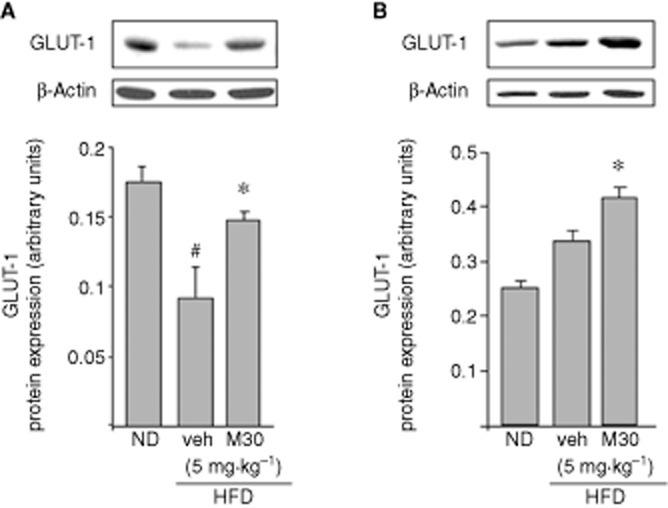
Effect of M30 on GLUT-1 levels in the hippocampus and frontal cortex of HFD mice. Representative Western blots of GLUT-1 in (A) hippocampus and (B) frontal cortex of mice fed with ND or HFD treated with vehicle or M30 (5 mg⋅kg−1). Graphs represent densitometry quantification of the lanes normalized to β-actin. Data are expressed as arbitrary units and represent means ± SEM (n = 7). #P < 0.05 versus ND mice; *P < 0.05 versus vehicle-treated HFD mice.
GLUT-1 and several other proteins that are involved in glucose transport and glycolysis are regulated, in part, by the transcription factor HIF-1α (Semenza, 2000). Previous studies have shown that one of the potential therapeutic effects of iron chelators is related to the inhibition of the iron-dependent HIF-PHDs that regulate HIF-1 stability, leading to transcriptional up-regulation of several protective genes, involved in glycolysis and glucose transport, survival, neurogenesis, iron metabolism and OS (Semenza, 2000; Siddiq et al., 2005). Thus, we next examined the effect of M30 on mRNA expression levels of HIF-1α and HIF-1α-dependent target genes (GLUT-1, GLUT-3, enolase-1, aldolase A, VEGF and HO-1 in the hippocampus of HFD mice). As shown in Figure 3A, HIF-1α protein levels were significantly increased in the hippocampus of M30-treated versus vehicle-treated HFD mice. Moreover, M30 up-regulated the mRNA expression levels of the HIF-1-related genes, GLUT-1, GLUT-3 and enolase-1 in the hippocampus of HFD mice compared with those in vehicle-treated HFD mice (Figure 3B). Additionally, M30 treatment increased mRNA expression levels of GLUT-1 and VEGF in the frontal cortex of HFD mice compared with vehicle-treated HFD mice (Figure 3C). No effect was evident on cortical mRNA levels of GLUT-3, enolase-1, aldolase A and HO-1.
Figure 3.

Effect of M30 on HIF-1α and HIF-1α-related genes in the hippocampus of HFD mice. (A) A representative Western blot and analysis of mRNA expression levels of HIF-1α in the hippocampus of mice fed with ND or HFD treated with vehicle or M30 (5 mg⋅kg−1). Western blot graphs represent densitometry quantification of the lanes, normalized to β-actin. (B) mRNA expression levels of GLUT-1, GLUT-3, enolase-1, aldolase A, VEGF and HO-1. (C) mRNA expression levels of GLUT-1 and VEGF. The amount of each mRNA product was measured by quantitative real-time RT-PCR and normalized to the housekeeping gene γ-tubulin. Data are expressed as arbitrary units and represent means ± SEM (n = 7). #P < 0.05 versus ND mice; *P < 0.05 versus vehicle-treated HFD mice.
The effect of M30 on cerebral expression levels of APP in HFD mice
Because the 5′ untranslated region of APP mRNA has been previously shown to possess a functional iron-responsive element (Rogers et al., 2002), we examined whether M30 could regulate cerebral holo-APP levels in the HFD mice. As shown in Figure 4, M30 treatment led to a significant reduction in holo-APP protein levels in the hippocampus of HFD mice, compared with vehicle-treated HFD mice, whereas no effect was evident on mRNA levels of APP in the different groups of mice (mRNA expression, arbitrary units; ND: 1.01 ± 0.009; HFD: 1.02 ± 0.07; HFD + M30 (5 mg⋅kg−1): 0.85 ± 0.05).
Figure 4.
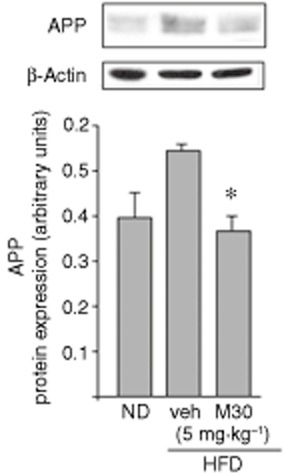
Effect of M30 on APP levels in the hippocampus of HFD mice. A representative Western blot of hippocampal APP; Western blot graphs represent densitometry quantification of the lanes normalized to β-actin. Data are expressed as arbitrary units and represent means ± SEM (n = 7). *P < 0.05 versus vehicle-treated HFD mice.
The effect of M30 on cerebral MAO-A and -B activities and amine metabolism in HFD mice
MAO-A and -B are the enzymes that catalyse the oxidative deamination of biogenic amines essential for the inactivation of monoaminergic neurotransmitters, such as DA, NA and 5-HT (Youdim and Bakhle, 2006). MAO activity dysfunction is linked with increased production of reactive oxygen species, associated with the pathology of age-related diseases and HFD (Shemyakov, 2001; Morrison et al., 2010). As M30 is a potent irreversible, brain-selective MAO-A and -B inhibitor in mouse models of neurodegenerative diseases (Gal et al., 2005; 2010,; Zheng et al., 2005a,b,; Avramovich-Tirosh et al., 2007; 2010,; Weinreb et al., 2010), we further examined the effect of the drug on MAO-A and -B activities in the cerebellum of mice fed with HFD. As shown in Figure 5A, the activity of both enzymes was significantly higher in the HFD-treated mice, compared with ND mice. However, M30 caused a marked inhibition of both MAO-A and -B activities in the cerebellum of HFD mice, compared with vehicle-treated HFD mice. Additionally, we examined the effect of M30 (5 mg⋅kg−1) on the levels of striatal amines in the HFD mice. As expected from MAO-A and -B inhibitors, M30 increased striatal levels of DA and markedly decreased levels of its metabolites, DOPAC and HVA (Figure 5B). Marked lower levels of 5-HT were observed in mice fed with a HFD versus ND mice, while treatment with M30 reversed this effect (Figure 5B).
Figure 5.

Effect of M30 on cerebral MAO-A and -B activities and striatal amine metabolism in HFD mice. (A) MAO-A and -B activities were determined in the cerebellum of mice fed with ND (control) or HFD treated with vehicle or M30 (5 mg⋅kg−1). Results represent MAO activity (% of control); means ± SEM. ND mice served as control. (B) Striatal DA, DOPAC, HVA, 5-HT, 5-HIAA and NA levels were determined via EC-HPLC analysis. Results represent means (pmol·mg−1) ± SEM (n = 7). #P < 0.05 versus ND mice; *P < 0.05 versus vehicle-treated HFD mice.
Previous studies have suggested that, among their other neuroprotective mechanisms, the propargyl-containing compounds can up-regulate antioxidant enzymes (Naoi and Maruyama, 2009). As shown in Figure 6, the transcript of catalase was reduced in the hippocampus and frontal cortex of vehicle-treated HFD mice, compared with ND mice. Administration of M30 (5 mg⋅kg−1) to HFD mice resulted in a significant increase in mRNA levels of catalase, compared with vehicle-treated HFD mice, both in the hippocampus and the frontal cortex (Figure 6).
Figure 6.
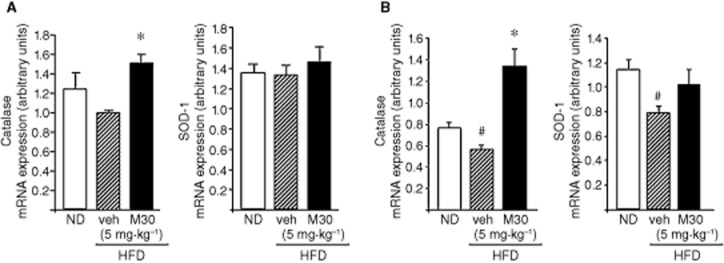
Effect of M30 on mRNA expression levels of the antioxidant enzymes, catalase and SOD-1 in the hippocampus and frontal cortex of HFD mice. The mRNA expression levels of catalase and SOD-1 in (A) hippocampus and (B) frontal cortex of mice fed with ND or HFD treated with vehicle or M30 (5 mg⋅kg−1) were measured by quantitative real-time RT-PCR. The amount of each product was normalized to the housekeeping gene γ-tubulin. Data are expressed as arbitrary units and represent means ± SEM (n = 7). #P < 0.05 versus ND mice; *P < 0.05 versus vehicle-treated HFD mice.
Effect of M30 on HIF-1α expression and HIF-1-related genes; MAO activity, expression of antioxidant enzymes and APP in the frontal cortex of ob/ob mice
Further evaluation of the effect of M30 in the brain of an insulin resistance animal genetic model was studied in the obese diabetic ob/ob Tg mice (leptin−/−). Figure 7A shows that this compound significantly increased the mRNA expression levels of HIF-1α in the frontal cortex of ob/ob Tg mice, as compared with vehicle-treated ob/ob Tg mice. Additionally, treatment with M30 increased the transcript levels of several HIF-1α-related glycolytic genes: GLUT-1, GLUT-3 and enolase-1 in the frontal cortex of ob/ob Tg mice. The mRNA expression levels of VEGF and HO-1 were markedly induced in the frontal cortex of M30-treated ob/ob Tg mice, compared with vehicle-treated ob/ob Tg mice, while a significant decline was observed in vehicle-treated ob/ob Tg, relative to non-Tg mice (Figure 7B).
Figure 7.
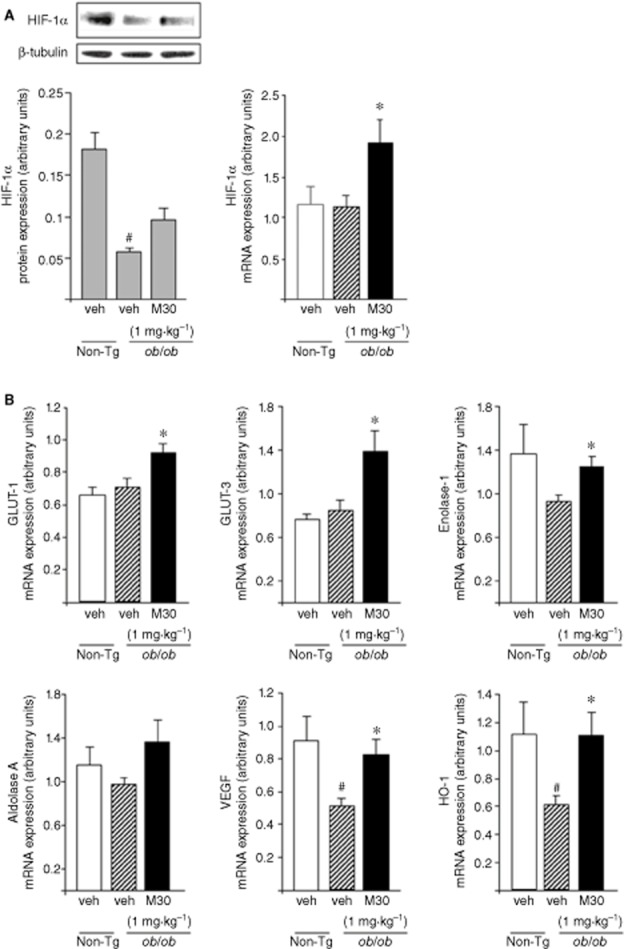
Effect of M30 on HIF-1α and HIF-1α-related genes in the frontal cortex of ob/ob mice. (A) A representative Western blot and mRNA expression levels of HIF-1α. Western blot graph represents densitometry quantification of the lanes normalized to β-tubulin. (B) mRNA expression levels of GLUT-1, GLUT-3, enolase-1, aldolase A, VEGF and HO-1 were measured by quantitative real-time RT-PCR. The amount of each mRNA product was normalized to the housekeeping gene Erk-2. Data are expressed in arbitrary units and represent means ± SEM (n = 8). #P < 0.05 versus non-Tg mice; *P < 0.05 versus vehicle-treated ob/ob Tg mice.
Further analysis revealed that M30 significantly reversed the elevated MAO-A activity in the cerebellum (Figure 8A) and increased mRNA expression levels of catalase in the frontal cortex of ob/ob Tg mice (Figure 8B). The levels of APP in the frontal cortex were markedly higher in the ob/ob than in the non-Tg mice, but were significantly reduced in M30-treated, compared with vehicle-treated ob/ob Tg mice (Figure 8C).
Figure 8.
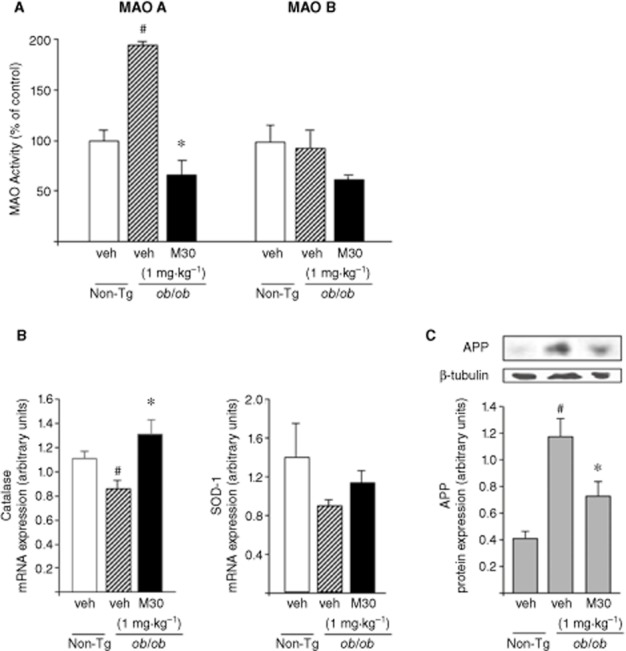
Effect of M30 on MAO-A and -B activities in the cerebellum, mRNA expression levels of antioxidant enzymes and APP levels in the frontal cortex of ob/ob Tg mice. (A) MAO-A and -B activities were determined in the cerebellum of ob/ob Tg mice. Results represent MAO activity (% of control non-Tg mice). (B) mRNA expression levels of catalase and SOD-1 in the frontal cortex of ob/ob Tg mice were measured by quantitative real-time RT-PCR. The amount of each product was normalized to the housekeeping gene γ-tubulin. Data are expressed as arbitrary units and represent means ± SEM (n = 8). #P < 0.05 versus non-Tg mice; *P < 0.05 versus vehicle-treated ob/ob Tg mice. (C) A representative Western blot of APP. Western blot graph represents densitometry quantification of the lanes normalized to β-tubulin. Data are expressed as arbitrary units and represent means ± SEM (n = 8). #P < 0.05 versus non-Tg mice; *P < 0.05 versus vehicle-treated ob/ob Tg mice.
Discussion
We have recently synthesized the multifunctional brain-permeable compound, M30, possessing propargyl MAO inhibitory and antioxidant-chelator moieties from the anti-Parkinsonian drug brain-selective, MAO-B inhibitor, rasagiline (Azilect) and the prototype iron chelator VK28 (Zheng et al., 2005a,b,). In the present study, we have extended our studies to examine the molecular mechanisms of M30, associated with neuroprotection, presumably derived from both the propargyl moiety, as well as the iron-chelating moiety of the compound in mice with HFD-induced obesity and insulin resistance. Previous studies have reported that animals that are genetically obese or else maintained on a HFD display insulin resistance within the CNS (Arase et al., 1988; Chavez et al., 1996). HFD produced insulin resistance in mouse brain, as demonstrated by failed activation of Akt and GSK-3β with ex vivo insulin stimulation and increased expression of serine-phosphorylated InsR substrate 1, a marker of insulin resistance (Arnold et al., 2014). The current findings demonstrate that chronic treatment with M30 reversed the reductions in the transcript levels of insulin, and protein levels of InsR and p-GSK-3β(Ser9) (inactivated form) in the hippocampus and frontal cortex of HFD mice. It is likely that this modulatory effect of M30 on the insulin/InsR/p-GSK-3β system could affect neuronal survival/neurogenesis and improve cognitive abilities, although direct evidence for the effects of insulin resistance and impaired glucose uptake in the brain causing specific cognitive impairments is currently lacking. These results are consistent with previous signalling studies conducted with M30 showing its regulatory effect on the Akt/GSK-3β phosphorylation pathways in cultured cortical neurons (Avramovich-Tirosh et al., 2010) and, in vivo, in the mouse brain (Kupershmidt et al., 2011). In addition, using the APP/PS1 mouse model of AD-type neuropathology, we have recently demonstrated that M30 produced a significant up-regulation in the insulin transcript and InsR expression and increased p-GSK-3β levels in the frontal cortex (Mechlovich et al., 2013). This modulatory effect of M30 on insulin signalling pathway may be associated, at least partly, with beneficial effects of the drug on various major characteristsics of AD, such as cognitive impairments, Aβ accumulation, senile plaque formation and τ hyperphosphorylation; Kupershmidt et al., 2012b).
An additional potential beneficial effect of M30, presumably derived from its iron-chelating potency, is the regulatory effect of the drug on the oxygen/iron-sensitive transcriptional factor, HIF-1α and several HIF-1α-related target genes. As iron is a cofactor required for HIF-1α breakdown, iron chelators can mimic hypoxia and induce the abundance of HIF-1α (Siddiq et al., 2005). In accordance, the current results demonstrated that M30 produced a significant up-regulation of HIF-1α expression and differentially induced the transcription of HIF-1-related protective genes in the hippocampus of HFD mice, such as those involved in glycolysis and glucose transport (GLUT-1, GLUT-3, enolase-1 and aldolase A), angiogenesis and neurogenesis (VEGF), and OS and anti-inflammation (HO-1), indicating activation of HIF-1α in the hippocampus. Similarly, we have observed that M30 increased the mRNA levels of GLUT-1, GLUT-3, and enolase-1, and attenuated the reduction in mRNA levels of VEGF and HO-1 in the frontal cortex of ob/ob Tg mice, compared with vehicle-treated ob/ob Tg mice. Thus, our data suggest that M30 treatment has a positive effect on HIF-1α and HIF-1α-related target genes in the brain of HFD and ob/ob Tg mice, which is in keeping with previous animal and in vitro studies with this compound (Kupershmidt et al., 2009; 2011,; Avramovich-Tirosh et al., 2010). Accordingly, in hepatocytes, activation of HIF-1α and induction of GLUT-1 and InsR were shown to be associated with the regulatory effect of deferoxamine on glucose metabolism, indicating that GLUT-1 was up-regulated directly following HIF-1α stabilization, whereas InsR induction appeared to be a secondary phenomenon regulated via increased glucose uptake and activation of glucose-sensitive elements, such as consensus sequences for HIF-1β (which is also the aryl hydrocarbon receptor nuclear transporter) present in the InsR promoter (Dongiovanni et al., 2008). Based on this finding, it may be suggested that in the brains of mice with obesity and insulin resistance the effect of M30 on up-regulating HIF-1α, followed by enhanced GLUT-1 expression and increased glucose uptake, can subsequently lead to enhanced InsR expression.
Regarding an additional regulatory effect that may be attributed to the iron-chelating moiety of M30, treatment with this compound significantly prevented the increase in protein levels of the AD full-length APP in the hippocampus of HFD mice, as well as in the frontal cortex of ob/ob Tg mice. In both mouse models, we observed elevated brain APP protein levels, further indicating a possible underlying linkage between DM and AD. In line with this, APP levels were increased in the brain of murine models of HFD obesity (Puig et al., 2012) and HF/high-cholesterol diet (Thirumangalakudi et al., 2008). In addition, obesity and caloric intake regulated the expression of APP in mononuclear cells (Ghanim et al., 2012), indicating that modulation of APP may serve as an additional marker to identify obesity-related changes (Puig et al., 2012).
The reduction in brain APP levels following M30 treatment in HFD and ob/ob Tg mice is consistent with our previous studies in a model of AD, APP/PS1 mice, and in vitro, showing the regulatory effect of M30 on APP, resulting in reduced APP expression (Avramovich-Tirosh et al., 2007; Kupershmidt et al., 2012b). This is presumably attributed to the iron-chelating moiety of M30, as the 5′UTR of APP encodes a stem loop functional iron-responsive element that reflects iron-dependent target for APP regulation. Indeed, M30 suppressed the translation of a luciferase reporter gene fused to the APP mRNA 5′UTR (Avramovich-Tirosh et al., 2007).
Another potentially neuroprotective target of M30 is MAO, because this compound is also a propargylamine derivative and thus, a potent irreversible brain-selective MAO inhibitor (Gal et al., 2005). In the present study, we found that MAO-A and -B activities were higher in the cerebellum of HFD mice, while M30 treatment significantly re-established the basal levels of both enzyme isoforms. In ob/ob Tg mice, M30 reversed the enhanced MAO-A activity and reduced MAO-B activity. In accordance with its MAO inhibitory properties, M30 administration increased DA, while significantly reducing levels of its metabolites, DOPAC and HVA, in the striatum of HFD mice. Indeed, only when both enzyme forms are inhibited does brain DA increase, whereas DOPAC and HVA decrease (Green et al., 1977). Similar effects of M30 on cerebral MAO-A and -B activities and on DA levels and its metabolites have been shown in other animal models of neurodegenerative diseases (Gal et al., 2005; Kupershmidt et al., 2012a).
Importantly, DA dysfunction has been identified in both obese individuals and HFD animal models (Huang et al., 2005; Lee et al., 2010). In addition, chronic HFD was shown to be associated with down-regulation of gene transcripts for DA D1 and D2 receptors and the DA transporter, further indicating that HFD may lead to an overall suppression of the DA system (Kaczmarczyk et al., 2013). Recently, it was shown that HFD increased MAO-A and -B activities in the hippocampus and hypothalamus (Kaczmarczyk et al., 2013), and induced an epigenetic dysregulation of the DA system (Vucetic et al., 2012), also linking HFD with alterations in DA-related parameters.
The increase in the MAO-A substrate, 5-HT, following M30 administration to HFD mice, and the decrease in its metabolite, 5-HIAA, reflected inhibition of MAO-A by M30. A similar trend was noted with the MAO-A substrate, NA, which plays a critical role in cognition and motivation, which are important factors in depression (Moret and Briley, 2011). These observations are of great importance as many cases of obesity are also associated with depression, further emphasizing the need of a multifunctional drug. Recent findings suggested that depression, associated with obesity, is due, in part, to impaired leptin activity in the hippocampus (Yamada et al., 2011). However, the precise mechanisms underlying the interaction between obesity and depression have not yet been elucidated (Roberts et al., 2003; Simon et al., 2006).
In addition, we observed a significant increase in mRNA levels of the antioxidant enzyme, catalase, in the hippocampus and frontal cortex of M30-treated, compared with vehicle-treated, HFD mice. M30 also up-regulated the transcript expression levels of catalase in the frontal cortex of ob/ob Tg mice. Because HF feeding increased various markers of OS in several brain regions (Morrison et al., 2010), this effect of M30 on cerebral catalase expression may be beneficial in reducing neurotoxic oxidative effects. This observation can be associated with the propargyl moiety of M30, as it has been previously shown that M30 and several other propargylamines, including rasagiline and deprenyl, all increased the activity of the anti-oxidative enzymes, catalase and SOD-1, in several brain regions relevant to dopaminergic transmission (Carrillo et al., 2000; Kupershmidt et al., 2011).
In conclusion, the data presented here demonstrate that, in the brain of HFD and ob/ob Tg mice, M30 possessing both the neuroprotective propargyl and antioxidant iron-chelating moieties, exhibits a range of beneficial regulatory effects that might be attributed to the multi-modal design paradigm of the drug. These include up-regulation of insulin/InsR/p-GSK-3β levels, activation of the HIF-1α pathway and HIF-1-related neuroprotective and glycolytic genes, down-regulation of APP levels, and MAO inhibition and anti-oxidation. It is suggested that all these favorable regulatory effects could act synergistically and with other molecular mechanisms to prevent or delay neurodegenerative processes caused by HFD-induced obesity and insulin resistance.
Acknowledgments
The authors gratefully acknowledge the support of the Alzheimer's Association (Chicago, USA) and Rappaport Family Research Institute, Technion-Israel Institute of Technology (Haifa, Israel).
Glossary
- 5-HIAA
5-hydroxyindoleacetic acid; AD, Alzheimer's disease
- APP
amyloid precursor protein
- BBB
blood–brain barrier
- DM
diabetes mellitus
- DOPAC
3,4-dihydroxyphenylacetic acid
- GLUT
glucose transporter
- GSK-3β
glycogen synthase kinase-3β
- GTT
glucose tolerance test
- HFD
high-fat diet
- HIF
hypoxia-inducible factor
- HO-1
haem oxygenase-1
- HVA
homovanillic acid
- InsR
insulin receptor
- MAO
monoamine oxidase
- ND
normal diet
- OS
oxidative stress
- PHD
prolyl-4-hydroxylases
- PS1
presenelin 1
- RT
reverse transcription
- SOD
superoxide dismutase
- T2DM
type 2 diabetes mellitus
- Tg
transgenic
- veh
vehicle
Author contributions
The work presented here was carried out in collaboration between all authors. D. M., T. A., O. B., O. W. and M. B. H. Y. defined the research theme. T. A., O. W. and M. B. H. Y. designed the methods and experiments. D. M., O. B. and O. W. performed the experiments. D. M. and O. B. analysed the data. D. M., T. A., O. B. and O. W. wrote the manuscript. All authors have revised and approved the manuscript.
Conflict of interest
M.B.H.Y. is CSO of Abital Pharma Pipeline Ltd. He is part owner of the company and has shares.
References
- Ahmad W. Overlapped metabolic and therapeutic links between Alzheimer and diabetes. Mol Neurobiol. 2013;47:399–424. doi: 10.1007/s12035-012-8352-z. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol. 2013a;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013b;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013c;170:17. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arase K, Fisler JS, Shargill NS, York DA, Bray GA. Intracerebroventricular infusions of 3-OHB and insulin in a rat model of dietary obesity. Am J Physiol. 1988;255(6 Pt 2):R974–R981. doi: 10.1152/ajpregu.1988.255.6.R974. [DOI] [PubMed] [Google Scholar]
- Arnold SE, Lucki I, Brookshire BR, Carlson GC, Browne CA, Kazi H, et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol Dis. 2014;67C:79–87. doi: 10.1016/j.nbd.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avramovich-Tirosh Y, Reznichenko L, Mit T, Zheng H, Fridkin M, Weinreb O, et al. Neurorescue activity, APP regulation and amyloid-beta peptide reduction by multi-functional brain permeable iron- chelating- antioxidants, M-30 and green tea polyphenol, EGCG. Curr Alzheimer Res. 2007;4:403–411. doi: 10.2174/156720507781788927. [DOI] [PubMed] [Google Scholar]
- Avramovich-Tirosh Y, Bar-Am O, Amit T, Youdim MB, Weinreb O. Up-regulation of hypoxia-inducible factor (HIF)-1alpha and HIF-target genes in cortical neurons by the multifunctional iron chelator anti-Alzheimer drug, M30. Curr Alzheimer Res. 2010;7:300–306. doi: 10.2174/156720510791162403. [DOI] [PubMed] [Google Scholar]
- Carrillo MC, Minami C, Kitani K, Maruyama W, Ohashi K, Yamamoto T, et al. Enhancing effect of rasagiline on superoxide dismutase and catalase activities in the dopaminergic system in the rat. Life Sci. 2000;67:577–585. doi: 10.1016/s0024-3205(00)00643-3. [DOI] [PubMed] [Google Scholar]
- Chavez M, Riedy CA, Van Dijk G, Woods SC. Central insulin and macronutrient intake in the rat. Am J Physiol. 1996;271(3 Pt 2):R727–R731. doi: 10.1152/ajpregu.1996.271.3.R727. [DOI] [PubMed] [Google Scholar]
- Correia SC, Santos RX, Carvalho C, Cardoso S, Candeias E, Santos MS, et al. Insulin signaling, glucose metabolism and mitochondria: major players in Alzheimer's disease and diabetes interrelation. Brain Res. 2012;1441:64–78. doi: 10.1016/j.brainres.2011.12.063. [DOI] [PubMed] [Google Scholar]
- De la Monte SM, Wands JR. Alzheimer's disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol. 2008;2:1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongiovanni P, Valenti L, Ludovica Fracanzani A, Gatti S, Cairo G, Fargion S. Iron depletion by deferoxamine up-regulates glucose uptake and insulin signaling in hepatoma cells and in rat liver. Am J Pathol. 2008;172:738–747. doi: 10.2353/ajpath.2008.070097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbay RA, Sutherland C, Gnudi L, Kahn BB, O'Brien RM, Granner DK, et al. Insulin regulation of phosphoenolpyruvate carboxykinase gene expression does not require activation of the Ras/mitogen-activated protein kinase signaling pathway. J Biol Chem. 1996;271:1890–1897. doi: 10.1074/jbc.271.4.1890. [DOI] [PubMed] [Google Scholar]
- Gal S, Zheng H, Fridkin M, Youdim MB. Multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases. In vivo selective brain monoamine oxidase inhibition and prevention of MPTP-induced striatal dopamine depletion. J Neurochem. 2005;95:79–88. doi: 10.1111/j.1471-4159.2005.03341.x. [DOI] [PubMed] [Google Scholar]
- Gal S, Zheng H, Fridkin M, Youdim MB. Restoration of nigrostriatal dopamine neurons in post-MPTP treatment by the multifunctional brain-permeable iron chelator-monoamine oxidase inhibitor drug, M30. Neurotox Res. 2010;17:15–27. doi: 10.1007/s12640-009-9070-9. [DOI] [PubMed] [Google Scholar]
- Ghanim H, Monte SV, Sia CL, Abuaysheh S, Green K, Caruana JA, et al. Reduction in inflammation and the expression of amyloid precursor protein and other proteins related to Alzheimer's disease following gastric bypass surgery. J Clin Endocrinol Metab. 2012;97:E1197–E1201. doi: 10.1210/jc.2011-3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein BJ. Insulin resistance as the core defect in type 2 diabetes mellitus. Am J Cardiol. 2002;90(5A):3G–10G. doi: 10.1016/s0002-9149(02)02553-5. [DOI] [PubMed] [Google Scholar]
- Green AR, Mitchell BD, Tordoff AF, Youdim MB. Evidence for dopamine deamination by both type A and type B monoamine oxidase in rat brain in vivo and for the degree of inhibition of enzyme necessary for increased functional activity of dopamine and 5-hydroxytryptamine. Br J Pharmacol. 1977;60:343–349. doi: 10.1111/j.1476-5381.1977.tb07506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunblatt E, Salkovic-Petrisic M, Osmanovic J, Riederer P, Hoyer S. Brain insulin system dysfunction in streptozotocin intracerebroventricularly treated rats generates hyperphosphorylated tau protein. J Neurochem. 2007;101:757–770. doi: 10.1111/j.1471-4159.2006.04368.x. [DOI] [PubMed] [Google Scholar]
- Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB J. 2004;18:902–904. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- Hooijmans CR, Graven C, Dederen PJ, Tanila H, van Groen T, Kiliaan AJ. Amyloid beta deposition is related to decreased glucose transporter-1 levels and hippocampal atrophy in brains of aged APP/PS1 mice. Brain Res. 2007;1181:93–103. doi: 10.1016/j.brainres.2007.08.063. [DOI] [PubMed] [Google Scholar]
- Hou WK, Xian YX, Zhang L, Lai H, Hou XG, Xu YX, et al. Influence of blood glucose on the expression of glucose trans-porter proteins 1 and 3 in the brain of diabetic rats. Chin Med J (Engl) 2007;120:1704–1709. [PubMed] [Google Scholar]
- Huang XF, Yu Y, Zavitsanou K, Han M, Storlien L. Differential expression of dopamine D2 and D4 receptor and tyrosine hydroxylase mRNA in mice prone, or resistant, to chronic high-fat diet-induced obesity. Brain Res Mol Brain Res. 2005;135:150–161. doi: 10.1016/j.molbrainres.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Kaczmarczyk MM, Machaj AS, Chiu GS, Lawson MA, Gainey SJ, York JM, et al. Methylphenidate prevents high-fat diet (HFD)-induced learning/memory impairment in juvenile mice. Psychoneuroendocrinology. 2013;38:1553–1564. doi: 10.1016/j.psyneuen.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn SE, Prigeon RL, Schwartz RS, Fujimoto WY, Knopp RH, Brunzell JD, et al. Obesity, body fat distribution, insulin sensitivity and Islet beta-cell function as explanations for metabolic diversity. J Nutr. 2001;131:354S–360S. doi: 10.1093/jn/131.2.354S. [DOI] [PubMed] [Google Scholar]
- Kupershmidt L, Weinreb O, Amit T, Mandel S, Carri MT, Youdim MB. Neuroprotective and neuritogenic activities of multimodal iron-chelating drugs in motor-neuron-like NSC-34 cells and transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 2009;23:3766–3779. doi: 10.1096/fj.09-130047. [DOI] [PubMed] [Google Scholar]
- Kupershmidt L, Weinreb O, Amit T, Mandel S, Bar-Am O, Youdim MB. molecular targets of the neuroprotective/neurorescue multimodal iron chelating drug M30 in the mouse brain. Neuroscience. 2011;189:345–358. doi: 10.1016/j.neuroscience.2011.03.040. [DOI] [PubMed] [Google Scholar]
- Kupershmidt L, Amit T, Bar-Am O, Youdim MB, Weinreb O. Neuroprotection by the multitarget iron chelator M30 on age-related alterations in mice. Mech Ageing Dev. 2012a;133:267–274. doi: 10.1016/j.mad.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Kupershmidt L, Amit T, Bar-Am O, Youdim MB, Weinreb O. The multi-target iron chelating-radical scavenging compound M30 possesses beneficial effects on major hallmarks of Alzheimer's disease. Antioxid Redox Signal. 2012b;17:860–877. doi: 10.1089/ars.2011.4279. [DOI] [PubMed] [Google Scholar]
- Lee AK, Mojtahed-Jaberi M, Kyriakou T, Astarloa EA, Arno M, Marshall NJ, et al. Effect of high-fat feeding on expression of genes controlling availability of dopamine in mouse hypothalamus. Nutrition. 2010;26:411–422. doi: 10.1016/j.nut.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008;582:359–364. doi: 10.1016/j.febslet.2007.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechlovich D, Amit T, Bar-Am O, Mandel S, Youdim MB, Weinreb O. The multi-target iron chelator, M30 Modulates HIF-1alpha-related glycolytic genes and insulin signaling pathway in the frontal cortex of APP/PS1 Alzheimer's disease mice. Curr Alzheimer Res. 2013;11:119–127. doi: 10.2174/1567205010666131212112529. [DOI] [PubMed] [Google Scholar]
- Moret C, Briley M. The importance of norepinephrine in depression. Neuropsychiatr Dis Treat. 2011;7(Suppl. 1):9–13. doi: 10.2147/NDT.S19619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison CD, Pistell PJ, Ingram DK, Johnson WD, Liu Y, Fernandez-Kim SO, et al. High fat diet increases hippocampal oxidative stress and cognitive impairment in aged mice: implications for decreased Nrf2 signaling. J Neurochem. 2010;114:1581–1589. doi: 10.1111/j.1471-4159.2010.06865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naoi M, Maruyama W. Functional mechanism of neuroprotection by inhibitors of type B monoamine oxidase in Parkinson's disease. Expert Rev Neurother. 2009;9:1233–1250. doi: 10.1586/ern.09.68. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig KL, Floden AM, Adhikari R, Golovko MY, Combs CK. Amyloid precursor protein and proinflammatory changes are regulated in brain and adipose tissue in a murine model of high fat diet-induced obesity. PLoS ONE. 2012;7:e30378. doi: 10.1371/journal.pone.0030378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RE, Deleger S, Strawbridge WJ, Kaplan GA. Prospective association between obesity and depression: evidence from the Alameda County Study. Int J Obes Relat Metab Disord. 2003;27:514–521. doi: 10.1038/sj.ijo.0802204. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer's amyloid precursor protein transcript. J Biol Chem. 2002;277:45518–45528. doi: 10.1074/jbc.M207435200. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Smith MA, Perry G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr Med Chem. 2001;8:721–738. doi: 10.2174/0929867013372922. [DOI] [PubMed] [Google Scholar]
- Scheepers A, Joost HG, Schurmann A. The glucose transporter families SGLT and GLUT: molecular basis of normal and aberrant function. JPEN J Parenter Enteral Nutr. 2004;28:364–371. doi: 10.1177/0148607104028005364. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Expression of hypoxia-inducible factor 1: mechanisms and consequences. Biochem Pharmacol. 2000;59:47–53. doi: 10.1016/s0006-2952(99)00292-0. [DOI] [PubMed] [Google Scholar]
- Shemyakov SE. Monoamine oxidase activity, lipid peroxidation, and morphological changes in human hypothalamus during aging. Bull Exp Biol Med. 2001;131:586–588. doi: 10.1023/a:1012371106766. [DOI] [PubMed] [Google Scholar]
- Siddiq A, Ayoub IA, Chavez JC, Aminova L, Shah S, LaManna JC, et al. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J Biol Chem. 2005;280:41732–41743. doi: 10.1074/jbc.M504963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon GE, Von Korff M, Saunders K, Miglioretti DL, Crane PK, van Belle G, et al. Association between obesity and psychiatric disorders in the US adult population. Arch Gen Psychiatry. 2006;63:824–830. doi: 10.1001/archpsyc.63.7.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A. 2010;107:7036–7041. doi: 10.1073/pnas.1000645107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirumangalakudi L, Prakasam A, Zhang R, Bimonte-Nelson H, Sambamurti K, Kindy MS, et al. High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J Neurochem. 2008;106:475–485. doi: 10.1111/j.1471-4159.2008.05415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucetic Z, Carlin JL, Totoki K, Reyes TM. Epigenetic dysregulation of the dopamine system in diet-induced obesity. J Neurochem. 2012;120:891–898. doi: 10.1111/j.1471-4159.2012.07649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb O, Amit T, Bar-Am O, Youdim MB. Rasagiline: a anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog Neurobiol. 2010;92:330–344. doi: 10.1016/j.pneurobio.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Yamada N, Katsuura G, Ochi Y, Ebihara K, Kusakabe T, Hosoda K, et al. Impaired CNS leptin action is implicated in depression associated with obesity. Endocrinology. 2011;152:2634–2643. doi: 10.1210/en.2011-0004. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Bakhle YS. Monoamine oxidase: isoforms and inhibitors in Parkinson's disease and depressive illness. Br J Pharmacol. 2006;147(Suppl. 1):S287–S296. doi: 10.1038/sj.bjp.0706464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman K, Ryu H, Hall D, O'Donovan K, Lin KI, Miller MP, et al. Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and erythropoietin. J Neurosci. 1999;19:9821–9830. doi: 10.1523/JNEUROSCI.19-22-09821.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Gal S, Weiner LM, Bar-Am O, Warshawsky A, Fridkin M, et al. multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases: in vitro studies on antioxidant activity, prevention of lipid peroxide formation and monoamine oxidase inhibition. J Neurochem. 2005a;95:68–78. doi: 10.1111/j.1471-4159.2005.03340.x. [DOI] [PubMed] [Google Scholar]
- Zheng H, Weiner LM, Bar-Am O, Epsztejn S, Cabantchik ZI, Warshawsky A, et al. Design, synthesis, and evaluation of bifunctional iron-chelators as potential agents for neuroprotection in Alzheimer's, Parkinson's, and other neurodegenerative diseases. Bioorg Med Chem. 2005b;13:773–783. doi: 10.1016/j.bmc.2004.10.037. [DOI] [PubMed] [Google Scholar]