

Abstract
Background and Purpose
Augmented Na+/Ca2+ exchanger (NCX) activity may play a crucial role in cardiac arrhythmogenesis; however, data regarding the anti-arrhythmic efficacy of NCX inhibition are debatable. Feasible explanations could be the unsatisfactory selectivity of NCX inhibitors and/or the dependence of the experimental model on the degree of Ca2+i overload. Hence, we used NCX inhibitors SEA0400 and the more selective ORM10103 to evaluate the efficacy of NCX inhibition against arrhythmogenic Ca2+i rise in conditions when [Ca2+]i was augmented via activation of the late sodium current (INaL) or inhibition of the Na+/K+ pump.
Experimental Approach
Action potentials (APs) were recorded from canine papillary muscles and Purkinje fibres by microelectrodes. NCX current (INCX) was determined in ventricular cardiomyocytes utilizing the whole-cell patch clamp technique. Ca2+i transients (CaTs) were monitored with a Ca2+-sensitive fluorescent dye, Fluo-4.
Key Results
Enhanced INaL increased the Ca2+ load and AP duration (APD). SEA0400 and ORM10103 suppressed INCX and prevented/reversed the anemone toxin II (ATX-II)-induced [Ca2+]i rise without influencing APD, CaT or cell shortening, or affecting the ATX-II-induced increased APD. ORM10103 significantly decreased the number of strophanthidin-induced spontaneous diastolic Ca2+ release events; however, SEA0400 failed to restrict the veratridine-induced augmentation in Purkinje-ventricle APD dispersion.
Conclusions and Implications
Selective NCX inhibition – presumably by blocking revINCX (reverse mode NCX current) – is effective against arrhythmogenesis caused by [Na+]i-induced [Ca2+]i elevation, without influencing the AP waveform. Therefore, selective INCX inhibition, by significantly reducing the arrhythmogenic trigger activity caused by the perturbed Ca2+i handling, should be considered as a promising anti-arrhythmic therapeutic strategy.
Table of Links
| TARGETS | LIGANDS |
| NCX | ATX-II |
| Forskolin | |
| SEA0400 | |
This Table lists key protein targets and ligands in this document, which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
The development of cardiac arrhythmias requires the concomitant existence of a trigger (i.e. extrasystoles, generated by large enough membrane potential oscillations, usually induced by marked perturbations in Ca2+i handling, leading to Ca2+i overload) and a substrate [i.e. large enough action potential duration (APD) dispersion between adjacent cells, typically caused by an uneven reduction in the efficacy of action potential (AP) repolarization] (Varro and Baczko, 2011). In physiological conditions, [Ca2+]i is tightly controlled via a delicate balance between Ca2+ fluxes (Eisner et al., 1998); consequently, Ca2+i overload is prevented and triggered events are rare. Furthermore, the high conduction and fast propagation of the electrical impulses and the homogeneous repolarization (i.e. small APD dispersion between adjacent cells) precede circular re-entry (Varro and Baczko, 2011).
The pivotal contribution of adverse shifts in the function of the Na+/Ca2+ exchanger (NCX) to arrhythmogenesis was clearly demonstrated in models of severe cardiac diseases, such as ischaemia/reperfusion injury (Schafer et al., 2001), heart failure (Pogwizd et al., 2001) and long-QT syndrome 3 (Nuyens et al., 2001), and were also suggested to contribute to the onset of atrial fibrillation (Lenaerts et al., 2009) and Torsades de Pointes (TdP) tachyarrhythmias (Farkas et al., 2009). Therefore, theoretically, selective NCX inhibition should represent a novel, promising tool against Ca2+-dependent arrhythmias.
Nonetheless, data in the literature regarding the efficacy and clinical perspective of NCX inhibition are controversial (Antoons et al., 2012). NCX inhibitors have shown anti-arrhythmic effects in heart rhythm disturbances evoked by ischaemia/reperfusion injury in vivo (Takahashi et al., 2003), in Langendorff-perfused hearts (Mukai et al., 2000; Elias et al., 2001; Schafer et al., 2001; Woodcock et al., 2001; Yamamura et al., 2001; Satoh et al., 2003; Morita et al., 2011) and in simulated ischaemia/reperfusion models (Watano et al., 1999; Nagy et al., 2004; Wongcharoen et al., 2006; Tanaka et al., 2007; Namekata et al., 2009). However, while SEA0400 was found to reduce the incidence (Nagy et al., 2004) or abolish the development of early afterdepolarizations (EADs) (Milberg et al., 2008), in guinea pig hearts it lacked efficacy against aconitine-induced arrhythmias (Amran et al., 2004). Furthermore, in Langendorff-perfused rat hearts, it even increased the incidence and duration of arrhythmias (Feng et al., 2006). Results from our related studies are also contradictory. SEA0400 did not reduce QTc following dofetilide application and failed to prevent the development of TdPs in rabbits (Farkas et al., 2008; 2009,), whereas in another study, it effectively suppressed EADs, without influencing APD (Nagy et al., 2004). In contrast, Milberg et al. found SEA0400 substantially shortened the APD and also reversed sotalol- or veratridine-induced TdPs (Milberg et al., 2008; 2012,). Recently, Jost et al. (2013) reported that ORM10103, a novel, more selective NCX blocker, suppressed pharmacologically-induced delayed afterdepolarization and EADs, confirming previous results with SEA0400 (Nagy et al., 2004).
Increased NCX activity induced by augmenting the level of Na+ [e.g. by increasing the INaL (late sodium current) or inhibiting the Na+/K+ pump] can lead to Ca2+i overload and subsequently abnormal automaticity (Antoons et al., 2012). Hence, in the present study we evaluated the efficacy of two NCX inhibitors, SEA0400 and the more selective ORM10103, against the arrhythmogenic consequences of the increased [Ca2+]i load induced by augmenting [Na+]i. Furthermore, as the efficacy of NCX inhibition may not necessarily be the same for the trigger and substrate sides of arrhythmogenesis, we also investigated the effect of NCX inhibition on the [Ca2+]i overload-induced shifts in AP kinetics and APD dispersion.
Methods
Experiments were carried out in isolated canine cardiomyocytes or cardiac multicellular preparations, harvested from 50 animals, in compliance with the Guide for the Care and Use of Laboratory Animals (USA NIH Publication No. 86–23, revised 1985). Protocols were approved by the Ethical Committee for Protection of Animals in Research of the University of Szeged, Hungary (Permit No. I-74-9/2009). Experimental settings and the protocols for anaesthesia, thoracotomy and isolation of ventricular cardiomyocytes were as described previously (Nagy et al., 2013).
Recording of APs in multicellular preparations
Isolated papillary muscles were obtained from the right ventricle. Ventricle-Purkinje fibre preparations, excised also from right ventricles with a free running Purkinje fibre, were used for dual electrode recordings. Multicellular preparations were mounted in a 40 mL glass chamber and perfused with Krebs–Henseleit solution at 37°C. The recording pipette was filled with 3 M KCl.
Monitoring [Ca2+]i transients in single ventricular cardiomyocytes
Cardiomyocytes were isolated from canine left ventricles using enzymatic protocols, as described in detail previously (Nagy et al., 2013). [Ca2+]i transients were monitored via a Ca2+-sensitive fluorescent dye, Fluo 4. Dye-loaded cells were mounted in a low volume imaging chamber (RC47FSLP, Warner Instruments, Hamden, CT, USA) and field-stimulated at a rate of 1 Hz, while continuously superfused with normal Tyrode's solution [containing (in mM): 144 NaCl, 0.4 NaH2PO4, 4 KCl, 0.53 MgSO4, 1.8 CaCl2, 5.5 glucose and 5 HEPES; pH was adjusted to 7.4 with NaOH]. Fluorescence measurements were performed on the stage of an Olympus IX 71 (Olympus Corporation, Tokyo, Japan) inverted fluorescence microscope. The Ca2+-sensitive dye was excited at 480 nm and the fluorescence emitted was detected at 535 nm. Optical signals were recorded by a photon counting photomultiplier module (Hamamatsu, model H7828; Hamamatsu Photonics Deutschland GmbH, Herrsching am Ammersee, Germany) and sampled at 1 kHz. Data acquisition and analysis were performed using a CAIRN Optoscan System (Cairn-Research Limited, Faversham Kent, England and Wales). Background fluorescence levels were recorded several times during each experiment and were used to correct raw fluorescence data.
Recording ion currents
Transmembrane currents were determined at 37°C using the whole-cell configuration of the patch clamp technique.
Late Na current (INaL)
Cardiomyocytes were perfused with a K-free, Cs-Tyrode's solution containing (in mM): NaCl 135, CsCl 10, CaCl2 1, MgCl2 1, BaCl2 0.2, NaH2PO4 0.33, TEACl 10, HEPES 10, glucose 10 (pH = 7.4), supplemented with 20 μM ouabain, and 1 μM nisoldipine in order to block the Na+/K+ pump and Ca2+ currents respectively. The pipette solution contained (in mM) CsOH 140, aspartic acid 75, TEACl 20, Mg-ATP 5, HEPES 10, NaCl 10, EGTA 20, CaCl2 10, at pH = 7.2. [Ca2+]i was set to 160 nM using WinMaxC (Patton et al., 2004). INaL was activated via 200 ms depolarizing pulses from −80 mV (holding potential) to −20 mV.
NCX current (INCX)
INCX was measured in Cs-Tyrode's solution supplemented with 50 μM lidocaine to inhibit INa. The internal solution was as above, except it contained 10 mM NaCl. INCX was determined using voltage ramps, from −40 mV holding potential to +60 mV then to −100 mV. Reverse and forward INCX were calculated at +40 and −80 mV respectively. Currents were first recorded in control solution, then in the presence of 1 μM SEA0400 or 10 μM ORM10103, finally 10 mM NiCl2 was also added. INCX was determined as the Ni2+-sensitive difference current.
Simultaneous determination of INaL and reverse mode INCX (revINCX)
For simultaneous determination of INaL and revINCX, the internal solution contained 15 mM NaCl. The Ca2+-activated Cl− current was eliminated by 100 μM niflumic acid. A 200 ms voltage step from −80 mV (holding potential) to −20 mV was used to determine INaL; a second step of the same duration (+40 mV) evoked revINCX. In the control group, first the total current was recorded, then the recording was repeated following the application of 1 μM veratridine and finally in the presence of 10 mM NiCl2 to completely block NCX. In the SEA0400 group, cells were first pretreated with 1 μM SEA0400 and then recordings were performed as above. In both groups, INCX was calculated relative to the full range defined by the control (maximum) and Ni2+-treated (minimum) currents.
Determination of the SEA0400- or ORM10103-sensitive current
To compare the SEA0400- and ORM10103-sensitive currents under normal conditions and following INaL activation, INaL, ICaL and INaK were not inhibited. A typical ventricular AP has been used as command waveform. The NCX-mediated charge was also calculated. In the control group, the SEA0400- and ORM10103-sensitive currents were calculated from the composite currents recorded before and after the application of either 1 μM SEA0400 or 10 μM ORM10103. In the anemone toxin II (ATX-II) group, following the recording of the steady-state current, first 2 nM ATX-II, then 1 μM SEA0400 or 10 μM ORM10103 were applied in order to increase INaL and inhibit INCX respectively. The SEA/ORM-sensitive NCX currents in this group were determined as the difference current between ATX-II alone and ATX-I I + SEA/ORM recordings.
Simultaneous determination of ICaL and forward mode INCX (fwdINCX)
Measurements were performed in Cs-Tyrode's solution. The internal solution contained only 5 mM NaCl in order to restrict the contribution of the reverse mode NCX activity to the current. The internal Ca2+ was not buffered. ICaL was activated by application of a voltage step (50 ms) from a holding potential of −80 to 0 mV. The tail current was determined at −80 mV. In the untreated group, the control current was compared with the current stimulated by 2 μM forskolin. In the ORM10103 group, the cells were first pretreated with 10 μM ORM10103 and then the forskolin challenge was repeated.
Materials
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). SEA0400 was synthesized in the Department of Pharmaceutical Chemistry, University of Szeged. ORM10103 was provided by Orion Pharma (Espoo, Finland). Stock solutions were stored at 4°C. All solutions were freshly prepared before the start of each experiment.
ATX-II or veratridine
In preliminary tests aimed to titrate optimal APD lengthening in multicellular samples, ATX-II (2 nM) and veratridine (1 μM) were effective; therefore, in this study, we used them alternately. The only difference observed was the somewhat slower development of the steady-state effect of veratridine. We usually used ATX-II, except for Purkinje-ventricle preparations, where only 0.5 μM veratridine was able to induce the appropriate level of AP dispersion, probably due to the decreased sensitivity of the endocardial samples to ATX-II compared with papillary muscles. Veratridine developed a strong effect on Purkinje strands as well.
For the intracellular calcium transient (CaT) measurements, similar to multicellular samples, 2 nM ATX-II was used. At this concentration – in contrast to veratridine – ATX-II enhanced the magnitude of [Ca2+]i transients without inducing abnormal decay kinetics. Such an effect of veratridine is probably a consequence of its stronger APD lengthening effect. While – as shown in Figure 6 – both drugs could be used to demonstrate the tight coupling between INaL and revNCX (reverse transport mode of the sodium–calcium exchanger), in the actual experimental conditions the use of 1 μM veratridine was preferred as it induced a more pronounced effect on INaL decay; thus, the subsequent enhancement of revINCX was also stronger. However, current/CaT and AP measurements were recorded on different sides of ventricles (left and right, respectively); NCX expression was found to be identical between ventricles (McDonald et al., 2000).
Figure 6.
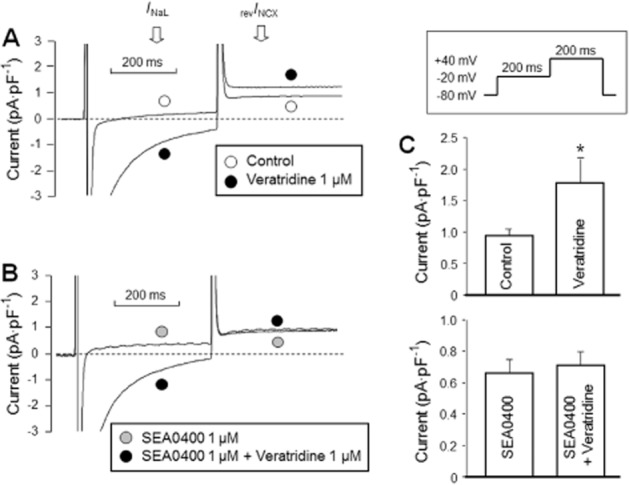
Effect of 1 μM SEA0400 on the veratridine-induced modulation of revINCX. Activation of INaL (determined at −20 mV) by 1 μM veratridine resulted in an enhanced revINCX (determined at +40 mV) (A), which was prevented by pretreatment with 1 μM SEA0400 (B, C). NCX currents were defined as the Ni2+-sensitive currents. The pulse protocol applied is displayed in the inset.
Concentrations of the inhibitors
IC50 values reported for SEA0400 in dog ventricular cardiomyocytes were 111 ± 43 and 108 ± 18 nM for the inward and outward NCX currents respectively (Birinyi et al., 2005). For ORM10103, these values were 780 nM for the inward and 960 nM for the reverse INCX (Jost et al., 2013). As our principal aim was to test the putative efficacy of INCX inhibition against arrhythmogenic Ca2+i elevation, we aimed to induce maximal INCX inhibition while not interfering significantly with any other currents present. SEA0400 was applied in 1 μM concentration. At this concentration, its inhibitory effect on ICa was 20% (Birinyi et al., 2005). ORM10103 has been applied in concentration of 10 μM in which it can exert its maximal inhibitory effect without influencing the Ca2+ current (Jost et al., 2013).
Statistics
Experimental data were compared and analysed using Student's t-test, or when needed, repeated measures anova with Bonferroni-corrected post hoc test. Differences were considered significant at P < 0.05.
Results
Activation of INaL by ATX-II
INaL was activated by 2 nM ATX-II, which is known to increase substantially the Na+ influx due to lengthening of the inactivation of INaL (Shryock et al., 2013). Application of 2 nM ATX-II significantly increased the amplitude of INaL measured at the end of the 200 ms depolarization (P < 0.05, n = 7/3; Figure 1A). The second ‘n’ is the number of experimental animals. APD90 was also increased (n = 5/5, P < 0.05; Figure 1B), as well as the amplitude of the [Ca2+]i transient (n = 6/3, P < 0.05; Figure 1C). Parallel to the enhancement of the [Ca2+]i transient, cell shortening was also enhanced by ATX-II (n = 6/3, P < 0.05; Figure 1D).
Figure 1.
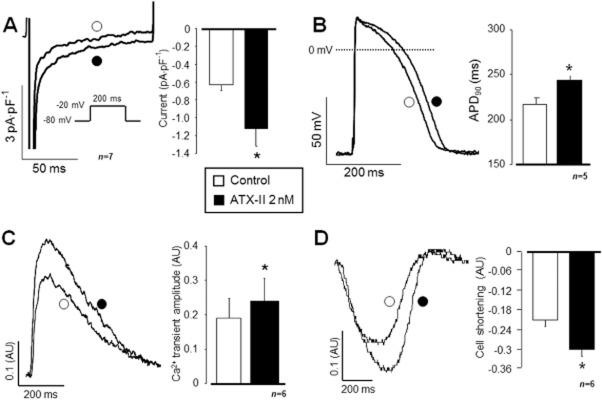
Effect of INaL activation on APD, [Ca2+]i transient and cell shortening in canine isolated ventricular myocytes. ATX-II 2 nM significantly enhanced the INaL current by apparently slowing its inactivation kinetics (A). As a consequence, APD was lengthened (B) and the amplitude of the [Ca2+]i transient was also increased (C), leading to an enhanced cell shortening (D). Columns and bars represent means ± SEM values; asterisks (*) denote significant differences from control (P < 0.05). AU represents arbitrary units.
The effects of 1 μM veratridine (not shown) were rather similar. At the end of a 200 ms depolarizing pulse to −20 mV, the magnitude of INaL increased from −0.70 ± 0.02 to −1.62 ± 0.17 pA·pF−1 (P < 0.05, n = 7/3); the APD90 was enhanced from 207 ± 7.6 to 276 ± 5.5 ms (P < 0.05, n = 5/5). Its application also increased the amplitude of the CaT from 0.16 ± 0.03 to 0.21 ± 0.04 AU (P < 0.05, n = 10/3).
Inhibition of NCX by SEA0400 and ORM10103
INCX was determined in cardiomyocytes superfused with modified Tyrode's solution as described in the Methods section. The pipette solution contained 10 mM NaCl, and [Ca2+]i was adjusted to ∼160 nM applying CaCl2 and EGTA as calculated by WinMaxC software (Patton et al., 2004). Figure 2A shows the blocking efficiency of 1 μM SEA0400 for the reverse and forward currents (n = 7/4, P < 0.05). In spite of this relatively high level of INCX inhibition, neither the APD90 (n = 5/5; Figure 2B), the amplitude of the [Ca2+]i transient (n = 5/3; Figure 2C), nor the half-relaxation time of the [Ca2+]i transient (301 ± 24 ms vs. 300 ± 20 ms) was affected by the application of 1 μM SEA0400. The magnitude of cell shortening was also unaffected (n = 5/3; Figure 2D).
Figure 2.
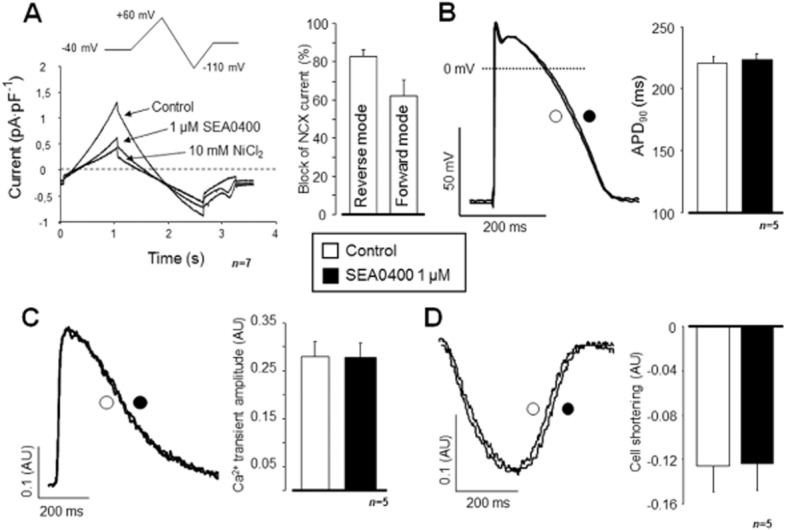
Effect of SEA0400 on INCX, APD and [Ca2+]i handling in canine isolated cardiac cells. INCX was determined using the conventional ramp protocol at +40 and −80 mV, respectively, defined as a Ni2+-sensitive current. SEA0400 1 μM substantially inhibited both the reverse and the forward mode activities of INCX (A). In contrast, neither APD90 (B) nor the [Ca2+]i transient (C) was influenced and the cell shortening (D) was also unaltered by 1 μM SEA0400.
Similar results were obtained with the novel NCX inhibitor compound ORM10103 (10 μM), which – as expected – caused a comparable, significant inhibition of INCX both in the reverse as well as in the forward mode operation (n = 6/2; Figure 3A). Again, in spite of the marked NCX blockade, neither the APD90 (n = 5/2; Figure 3B) nor the amplitude of the [Ca2+]i transient (n = 8/3; Figure 3C) were altered by ORM10103, and there was no change in the magnitude of cell shortening either (Figure 3D). However, in contrast to the results obtained with SEA0400, a small but statistically significant increase could be observed in the half-relaxation time (292 ± 22 ms vs. 304 ± 23 ms, P < 0.05, n = 8/3).
Figure 3.
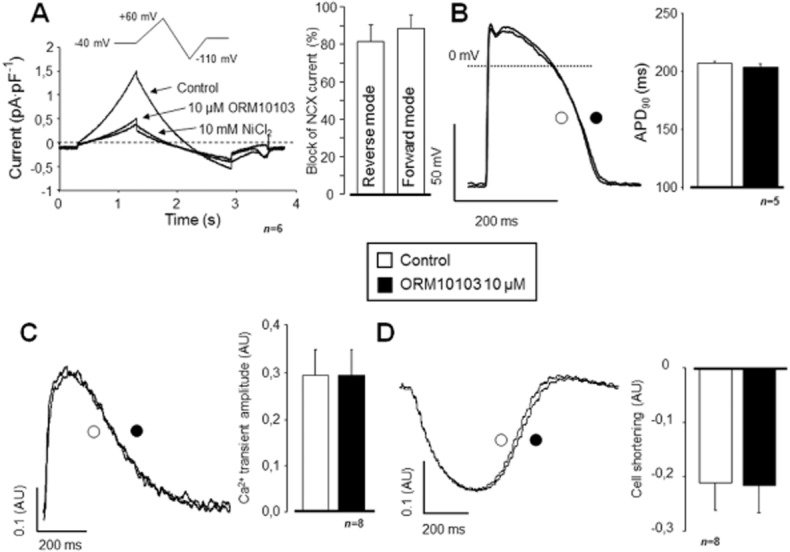
Effect of NCX inhibition by ORM10103 on INCX, APD and [Ca2+]i. Using the conventional ramp protocol, 10 μM ORM10103 efficiently inhibited INCX in both transport modes of the exchanger (A). Like SEA0400, ORM10103 also failed to modulate APD90 (B), the amplitude of the [Ca2+]i transient (C) or cell shortening (D).
Selective NCX inhibition eliminates the ATX-II-induced increase in [Ca2+]i transient amplitude and cell shortening
As shown previously, administration of 2 nM ATX-II significantly increased the amplitude of the [Ca2+]i transient (n = 6/3, P < 0.05). This elevation was fully reversed by superfusion with 1 μM SEA0400 (n = 6/3, P < 0.05; Figure 4A). The same pattern was observed in the case of cell shortening (control: −0.15 ± 0.01; ATX-II: −0.23 ± 0.01; and ATX-II + SEA0400: −0.14 ± 0.01 AU, n = 6/3, P < 0.05). When SEA0400 was applied first, it fully prevented the ATX-II-induced increase in the amplitude of the [Ca2+]i transient (n = 5/2; Figure 4B). Similarly, no changes were evoked by ATX-II in the magnitude of cell shortening after pretreatment with SEA0400 (−0.13 ± 0.03, −0.12 ± 0.02 and −0.13 ± 0.03 AU, n = 6/2).
Figure 4.
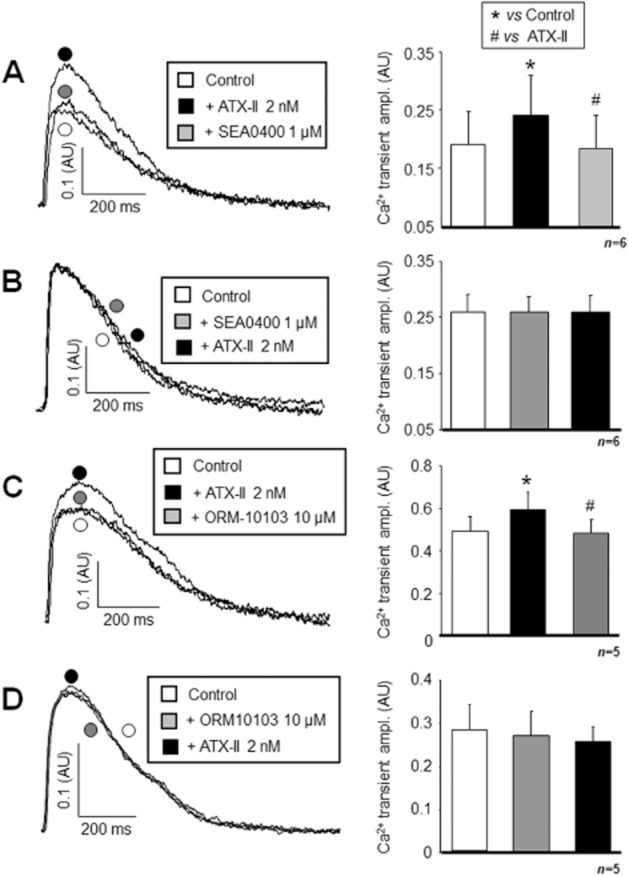
Demonstration of the efficacy of NCX inhibition against the INaL activation-induced [Ca2+]i load. If applied first, 2 nM ATX-II significantly enhanced the magnitude of the [Ca2+]i transient, but this increase was diminished by subsequent application of 1 μM SEA0400 (A). In contrast, when 1 μM SEA0400 was applied as a pretreatment, the secondary administration of ATX-II failed to enhance the [Ca2+]i transient (B). The same pattern of results was obtained with 10 μM ORM10103 (C and D respectively). The ATX-II-induced enhancement of the Ca2+i transient was reversed by ORM10103 (C). Statistical significances were verified by repeated measures anova plus Bonferroni post hoc test. Columns and bars are means ± SEM. *Denote significant differences from control, while #denote significant differences from the ATX-II-treated group (P < 0.05).
Essentially identical results were obtained with 10 μM ORM10103. It reversed the ATX-II-induced increase in the [Ca2+]i transient amplitude (n = 5/2, P < 0.05; Figure 4C), or alternatively, it prevented its elevation by ATX-II when ORM10103 was applied as a pretreatment (n = 5/2; Figure 4D). The magnitude of the cell shortening followed the shifts in [Ca2+]i both in the case of cumulative application (control: −0.24 ± 0.07; ATX-II: −0.31 ± 0.09; and ATX-II + ORM10103: −0.23 ± 0.07 AU, n = 5/2, P < 0.05) and in the case of pretreatment (−0.21 ± 0.07, −0.22 ± 0.07 and −0.21 ± 0.08 AU, respectively, n = 5/2).
Suppression of the arrhythmogenic diastolic [Ca2+]i release induced by ORM10103
In this set of experiments, the efficacy of partial INCX inhibition against arrhythmogenic diastolic [Ca2+]i release events, evoked by a combination of strophanthidin treatment and burst stimulation, was evaluated. Three randomly selected groups of cells were investigated: the first group was untreated, the second group was treated with 1 μM strophanthidin, whereas the third group was pretreated with 10 μM ORM10103 before the superfusion with strophanthidin. Diastolic [Ca2+]i release events were evoked by short rapid (2 Hz) pacing and were counted during the subsequent resting period of 2 min. Application of 1 μM strophanthidin significantly increased the amplitude of the [Ca2+]i transients at 1 Hz (n = 10/4, P < 0.05; Figure 5A). While ORM10103 pretreatment alone had no influence on the [Ca2+]i transient, the strophanthidin-induced [Ca2+]i load was largely reduced by ORM10103 pretreatment (n = 10/4; Figure 5B). The relative increase in [Ca2+]i transient amplitude was higher with strophanthidin alone compared to the increase after ORM1013 pretreatment (Figure 5C). Furthermore, while in the untreated group, spontaneous diastolic [Ca2+]i release events were absent (Figure 5D, upper trace), a large number of releases could be observed in response to 1 μM strophanthidin (Figure 5D, middle trace), which was significantly reduced in the presence of 10 μM ORM10103 (Figure 5D, lower trace, and Figure 5E).
Figure 5.
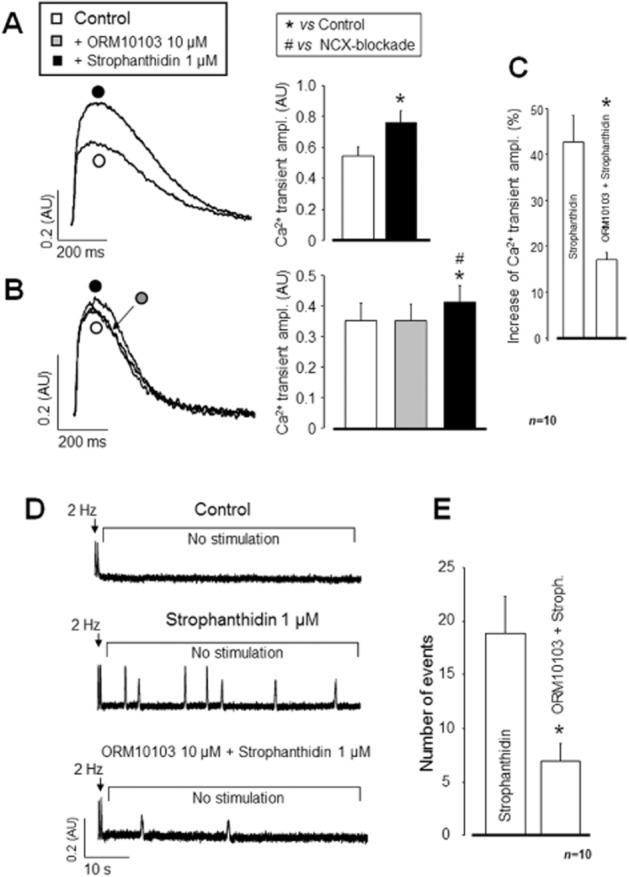
Anti-arrhythmic efficacy of ORM10103 during a strophanthidin challenge. Inhibition of the transport activity of the Na+/K+ pump with 1 μM strophanthidin resulted in significantly enhanced [Ca2+]i transient (A). Pretreatment with 10 μM ORM10103 markedly reduced the effect of the subsequently applied strophanthidin (B, C). In the absence of strophanthidin (control), spontaneous diastolic Ca2+ release was not observed following a short period of rapid (2 Hz) pacing (D, upper trace). In contrast, rapid pacing induced multiple arrhythmogenic Ca2+ release events, presumably resulting in delayed afterdepolarization in the presence of 1 μM strophanthidin (D, middle trace). Following 10 μM ORM10103 pretreatment, the same strophanthidin challenge was much less effective at evoking spontaneous Ca2+ release (D, lower trace, and E). In (B), the statistical significance level was verified by repeated measures anova plus Bonferroni post hoc test; *denote significant differences from control, while #denote significant differences from the ORM10103-treated group (P < 0.05). In the case of (C) and (E), *indicate significant differences from the strophanthidin-treated group.
Inhibition of INaL induced enhancement of revINCX by SEA0400
In this set of experiments, simultaneous determination of INaL and revINCX was measured, as shown in the inset of Figure 6. Activation of revINCX was supported by buffering [Ca2+]i to 160 nM and by adding 15 mM Na+ to the pipette solution.
When applying selective INCX inhibition (SEA0400 pretreatment) before the veratridine superfusion, the close relationship between INaL and revINCX was largely uncoupled. Furthermore, 1 μM SEA0400 alone did not influence the magnitude of INaL, but the veratridine-induced secondary increase in INCX due to increased Na+ load was abolished by SEA0400. The amplitude of revINCX was 0.93 ± 0.24 pA·pF−1 in control, which rose to 1.79 ± 0.43 pA·pF−1 in the presence of 1 μM veratridine (Figure 6A). Pretreatment with SEA0400 reduced the secondary increase in INCX (n = 8/4, Figure 6B and C). Some experiments, performed using 2 nM ATX-II instead of veratridine in combination with SEA0400, gave principally identical results (not shown).
SEA0400 and ORM10103 failed to eliminate the APD lengthening effect of ATX-II
While selective inhibition of INCX by either SEA0400 or ORM10103 was found to be quite effective against the ATX-II-induced increase in cellular Ca2+ load, it completely failed to suppress the significant lengthening of APD caused by ATX-II. Application of 1 μM SEA0400 either before or following the ATX-II treatment had no effect on APD (Figure 7A and B). Similar to SEA0400, the more selective ORM10103 was also ineffective against the ATX-II-induced APD lengthening. Independently of the sequence of application, it failed to prevent or even reduce significantly the APD lengthening effect of ATX-II (Figure 7C and D).
Figure 7.
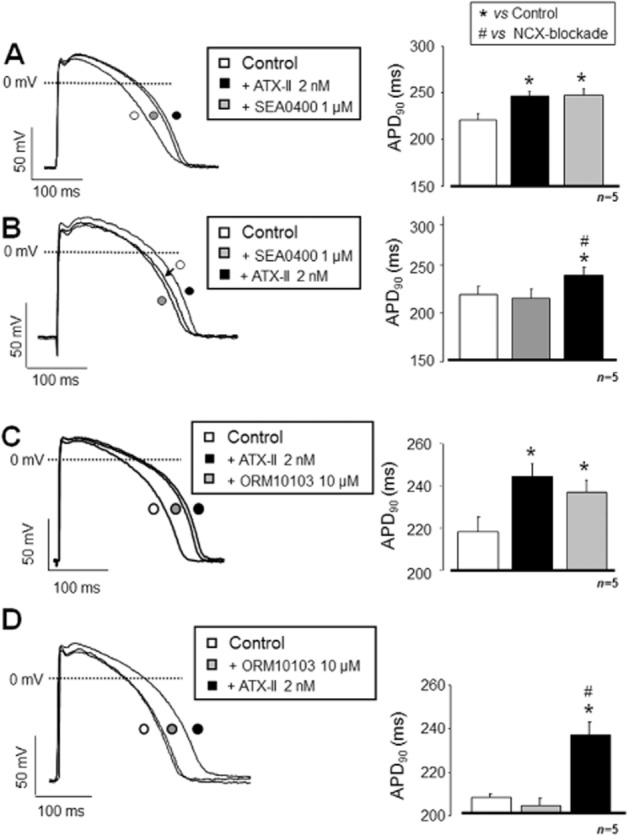
NCX inhibition has no effect on the INaL activation-induced APD lengthening. The experimental protocol was identical to that used in Figure 4. In contrast to [Ca2+]i transient measurements, NCX inhibition was ineffective against the INaL activation-induced APD90 prolongation. Neither SEA0400 (A, B) nor ORM10103 (C, D) reversed or even substantially decreased the lengthening of APD, leading frequently to generation of EADs. Statistical significances were verified using repeated measure anova plus Bonferroni post hoc test. Columns and bars represent means ± SEM. *Denote significant differences from control, while #from the group pretreated with either SEA0400 or ORM10103 (P < 0.05).
Effect of SEA0400 on the veratridine-induced APD dispersion in ventricle-Purkinje preparations
The effectiveness of INCX inhibition against the INaL activation-induced increase in AP dispersion was monitored in multicellular preparations, that is, in a piece of ventricular myocardial tissue coupled to the corresponding Purkinje fibre. The APD90 dispersion between Purkinje and ventricle was determined in two randomly composed experimental groups using dual microelectrodes. In the first group, only veratridine was applied, whereas in the second group, the preparations were pretreated with 1 μM SEA0400 before the exposure to veratridine. Under control conditions, AP dispersion was significantly increased by superfusion with 0.5 μM veratridine (Figure 8A and C). The control APD90 dispersion level was unaltered by pretreatment with 1 μM SEA0400; however, the application of 0.5 μM veratridine caused an increase in APD dispersion identical to that observed without SEA0400 (Figure 8B and C). The increase in AP dispersion following veratridine treatment, under control conditions, was unaltered by 1 μM SEA0400 pretreatment (n = 6/6; Figure 8D).
Figure 8.
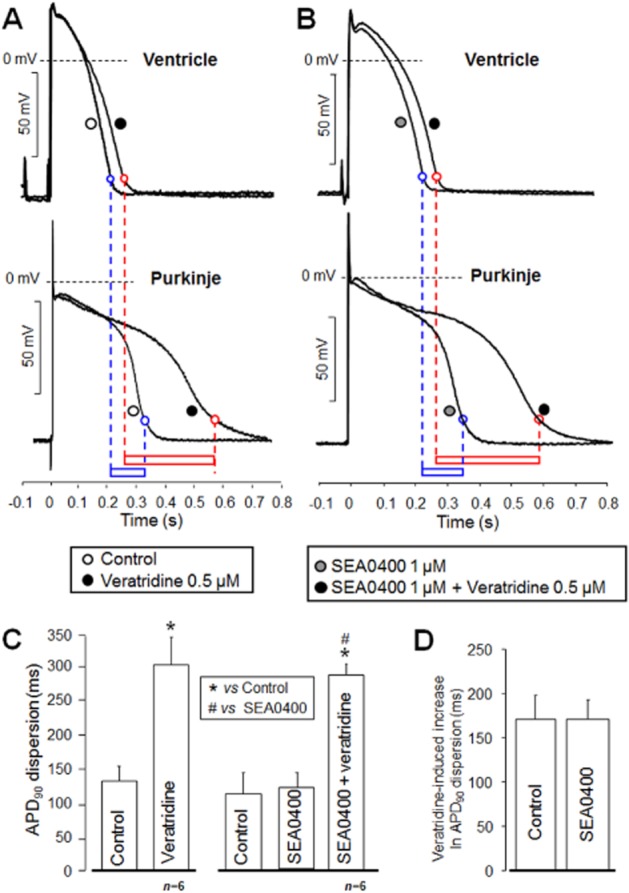
NCX inhibition is ineffective against increased AP dispersion induced by activation of INaL. In these experiments, ventricular and Purkinje APs were simultaneously determined using two sharp microelectrodes. Ventricle-Purkinje dispersion was defined as the difference between the corresponding APD90 values. Dispersion measured in the control group (A, lower box) was largely increased by 0.5 μM veratridine (A, upper box). Neither APD was significantly affected by pretreatment with 1 μM SEA0400 (B). In addition, dispersion in the presence of SEA0400 and its veratridine-induced increase (B, lower and upper boxes, respectively) were practically the same as those determined in the control group. Average values of dispersion obtained in the control and SEA0400 pretreated groups before and after veratridine are summarized in (C) and (D) respectively. *Denote significant differences from control, while #) denote significant differences from the group pretreated with SEA0400 (P < 0.05).
Estimation of INCX kinetics under normal conditions and after activation of INaL by SEA0400 and ORM10103
To further explore the relationship between INaL activation and INCX, we attempted to characterize the kinetics of INCX during an AP either under normal conditions or following enhanced intracellular Na+ as a result of the activation of INaL. In these experiments, Na+ concentration in the pipette was set to 15 mM and Ca2+ movements were unbuffered. An AP-like command voltage pulse was applied to mimic the physiological situation. When 1 μM SEA0400 was used (as shown in Figure 9), only a small amplitude of revINCX (carrying a charge of 4.1 ± 2.8 fC·pF−1) could be observed in the early phase of the AP. The reverse mode activity of INCX was switched to a much larger forward INCX carrying −105 ± 16 fC·pF−1, the estimated net charge was −101 ± 16 fC·pF−1. Following activation of INaL by 2 nM ATX, both the reverse mode (outward current) and the forward mode activities (inward current) of NCX were markedly enhanced (to 61 ± 12 and −180 ± 27 fC·pF−1, respectively, when characterized by the transferred charge), resulting in a net charge transfer of −119 ± 19 fC·pF−1 during the whole AP. Importantly, following INaL activation, both the reverse and the forward components of INCX were substantially elevated compared with the control group, whereas the net current was only moderately enhanced (−101 ± 16 fC·pF−1 vs. −119 ± 19 fC·pF−1, NS, n = 5/4 in both groups).
Figure 9.

Estimation of INCX as a SEA0400- or ORM10103-sensitive current during an AP. In order to estimate the kinetics of NCX during a representative AP, (A) SEA0400 and ORM10103 were applied, and the NCX current was estimated as a subtracted current. Under control conditions, only negligible (outward) revINCX could be observed during the early phase of the AP. The current rapidly turned into (inward) fwdINCX (see B and C, open squares). Application of 2 nM ATX-II (B and C, filled square) induced an apparent increase in the composite current; thus, after current subtraction, the amplitude of the reverse component was enhanced, the reversal time delayed (see also the bar graphs in D, left for SEA0400 right for ORM10103). The amplitude of the forward component was also enhanced (bar graphs in E, left for SEA0400 right for ORM10103). Note that the net charge carried by a current component may change differently from the amplitude of the respective peak current.
Under control conditions, the magnitudes and kinetics of the INCX components calculated following the application of 10 μM ORM10103 were similar to those determined after the application of SEA0400 (reverse: 11.94 ± 4.9 and forward: −65.65 ± 20.8 fC·pF−1; net charge: −53.71 ± 16.5 fC·pF−1, n = 8/5). In sharp contrast, following the application of 2 nM ATX, both the magnitudes and the kinetics of the ORM-sensitive current components were markedly different from those recorded with SEA0400 (reverse: 32.63 ± 8.1 forward: −164.2 ± 32.7 fC·pF−1; net charge: −147.86 ± 42.84 fC·pF−1, n = 8/5). Indeed, the difference between net charges was found to be statistically significant (−53.71 ± 16.5 fC·pF−1 vs. −147.86 ± 42.84 fC·pF−1).
It should be noted that in the case of SEA0400, this experimental arrangement may have serious limitations in estimating INCX components quantitatively as it cannot effectively inhibit INCX without inhibiting ICa (at 1 μM about 20%) (Birinyi et al., 2005). In contrast, ORM10103 had no marked effect on ICa (Jost et al., 2013). Furthermore, the AP lengthening effect of INaL activation was not taken into account in calculations, consequently the amount of extra Ca2+ entry via ICaL during the longer plateau phase was ignored.
ORM10103 10 μM failed to prevent forskolin-induced tail current enhancement
In this set of experiments, the Ca2+-activated fwdNCX (forward transport mode of the NCX) current was determined, as shown in the inset of Figure 10. The activation of fwdNCX was enhanced by unbuffered intracellular Ca2+, low Na+ in the pipette solution (5 mM), hyperpolarized membrane potential (−80 mV) and forskolin (2 μM)-stimulated Ca2+ influx.
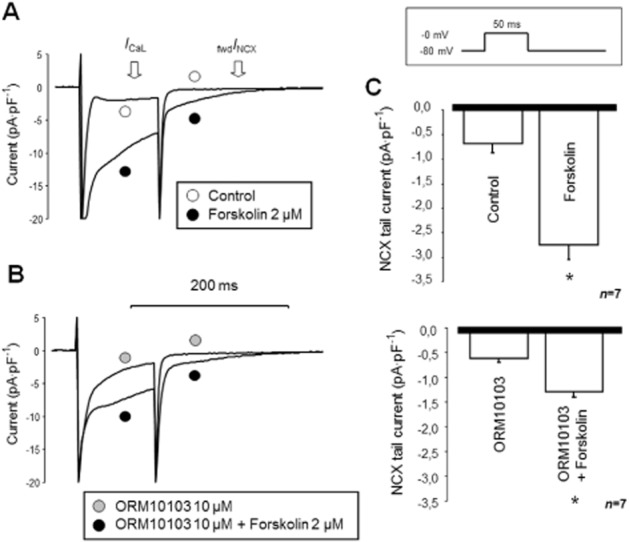
Estimation of fwdINCX inhibition by 10 μM ORM10103 during Ca2+i load. In the ORM10103-untreated group (A), compared with control, 2 μM forskolin markedly enhanced the ICa and the NCX tail current (C, upper panel). Pretreatment with 10 μM ORM10103 had no apparent effect on the baseline current (the baseline curve is not shown), but 2 μM forskolin still significantly increased both currents (B, C).
As shown in Figure 10A and C, in the untreated group, the magnitude of the tail current was significantly increased following the application of forskolin (P < 0.05, n = 7/3). In the other group, 10 μM ORM1003 was applied before the exposure to 2 μM forskolin to test the efficacy of ORM in preventing the secondary increase in fwdINCX current (Figure 10B and C). The ORM10103 per se did not change the magnitude of NCX tail current (−0.71 ± 0.1 pA·pF−1 vs. −0.63 ± 0.1 pA·pF−1, NS, n = 7; not shown in Figure 10B). Following the application of ORM10103, the forskolin challenge was repeated. While the increase in the tail current was still significant (−0.63 ± 0.1 pA·pF−1 vs. 1.34 ± 0.58 pA·pF−1, P < 0.05, n = 7/3), the quantitative comparison of the two groups clearly showed that in the presence of ORM10103, the increase was significantly lower than in the untreated group (−2.75 ± 0.3 pA·pF−1 vs. 1.34 ± 0.58 pA·pF−1, P < 0.05, n = 7/3). Furthermore, the activation of the ICa (measured at the end of the 50 ms pulse) also had a tendency to decrease (−5.35 ± 0.6 pA·pF−1 vs. −3.72 ± 0.5 pA·pF−1, NS, n = 7/3).
Discussion
In the present study, the potential anti-arrhythmic effect of selective INCX inhibition in conditions of [Na+]i elevation-induced Ca2+i load was evaluated using the most selective NCX inhibitors available. SEA0400 is well characterized: it has good or at least reasonable selectivity, although it exhibits a moderate inhibitory effect on ICa (Birinyi et al., 2005). A concentration of 1 μM SEA0400 was the first ‘selective’ INCX inhibitor widely used in corresponding studies. Recently, the novel, more selective INCX inhibitor, ORM10103, was reported to have EC50 values of 0.78/0.96 μM for forward/reverse INCX respectively (Jost et al., 2013). ORM10103 even at a high concentration (10 μM) has no detectable effect on further transmembrane currents, except for minimal suppression of IKr (Jost et al., 2013). In this study, SEA0400 and ORM10103 were applied alternately. Since both the increase in [Na+]i-generated Ca2+ load via revNCX transport and the increase in fwdNCX activity may significantly contribute to APD prolongation, NCX inhibition is expected to be anti-arrhythmic in this model.
In the first part of the study, the basic pharmacological effects of the applied agents (ATX-II, SEA0400 and ORM10103) were tested on the corresponding membrane current and a few important physiological parameters, such as APD, CaT amplitude and cell shortening (Figures 3). In the second set of experiments, the effects of selective INCX inhibition on [Ca2+]i homeostasis were evaluated under ‘close to physiological’ conditions (Figure 4), in a highly arrhythmogenic state (Figure 5). The results summarized in Figure 6 may provide a feasible explanation for the data presented in Figures 4 and 5. In the third part of this study, the effects of selective INCX inhibition on AP parameters were investigated under conditions corresponding to studies on [Ca2+]i homeostasis (Figures 9). In the final part of the study, we investigated the effect of ‘selective’ fwdINCX inhibition in order to explore its anti-arrhythmic mechanism and inability to modulate physiological CaT. To characterize these effects, two principal experimental arrangements were applied. (i) If the INa activator was applied before the INCX blockade, the question was whether selective NCX inhibition is able to reverse the INaL or Na+/K+ pump-induced APD prolongation or [Ca2+]i rise. (ii) When the NCX inhibitor was applied first, the question was whether the inhibition is able to prevent the arrhythmogenic effects of INaL activation. Our results indicate that while selective INCX inhibition satisfactorily reversed and prevented the Na+-induced [Ca2+]i rise, it had no apparent effect on the prolonged APD nor was the increased ventricle-Purkinje fibre APD90 dispersion reduced. The [Ca2+]i data directly support the potential anti-arrhythmic efficacy of INCX inhibition, which is thought to reduce abnormal automaticity due to Ca2+ overload (‘trigger side’) without apparently influencing the ‘substrate side’ or the repolarization and dispersion of repolarization, even when this was augmented by pharmacological means.
INCX inhibition failed to modulate basic electrophysiological parameters in isolated cardiomyocytes
Under control conditions – in spite of the observed marked INCX inhibition – no apparent effect of either SEA0400 or ORM10103 on CaT and AP parameters was observed. These results are in line with our previous work (Birinyi et al., 2008) and that of others using SEA0400 (Amran et al., 2004). However, at present, it is not clear why these NCX inhibitors failed to modulate the parameters of AP, CaT and contractility. Compared with previous studies (Weber et al., 2002; 2003,), our data seem to support the notion that under normal conditions, only a small net INCX is evoked during the AP. Consequently, the AP modulator effect of this small current may be fully compensated for by other repolarizing transmembrane currents, that is, by a strong repolarization reserve of the cardiomyocytes (Biliczki et al., 2002). In the normal situation, the reduction in Ca2+ efflux via fwdINCX inhibition failed to induce a net [Ca2+]i rise. The reason may be either a concomitant reduction in revINCX (thus preventing the Ca2+i gain during depolarization) or the unblocked fraction of fwdINCX is large enough to generate the Ca2+ efflux required for normal relaxation, or both. The lack of effect of both inhibitors may contradict the hypothesis (Hobai and O'Rourke, 2004) that INCX inhibition could, indeed, be a highly effective pharmacological tool to enhance cardiac contractility, which can be exploited for the treatment of heart failure.
NCX inhibition reverses or prevents the INaL-induced [Ca2+]i rise
Both NCX inhibitors effectively reduced previously increased [Ca2+]i and enhanced cell shortening or, alternatively, prevented the INaL-mediated rise in [Ca2+]i. If ATX-II was added first, the increased Na+ influx might promote revNCX activity, leading to a rise in [Ca2+]i and augmented CaTs. Subsequent application of an INCX inhibitor reduced the enhanced revINCX and limited any further Ca2+ influx and subsequent elevation of [Ca2+]i. The apparent failure of INaL activation to increase [Ca2+]i following NCX blockade clearly indicates the dominance of the reverse mode inhibition in this condition.
Partial NCX inhibition by ORM10103 reduces the incidence of diastolic Ca2+ release
Data shown in Figure 4 support a direct anti-arrhythmic effect of selective INCX inhibition via modulation of [Ca2+]i. This hypothesis has been tested by pharmacological inhibition of the Na+/K+ pump by strophanthidin, known to cause AP shortening (Pueyo et al., 2010), increased [Na+]i and subsequent SR Ca2+ overload, leading to arrhythmogenic Ca2+ release events (Satoh et al., 2000). ORM10103 substantially reduced the strophanthidin-induced rise in [Ca2+]i and significantly decreased the number of pacing-induced diastolic releases. This effect may be a consequence of a major reduction in the revNCX-mediated SR Ca2+ overload. The beneficial effect of NCX inhibition under these conditions – considering also the previous results – could be best explained by the observation that perturbation of Ca2+i handling in this case was mediated only by revNCX.
Direct coupling between INaL and Ca2+ handling via revINCX
To better understand the results shown in Figures 4 and 5, additional experiments were performed. The efficacy of INCX inhibition against the INaL-mediated increase in [Ca2+]i can be satisfactorily explained by supposing the existence of a ‘revNCX-mediated direct coupling’ between Na+ influx and [Ca2+]i rise, as also suggested by previous studies (Despa et al., 2002; Verdonck et al., 2004). To better characterize this hypothetical crosstalk, a tight interaction was modelled using a two-step protocol, as shown in Figure 6. The veratridine-induced INa increase led to a net gain in [Na+]i, subsequently shifting the reversal potential of NCX towards the more negative values. At the test potential of +40 mV, this shift favours Ca2+ influx via the enhanced revINCX, simultaneously generating a large outward current. In an additional set of experiments, this secondary increase in revINCX was fully abolished by pretreatment with 1 μM SEA0400. In summary, these data support a pivotal role of the INaL activation-coupled revINCX increase in cardiac arrhythmogenesis and may explain the beneficial anti-arrhythmic effects of NCX inhibition (see Figures 4 and 5).
Failure of selective NCX inhibition to modulate ventricular APD and reduce ventricle-Purkinje repolarization dispersion
In contrast to the CaT data, selective NCX inhibition failed to modulate APD – either as pretreatment or following the exposure to ATX-II (Figure 7). NCX inhibition also failed to protect or even reduce the INaL-induced large increase in ventricle-Purkinje APD dispersion (Figure 8). The NCX inhibition-induced uncoupling between INaL and Ca2+i handling may also be effective under these conditions; however, it seems to exert only a minor effect on APD. Furthermore, considering the similar ineffectiveness of these inhibitors on APD, we concluded that ORM10103 should have negligible effect on increased APD dispersion. This observation contradicts the results of Milberg et al. (2008) but supports the findings of Farkas et al. (2008; 2009). The reason for the discrepancy between our present findings and those of Milberg et al. is unclear. It could be due to different experimental conditions, methods of investigation and/or species differences. Hence, the elusive nature of the inhibitory effect of SEA0400 on ICa and NCX needs to be elucidated in further experiments.
The reason for the lack of effect on APD seems to be rather complex. Theoretically, the lengthening of APD following the enhancement of INaL may have two sources: (i) a direct source, that is, the effect of the increased INaL on APD, and (ii) an indirect source, that is, the role of INCX in defining the actual APD. The reasonable assumption that inhibition of NCX has no direct effect on INaL may partially explain its failure to counteract the prolongation of the APD. Also, direct estimation of INCX kinetics during an AP is complicated following the application of ATX-II. In this study, we approximated INCX as the SEA0400- or ORM10103-sensitive current (see Figure 9). In both cases, following the activation of INaL, both revINCX and fwdINCX were likely to be enhanced. The concomitant inhibition of both currents and the relatively small INCX found in the range of APD90 suggest the limited contribution of this current to ventricular repolarization under normal conditions. Interestingly, the kinetics of the ORM10103-sensitive and SEA0400-sensitive currents were moderately different following the application of ATX. As this protocol has major limitations, care should be taken when reaching a conclusion. We assume that the observed discrepancy is primarily a consequence of the higher selectivity of ORM10103 and that the ORM10103-sensitive current may better approximate the real kinetics of the NCX current.
Taken together with our CaT and AP data, an alternative explanation may also be feasible. Following INaL activation, parallel to the APD prolongation, [Ca2+]i also seemed to increase. However, subsequent inhibition of NCX reduced [Ca2+]i without an apparent effect on APD. It is possible that a primary reduction in CaT via negative feedback prolongs ICa inactivation and subsequently lengthens APD. Therefore, if NCX inhibition had any direct effect on APD (i.e. INCX-mediated reduction), it would be largely reduced by this indirect mechanism (i.e. ICaL-mediated prolongation). NCX inhibition, following the INaL-induced rise in [Ca2+]i, may have two parallel, but opposite effects on APD: directly, it may shorten APD via inhibition of INCX, and indirectly – due to its reducing effect on [Ca2+]i and the subsequent modulation of ICa kinetics – it may also prolong it. Consequently, the actual balance of these two counteracting effects may intimately influence the overall effect of INCX inhibition on the APD; this is hard to predict, and may significantly differ in various arrhythmia models and species. Indeed, this complex relationship may explain the reduction in APD induced by SEA0400, which was observed following a sotalol/veratridine challenge (Milberg et al., 2008), and – under rather similar experimental conditions – the increased incidence of TdP in Langendorff-perfused rabbit hearts following dofetilide treatment (Farkas et al., 2008; 2009,).
The anti-arrhythmic efficacy of ORM10103 may be a consequence of revINCX inhibition
In order to clarify the anti-arrhythmic action of ORM10103, the experiments summarized in Figure 10 were aimed at investigating the consequences of ‘selective’ fwdINCX inhibition. We found that under normal conditions (i.e. without stimulation of the Ca2+ influx with forskolin), 10 μM ORM10103 did not inhibit the tail current determined at −80 mV, which may be considered as fwdINCX. This result seems to contradict the data shown in Figure 3A, determined using the ‘traditional’ ramp protocol and may indicate that NCX inhibition during intact Ca2+ cycling provides more realistic information about its inhibitory efficacy. This result may also help to properly interpret the apparent failure of NCX inhibitors to modulate normal Ca2+ transient kinetics (Figure 3B). Nonetheless, we also found a statistically significant effect of ORM10103 on fwdINCX during Ca2+i load (Figure 10A–C) since the compound reduced the forskolin-induced increase in NCX tail current. This finding further emphasizes the importance of revINCX inhibition in the anti-arrhythmic efficacy of ORM10103, but it also justifies the need for additional experimental work.
Computer simulations of NCX current during an AP
A number of modelling studies aimed to simulate the kinetics of INCX during an AP. In spite of the experimental and simulation efforts, there is no consistent agreement about the reversal point of the NCX current during a ventricular AP. The guinea pig model of Luo and Rudy (1994) predicted a relatively long period for reverse mode activity (∼100 ms). Similar results were published for the guinea pig model of Faber and Rudy (2000). In contrast, the rabbit model developed by Weber et al. (2002) predicted a much shorter period of reverse activity (∼10 ms). Similarly, in a human ventricular AP model developed by Grandi et al. (2009), an inward NCX current was present during most of the plateau phase. In contrast, in the canine AP models described by Armoundas et al. (2003) and Greenstein et al. (2006), the reversal point was calculated to follow the plateau phase and the NCX was suggested to carry primarily outward current during the AP. Although interspecies differences (AP shape, Ca2+i and Na+i levels) may play an important role in the diversity of data, it should also be emphasized that even results derived from models designed for the same species (e.g. in guinea pig models) may differ significantly (Noble et al., 1991; 1998; Faber and Rudy, 2000). The large diversity of the simulated results may primarily be a consequence of the inconsistent interpretation of submembrane Ca2+ and Na+ movements and membrane potential dynamics, which are the main modulators of NCX activity.
Regarding the function of NCX inhibition on APD, Li and Rudy (2011) concluded that at a high pacing rate, in Purkinje and ventricular cells, INaK-mediated [Na+]i accumulation is the most important factor causing APD shortening. However, in Purkinje cells, INCX and INaL are also important contributors (Li and Rudy, 2011). In the computer model of NCX knockout mice by Sarai et al. (2006), the inward current decreased and the APD slightly shortened following 70% inhibition of INCX. In the study published by Amran et al. (2004), an integrated mathematical model was applied to investigate whether the NCX plays a role in aconitine-induced arrhythmias. The model well mimicked the aconitine-induced membrane oscillations; however, the fit was made worse by SEA0400 and the study concluded that the oscillations could not be suppressed by NCX inhibition.
Our present experimental results seem to support the simulated results from the Weber model (Weber et al., 2002), which predicted a predominantly inward INCX with a relatively short period of reverse mode activity during AP and the assumption that the NCX reversal point is within the first 10 ms of the AP.
Conclusions and perspectives
Our present data support the hypothesis that selective, partial NCX inhibition may, by restricting the Na+-induced [Ca2+]i elevation, have an anti-arrhythmic effect; this protective effect is mediated primarily by its inhibitory effect on revINCX. Therefore, NCX inhibition can be considered as a promising therapeutic strategy against Ca2+ overload-induced, revNCX-mediated cardiac arrhythmias.
Acknowledgments
We would like to thank Attila Farkas, MD, PhD, for his professional help in editing the figures. This work was supported by the Postdoctoral Programme of Hungarian Academy of Sciences (for N. N.); Richter Gedeon Talentum Foundation (for A. K.); and by the European Union and the State of Hungary, co-financed by the European Social Fund in the framework of TÁMOP 4.2.4. A/2-11-1-2012-0001 ‘National Excellence Program’. Grants were received from the Hungarian Scientific Research Fund (NK-104331 and NN-109904), the National Office for Research and Technology-Baross Programmes (REG-DA-09-2-2009-0115-NCXINHIB), the National Development Agency and co-financed by the European Regional Fund (TÁMOP-4.2.2A-11/1/KONV-2012-0073 and TÁMOP-4.2.2.A-11/1/KONV-2012-0060), the HU-RO Cross-Border Cooperation Programmes (HURO/1001/086/2.2.1 HURO-TWIN) and the Hungarian Academy of Sciences.
Glossary
- [Ca2+]i
intracellular calcium
- AP
action potential
- APD
action potential duration
- ATX-II
anemone toxin II
- CaT
intracellular calcium transient
- EAD
early afterdepolarization
- fwdNCX
forward transport mode of the sodium–calcium exchanger
- fwdINCX
forward mode NCX current
- INaL
late sodium current
- ORM10103
5-nitro-2-(2-phenylchroman-6-yloxy)pyridine
- revNCX
reverse transport mode of the sodium–calcium exchanger
- revINCX
reverse mode NCX current
- SEA0400
2-(4-((2,5-difluorophenyl)methoxy)phenoxy)-5-ethoxy-aniline
- TdP
Torsades de Pointes
Author contributions
N. N. was responsible for the concept of the study, arrangement of the experimental protocols, ion current (Figures 1A, 2A, 6A–B and 9A–B) and measurements of APs (Figures 1B, 2B, 3B, 5 and 8), data analysis and preparation of the manuscript and most figures. A. K. performed fluorescence and cell shortening measurements, and processed and analysed the data obtained from these measurements. Z. K. was responsible for the NCX current measurements with ORM10103 (Figure 3A). Á. S. contributed to all current measurements (except Figure 3A). J. S. contributed to current measurements for the revised manuscript (Figures 9 and 10). P. P. and J. L. were responsible for the pre-clinical characterization of ORM10103 and provided the compound. K. A. arranged the experimental protocols and analysed the data. L. V. analysed the data in Figure 3A. P. P. N. prepared and supervised the manuscript and figures. J. G. P. supervised the manuscript and figures. A. V. provided the grant support of the study, contributed to the concept, and supervised the manuscript and figures. A. T. contributed to the concept of the study, organized and controlled the experimental work, supported technical background, and prepared and supervised the manuscript and figures.
Conflict of interest
P. P. and J. L. are full-time employees at Orion Pharma.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14:Transporters. Br J Pharmacol. 2013;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amran MS, Hashimoto K, Homma N. Effects of sodium-calcium exchange inhibitors, KB-R7943 and SEA0400, on aconitine-induced arrhythmias in guinea pigs in vivoin vitro, and in computer simulation studies. J Pharmacol Exp Ther. 2004;310:83–89. doi: 10.1124/jpet.104.066951. [DOI] [PubMed] [Google Scholar]
- Antoons G, Willems R, Sipido KR. Alternative strategies in arrhythmia therapy: evaluation of Na/Ca exchange as an anti-arrhythmic target. Pharmacol Ther. 2012;134:26–42. doi: 10.1016/j.pharmthera.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL, O'Rourke B. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res. 2003;93:46–53. doi: 10.1161/01.RES.0000080932.98903.D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biliczki P, Virag L, Iost N, Papp JG, Varro A. Interaction of different potassium channels in cardiac repolarization in dog ventricular preparations: role of repolarization reserve. Br J Pharmacol. 2002;137:361–368. doi: 10.1038/sj.bjp.0704881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birinyi P, Acsai K, Banyasz T, Toth A, Horvath B, Virag L, et al. Effects of SEA0400 and KB-R7943 on Na+/Ca2+ exchange current and L-type Ca2+ current in canine ventricular cardiomyocytes. Naunyn Schmiedebergs Arch Pharmacol. 2005;372:63–70. doi: 10.1007/s00210-005-1079-x. [DOI] [PubMed] [Google Scholar]
- Birinyi P, Toth A, Jona I, Acsai K, Almassy J, Nagy N, et al. The Na+/Ca2+ exchange blocker SEA0400 fails to enhance cytosolic Ca2+ transient and contractility in canine ventricular cardiomyocytes. Cardiovasc Res. 2008;78:476–484. doi: 10.1093/cvr/cvn031. [DOI] [PubMed] [Google Scholar]
- Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na(+) concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Trafford AW, Diaz ME, Overend CL, O'Neill SC. The control of Ca release from the cardiac sarcoplasmic reticulum: regulation versus autoregulation. Cardiovasc Res. 1998;38:589–604. doi: 10.1016/s0008-6363(98)00062-5. [DOI] [PubMed] [Google Scholar]
- Elias CL, Lukas A, Shurraw S, Scott J, Omelchenko A, Gross GJ, et al. Inhibition of Na+/Ca2+ exchange by KB-R7943: transport mode selectivity and antiarrhythmic consequences. Am J Physiol Heart Circ Physiol. 2001;281:H1334–H1345. doi: 10.1152/ajpheart.2001.281.3.H1334. [DOI] [PubMed] [Google Scholar]
- Faber GM, Rudy Y. Action potential and contractility changes in [Na(+)](i) overloaded cardiac myocytes: a simulation study. Biophys J. 2000;78:2392–2404. doi: 10.1016/S0006-3495(00)76783-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas AS, Acsai K, Nagy N, Toth A, Fulop F, Seprenyi G, et al. Na(+)/Ca(2+) exchanger inhibition exerts a positive inotropic effect in the rat heart, but fails to influence the contractility of the rabbit heart. Br J Pharmacol. 2008;154:93–104. doi: 10.1038/bjp.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas AS, Makra P, Csik N, Orosz S, Shattock MJ, Fulop F, et al. The role of the Na+/Ca2+ exchanger, I(Na) and I(CaL) in the genesis of dofetilide-induced torsades de pointes in isolated, AV-blocked rabbit hearts. Br J Pharmacol. 2009;156:920–932. doi: 10.1111/j.1476-5381.2008.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng NC, Satoh H, Urushida T, Katoh H, Terada H, Watanabe Y, et al. A selective inhibitor of Na+/Ca2+ exchanger, SEA0400, preserves cardiac function and high-energy phosphates against ischemia/reperfusion injury. J Cardiovasc Pharmacol. 2006;47:263–270. doi: 10.1097/01.fjc.0000202561.69291.ac. [DOI] [PubMed] [Google Scholar]
- Grandi E, Pasqualini FS, Pes C, Corsi C, Zaza A, Severi S. Theoretical investigation of action potential duration dependence on extracellular Ca2+ in human cardiomyocytes. J Mol Cell Cardiol. 2009;46:332–342. doi: 10.1016/j.yjmcc.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Greenstein JL, Hinch R, Winslow RL. Mechanisms of excitation-contraction coupling in an integrative model of the cardiac ventricular myocyte. Biophys J. 2006;90:77–91. doi: 10.1529/biophysj.105.065169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobai IA, O'Rourke B. The potential of Na+/Ca2+ exchange blockers in the treatment of cardiac disease. Expert Opin Investig Drugs. 2004;13:653–664. doi: 10.1517/13543784.13.6.653. [DOI] [PubMed] [Google Scholar]
- Jost N, Nagy N, Kohajda Z, Horvath A, Corici C, Acsai K, et al. ORM-10103, a novel specific inhibitor of the sodium/calcium exchanger, decreases early and delayed afterdepolarization in the canine heart. Br J Pharmacol. 2013;170:768–778. doi: 10.1111/bph.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaerts I, Bito V, Heinzel FR, Driesen RB, Holemans P, D'Hooge J, et al. Ultrastructural and functional remodeling of the coupling between Ca2+ influx and sarcoplasmic reticulum Ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circ Res. 2009;105:876–885. doi: 10.1161/CIRCRESAHA.109.206276. [DOI] [PubMed] [Google Scholar]
- Li P, Rudy Y. A model of canine Purkinje cell electrophysiology and Ca(2+) cycling: rate dependence, triggered activity, and comparison to ventricular myocytes. Circ Res. 2011;109:71–79. doi: 10.1161/CIRCRESAHA.111.246512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ Res. 1994;74:1071–1096. doi: 10.1161/01.res.74.6.1071. [DOI] [PubMed] [Google Scholar]
- McDonald RL, Colyer J, Harrison SM. Quantitative analysis of Na+-Ca2+ exchanger expression in guinea-pig heart. Eur J Biochem. 2000;267:5142–5148. doi: 10.1046/j.1432-1327.2000.01579.x. [DOI] [PubMed] [Google Scholar]
- Milberg P, Pott C, Fink M, Frommeyer G, Matsuda T, Baba A, et al. Inhibition of the Na+/Ca2+ exchanger suppresses torsades de pointes in an intact heart model of long QT syndrome-2 and long QT syndrome-3. Heart Rhythm. 2008;5:1444–1452. doi: 10.1016/j.hrthm.2008.06.017. [DOI] [PubMed] [Google Scholar]
- Milberg P, Pott C, Frommeyer G, Fink M, Ruhe M, Matsuda T, et al. Acute inhibition of the Na(+)/Ca(2+) exchanger reduces proarrhythmia in an experimental model of chronic heart failure. Heart Rhythm. 2012;9:570–578. doi: 10.1016/j.hrthm.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Morita N, Lee JH, Bapat A, Fishbein MC, Mandel WJ, Chen PS, et al. Glycolytic inhibition causes spontaneous ventricular fibrillation in aged hearts. Am J Physiol Heart Circ Physiol. 2011;301:H180–H191. doi: 10.1152/ajpheart.00128.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai M, Terada H, Sugiyama S, Satoh H, Hayashi H. Effects of a selective inhibitor of Na+/Ca2+ exchange, KB-R7943, on reoxygenation-induced injuries in guinea pig papillary muscles. J Cardiovasc Pharmacol. 2000;35:121–128. doi: 10.1097/00005344-200001000-00016. [DOI] [PubMed] [Google Scholar]
- Nagy N, Acsai K, Kormos A, Sebok Z, Farkas AS, Jost N, et al. [Ca2+]i-induced augmentation of the inward rectifier potassium current (IK1) in canine and human ventricular myocardium. Pflugers Arch. 2013;465:1621–1635. doi: 10.1007/s00424-013-1309-x. [DOI] [PubMed] [Google Scholar]
- Nagy ZA, Virag L, Toth A, Biliczki P, Acsai K, Banyasz T, et al. Selective inhibition of sodium-calcium exchanger by SEA-0400 decreases early and delayed after depolarization in canine heart. Br J Pharmacol. 2004;143:827–831. doi: 10.1038/sj.bjp.0706026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekata I, Tsuneoka Y, Takahara A, Shimada H, Sugimoto T, Takeda K, et al. Involvement of the Na(+)/Ca(2+) exchanger in the automaticity of guinea-pig pulmonary vein myocardium as revealed by SEA0400. J Pharmacol Sci. 2009;110:111–116. doi: 10.1254/jphs.08159sc. [DOI] [PubMed] [Google Scholar]
- Noble D, Noble SJ, Bett GC, Earm YE, Ho WK, So IK. The role of sodium-calcium exchange during the cardiac action potential. Ann N Y Acad Sci. 1991;639:334–353. doi: 10.1111/j.1749-6632.1991.tb17323.x. [DOI] [PubMed] [Google Scholar]
- Noble D, Varghese A, Kohl P, Noble P. Improved guinea-pig ventricular cell model incorporating a diadic space, IKr and IKs, and length- and tension-dependent processes. Can J Cardiol. 1998;14:123–134. [PubMed] [Google Scholar]
- Nuyens D, Stengl M, Dugarmaa S, Rossenbacker T, Compernolle V, Rudy Y, et al. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med. 2001;7:1021–1027. doi: 10.1038/nm0901-1021. [DOI] [PubMed] [Google Scholar]
- Patton C, Thompson S, Epel D. Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium. 2004;35:427–431. doi: 10.1016/j.ceca.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42:D1098–D1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- Pueyo E, Husti Z, Hornyik T, Baczko I, Laguna P, Varro A, et al. Mechanisms of ventricular rate adaptation as a predictor of arrhythmic risk. Am J Physiol Heart Circ Physiol. 2010;298:H1577–H1587. doi: 10.1152/ajpheart.00936.2009. [DOI] [PubMed] [Google Scholar]
- Sarai N, Kobayashi T, Matsuoka S, Noma A. A simulation study to rescue the Na+/Ca2+ exchanger knockout mice. J Physiol Sci. 2006;56:211–217. doi: 10.2170/physiolsci.RP005006. [DOI] [PubMed] [Google Scholar]
- Satoh H, Ginsburg KS, Qing K, Terada H, Hayashi H, Bers DM. KB-R7943 block of Ca(2+) influx via Na(+)/Ca(2+) exchange does not alter twitches or glycoside inotropy but prevents Ca(2+) overload in rat ventricular myocytes. Circulation. 2000;101:1441–1446. doi: 10.1161/01.cir.101.12.1441. [DOI] [PubMed] [Google Scholar]
- Satoh H, Mukai M, Urushida T, Katoh H, Terada H, Hayashi H. Importance of Ca2+ influx by Na+/Ca2+ exchange under normal and sodium-loaded conditions in mammalian ventricles. Mol Cell Biochem. 2003;242:11–17. [PubMed] [Google Scholar]
- Schafer C, Ladilov Y, Inserte J, Schafer M, Haffner S, Garcia-Dorado D, et al. Role of the reverse mode of the Na+/Ca2+ exchanger in reoxygenation-induced cardiomyocyte injury. Cardiovasc Res. 2001;51:241–250. doi: 10.1016/s0008-6363(01)00282-6. [DOI] [PubMed] [Google Scholar]
- Shryock JC, Song Y, Rajamani S, Antzelevitch C, Belardinelli L. The arrhythmogenic consequences of increasing late INa in the cardiomyocyte. Cardiovasc Res. 2013;99:600–611. doi: 10.1093/cvr/cvt145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Takahashi T, Suzuki T, Onishi M, Tanaka Y, Hamano-Takahashi A, et al. Protective effects of SEA0400, a novel and selective inhibitor of the Na+/Ca2+ exchanger, on myocardial ischemia-reperfusion injuries. Eur J Pharmacol. 2003;458:155–162. doi: 10.1016/s0014-2999(02)02732-2. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Shimada H, Namekata I, Kawanishi T, Iida-Tanaka N, Shigenobu K. Involvement of the Na+/Ca2+ exchanger in ouabain-induced inotropy and arrhythmogenesis in guinea-pig myocardium as revealed by SEA0400. J Pharmacol Sci. 2007;103:241–246. doi: 10.1254/jphs.fp0060911. [DOI] [PubMed] [Google Scholar]
- Varro A, Baczko I. Cardiac ventricular repolarization reserve: a principle for understanding drug-related proarrhythmic risk. Br J Pharmacol. 2011;164:14–36. doi: 10.1111/j.1476-5381.2011.01367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdonck F, Mubagwa K, Sipido KR. Na(+)] in the subsarcolemmal ‘fuzzy’ space and modulation of [Ca(2+)](i) and contraction in cardiac myocytes. Cell Calcium. 2004;35:603–612. doi: 10.1016/j.ceca.2004.01.014. [DOI] [PubMed] [Google Scholar]
- Watano T, Harada Y, Harada K, Nishimura N. Effect of Na+/Ca2+ exchange inhibitor, KB-R7943 on ouabain-induced arrhythmias in guinea-pigs. Br J Pharmacol. 1999;127:1846–1850. doi: 10.1038/sj.bjp.0702740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CR, Piacentino V, 3rd, Ginsburg KS, Houser SR, Bers DM. Na(+)-Ca(2+) exchange current and submembrane [Ca(2+)] during the cardiac action potential. Circ Res. 2002;90:182–189. doi: 10.1161/hh0202.103940. [DOI] [PubMed] [Google Scholar]
- Weber CR, Piacentino V, 3rd, Houser SR, Bers DM. Dynamic regulation of sodium/calcium exchange function in human heart failure. Circulation. 2003;108:2224–2229. doi: 10.1161/01.CIR.0000095274.72486.94. [DOI] [PubMed] [Google Scholar]
- Wongcharoen W, Chen YC, Chen YJ, Chang CM, Yeh HI, Lin CI, et al. Effects of a Na+/Ca2+ exchanger inhibitor on pulmonary vein electrical activity and ouabain-induced arrhythmogenicity. Cardiovasc Res. 2006;70:497–508. doi: 10.1016/j.cardiores.2006.02.026. [DOI] [PubMed] [Google Scholar]
- Woodcock EA, Arthur JF, Harrison SN, Gao XM, Du XJ. Reperfusion-induced Ins(1,4,5)P(3) generation and arrhythmogenesis require activation of the Na(+)/Ca(2+) exchanger. J Mol Cell Cardiol. 2001;33:1861–1869. doi: 10.1006/jmcc.2001.1450. [DOI] [PubMed] [Google Scholar]
- Yamamura K, Tani M, Hasegawa H, Gen W. Very low dose of the Na(+)/Ca(2+) exchange inhibitor, KB-R7943, protects ischemic reperfused aged Fischer 344 rat hearts: considerable strain difference in the sensitivity to KB-R7943. Cardiovasc Res. 2001;52:397–406. doi: 10.1016/s0008-6363(01)00409-6. [DOI] [PubMed] [Google Scholar]