

Abstract
Background and Purpose
Anti-retrovirals have improved and extended the life expectancy of patients with HIV. However, as this population ages, the prevalence of cognitive changes is increasing. Aberrant activation of kinases, such as receptor tyrosine kinases (RTKs) and cyclin-dependent kinase 5 (CDK5), play a role in the mechanisms of HIV neurotoxicity. Inhibitors of CDK5, such as roscovitine, have neuroprotective effects; however, CNS penetration is low. Interestingly, tyrosine kinase inhibitors (TKIs) display some CDK inhibitory activity and ability to cross the blood–brain barrier.
Experimental Approach
We screened a small group of known TKIs for a candidate with additional CDK5 inhibitory activity and tested the efficacy of the candidate in in vitro and in vivo models of HIV-gp120 neurotoxicity.
Key Results
Among 12 different compounds, sunitinib inhibited CDK5 with an IC50 of 4.2 μM. In silico analysis revealed that, similarly to roscovitine, sunitinib fitted 6 of 10 features of the CDK5 pharmacophore. In a cell-based model, sunitinib reduced CDK5 phosphorylation (pCDK5), calpain-dependent p35/p25 conversion and protected neuronal cells from the toxic effects of gp120. In glial fibrillary acidic protein-gp120 transgenic (tg) mice, sunitinib reduced levels of pCDK5, p35/p25 and phosphorylated tau protein, along with amelioration of the neurodegenerative pathology.
Conclusions and Implications
Compounds such as sunitinib with dual kinase inhibitory activity could ameliorate the cognitive impairment associated with chronic HIV infection of the CNS. Moreover, repositioning existing low MW compounds holds promise for the treatment of patients with neurodegenerative disorders.
Tables of Links
| TARGETS |
|---|
| Catalytic receptorsa |
| VEGF receptor, VEGFR-2 |
| Enzymesb |
| CDKs |
| CDK5 |
| Transportersc |
| ABCB1 (P-glycoprotein) |
| ABCG2 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013a,b,c,,).
| LIGANDS |
|---|
| Dasatinib |
| Erlotinib |
| Lapatinib |
| PD-98059 |
| Roscovitine (seliciclib) |
Introduction
Modern antiretroviral therapy (ART) regimens can effectively suppress HIV viral load, and promote immune recovery with dramatic improvements in clinical symptoms and increased life expectancy (Chambers et al., 2014). For this reason, the ageing population represents one of the fastest growing groups with HIV (CDC, 2005, 2007; Scott et al., 2011), as it is estimated that by the year 2015, over half of people living in the United States with HIV will be over the age of 50 (CDC, 2005, 2007; Smith, 2005). In spite of the clinical efficacy of ART, some studies have shown that active or silenced HIV can persist in the CNS for long periods (Bell, 2004). The chronic presence of HIV in the brain might be associated with neurodegenerative changes that contribute to age-related cognitive changes (Diesing et al., 2002; Gonzalez-Scarano and Martin-Garcia, 2005; Crews et al., 2009). Remarkably, in aged patients with HIV, the prevalence of cognitive changes has increased and could represent an important morbidity factor in this patient population (Grant et al., 1995; Sacktor et al., 2002; McArthur et al., 2003; Chambers et al., 2014).
In patients with HIV, neurodegeneration is characterized by synaptic and dendritic damage (Masliah et al., 1992) accompanied by loss of selective neuronal populations and neuro-inflammatory pathology (Gonzalez-Scarano and Martin-Garcia, 2005; Kaul and Lipton, 2006; Lindl et al., 2010). The mechanisms by which secreted HIV proteins lead to neurodegeneration in patients with HIV are the subject of intense investigation (Ellis et al., 2007). Diverse pathways have been implicated in this process, including activation of apoptosis (Kaul et al., 2001; Garden et al., 2002), calcium dysregulation (Lipton and Rosenberg, 1994; Nath, 2002; Mattson et al., 2005), mitochondrial damage (Norman et al., 2008), autophagy dysfunction (Alirezaei et al., 2008; Zhou et al., 2011; Fields et al., 2013), oxidative stress (Ozdener, 2005), excitotoxicity (Lipton and Rosenberg, 1994) and neuro-inflammation. Moreover, abnormal kinase activation triggered by HIV products might also contribute to neuronal cell death. For example, HIV-associated alterations in JNK3, double-stranded RNA-activated PK (Alirezaei et al., 2007), glycogen synthase kinase-3β (GSK3β) (Maggirwar et al., 1999; Dewhurst et al., 2007; Crews et al., 2009), and receptor and non-receptor tyrosine kinases (RTKs) pathways have been reported. HIV-1 Tat binds to growth factor receptor-bound protein 2 (Grb2), an adaptor protein that contains two SH3 domains involved in RTK activity (Rom et al., 2011). Chronic HIV and SIV infection also dysregulate the RTKs recepteur d'orgine nantais (Cary et al., 2013), VEGF receptor (VEGFR; Rasheed et al., 2009) and PDGF signalling (Bethel-Brown et al., 2012).
Additionally, alterations in cyclin-dependent kinase 5 (CDK5) (Wang et al., 2007; Crews et al., 2009) have been linked to HIV-associated neurocognitive disorders (HAND). In neurodegenerative disorders such as Alzheimer's disease (AD), CDK5 hyperactivation is associated with elevated intracellular calcium that activates calpain-I, which in turn cleaves the CDK5 activator protein p35, generating the more potent product p25 (Lee et al., 2000). Activation of CDK5 by p25 results in aberrant phosphorylation of neuronal substrates (Ahlijanian et al., 2000) and neuronal cell death (Cicero and Herrup, 2005; Lopes et al., 2010). Recent studies have shown that in patients with HIV-associated cognitive alterations, as well as in transgenic (tg) models of HIV protein toxicity, CDK5 is hyperactivated, resulting in neurodegeneration with hyperphosphorylation of the transcription factor MEF2 and the microtubule-associated tau protein (Masliah et al., 2004; Wang et al., 2007; Patrick et al., 2011). Moreover, intracerebral infusion of the CDK5 inhibitor roscovitine ameliorated the behavioural and degenerative pathology in HIV-gp120 tg mice (Patrick et al., 2011). Although roscovitine has demonstrated some therapeutic potential as a neuroprotective compound in models of HAND and stroke (Menn et al., 2010), one issue about this compound is the limited bioavailability to the CNS when delivered systemically. Previous studies have shown that roscovitine concentrations in the brain range from 15 to 30% of that observed in plasma and this compound is eliminated rapidly from both blood and brain (Vita et al., 2005).
The objective of the present study was to investigate a small group of known tyrosine kinase inhibitors (TKIs) that can cross the blood–brain barrier (BBB), with the goal of identifying compounds with additional CDK5 blocking activity for testing in in vitro and in vivo models of HIV-gp120 neurotoxicity. TKIs have been shown to penetrate the BBB and some affect several kinases, including members of the CDK family (Zapata-Torres et al., 2004; Ling et al., 2007; Wang et al., 2010; Tang et al., 2013). Given that in HIV neurotoxicity, both RTKs and CDK5 are deregulated, therapeutic strategies using compounds with dual activity targeting both these pathways might be particularly advantageous. Applying molecular modelling methods coupled with validation in in vitro kinase assays, we found that sunitinib – an orally effective, low MW, multi-targeted TKI approved for the treatment of renal cell carcinoma (RCC) and gastrointestinal stromal tumours – also inhibited CDK5 with an IC50 similar to that of roscovitine. In cell-based assays and animal models, sunitinib was neuroprotective.
Methods
In vitro CDK5 activity assay
CDK5 kinase activity was determined with the substrate-binding ATP-based assay. A known CDK5 inhibitor, roscovitine, was included as a positive control. Compounds were added to a mixture containing kinase assay buffer (SignalChem, BC, Canada), 100 μM of CDC2 PK peptide substrate (Promega Corporation, Madison, WI, USA) and 125 ng of CDK5/p35 active kinase (SignalChem). Reactions were incubated for 10–20 min before ATP. Kinase-glo reagent (Promega Corporation) was then added and luminescence signals were read on the DTX880 Microplate reader (Beckman Coulter, Inc., Brea, CA, USA).
CDK5 molecular pocket analysis and pharmacophore development
Briefly, similarly to strategies used previously by Mapelli et al. (2005), we utilized the X-ray crystal structure of the human CDK5-p25/(R)-roscovitine complex (pdb ID 1UNL) to develop a pharmacophore based on the accessibility to small molecules. Using the program MOE2012.10 (Chemical Computing Group, Montreal, Canada), we generated a molecular surface that encompassed the binding pocket and then scrutinized all residues in the vicinity of this pocket that could make contacts. These residues that included Ile10, Val18, Lys33, Asp86, Lys89, Cys83, Gln130 and Asn144 were used for the development of the pharmacophore. For all selected interacting atoms of the pocket, we generated 10 pharmacophore centres, using MOE2012.10 Pharmacophore Editor. To fit the developed pharmacophore in this pocket, we applied restrictions for pharmacophore space, using excluded volume features that allowed a pharmacophore search of bound compounds. To investigate if the selected TKI binds similarly to the RTK and the CDK pocket we performed a molecular modelling study (ICM software, MolSoft LLC, San Diego, USA) for the compound in both the VEGF receptor 2 (VEGFR-2) (http://www.rcsb.org/pdb/explore/explore.do?pdbId=2xir) and CDK5 (http://www.rcsb.org/pdb/explore/explore.do?pdbId=1ung) using the crystal structure data from the protein databases. We also performed calculations of the binding parameters including the binding energy scores and related binding parameters.
Treatment of neuronal precursor cells (NPCs) with gp120 and sunitinib
For these experiments, adult rat hippocampal NPCs were selected because these cells display features similar to those observed in patients with HAND (Okamoto et al., 2007; Lee et al., 2011). The cells were grown at 37°C in humidified air with 5% CO2 in DMEM (Life Technologies, Carlsbad, CA, USA) containing 5% FBS and 1% penicillin/streptomycin. Cells were plated onto glass coverslips and after 2 h cells were treated for 24 h with gp120 [HIV-1SF162 gp120, National Institutes of Health (NIH) AIDS Reagent Program #7363] at a concentration of 200 μg·mL−1 combined with increasing concentrations (0.1, 1, 10 μM) of sunitinib (Calbiochem, San Diego, CA, USA) in a final volume of 1 mL. For immunoblot analysis, cells were washed with PBS and lysates were harvested with 80 μL of lysis buffer containing fresh protease and phosphatase inhibitors (EMD Millipore, Temecula, CA, USA).
Animals
All animal care and experimental procedures complied with NIH recommendations and were approved by the Institutional Animal Care and Use Committee at the University of California at San Diego. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 40 animals were used in the experiments described here.
Generation of glial fibrillary acidic protein (GFAP)-gp120 tg mice and sunitinib treatment
Tg mice expressing high levels of gp120 under the control of the GFAP promoter were used (Toggas et al., 1994); these were male C57/BL6 mice from our own animal facility. These mice develop neurodegeneration (Toggas et al., 1994), and show increased CDK5 activation. As previously described (Crews et al., 2010), 12-month-old non-tg and gp120 tg animals (a total of 40 mice; n = 10 per group) received daily i.p. injections with saline (vehicle) alone or sunitinib (Calbiochem) at a dose of 10 mg·kg−1 for 4 weeks. Exposure levels in blood and CNS were evaluated at nine time points in wild-type (C57BL6) mice (n = 27) that received a single i.v. injection of sunitinib at 10 mg·kg−1. Pharmacokinetics for sunitinib were determined by HPLC-MS (SAI-Life Sciences, Ltd., Hyderabad, India).
Tissue processing
Mice (C57/BL6 males, 12 months old, from our own animal facility) were anaesthetized with chloral hydrate (400 mg kg−1) and flush-perfused transcardially with 0.9% saline. Brains were removed and divided sagittally. One hemibrain was post-fixed in phosphate-buffered 4% paraformaldehyde at 4°C for 48 h and sectioned at 40 μm with a Vibratome 2000 (Leica, Buffalo Grove, IL, USA), while the other hemibrain was snap frozen and stored at −70°C for protein analysis.
Immunoblot analysis
Cell lysates or brain homogenates were loaded (20 μg total protein per lane), separated on 4–12% Bis-Tris NuPAGE gels (Life Technologies) and separated by electrophoresis in 5% running buffer. Gels were transferred onto Immobilon-P 0.45 μm membranes (Millipore, Billerica, MA, USA) using NuPAGE transfer buffer (Life Technologies). Membranes were incubated with rabbit polyclonal antibodies against CDK5 (1:500, C-8, Santa Cruz Biotechnology, Santa Cruz, CA, USA), phosphorylated CDK5 (Tyr15, 1:500, Santa Cruz Biotechnology) and p35/p25 (1:500, C-19, Santa Cruz Biotechnology). Blots were also probed with mouse monoclonal antibodies against total tau protein (1:1000, Sigma-Aldrich, Saint Louis, MO, USA), phosphorylated tau protein (p-tau), PHF1 and CP13 1:1000, generously provided by Dr Peter Davies, Albert Einstein College of Medicine), AT270 (pThr181, Thermo Fisher Scientific, Waltham, MA, USA), AT8 (pSer202 + Thr205, Thermo Fisher Scientific), β-III Tubulin (1:5000, clone Tuj1, Covance, Princeton, NJ, USA), or rabbit monoclonal antibody against total tau protein (1:1000, Dako, Carpinteria, CA, USA), collapsin response mediator protein-2 (CRMP2), and pCRMP2 (1:1000, Abcam, Cambridge, UK). Bands were visualized with enhanced chemiluminescence reagent (ECL, PerkinElmer, Waltham, MA, USA) and analysed with the VersaDoc gel imaging system and Quantity One software (Bio-Rad, Carpinteria, CA, USA). Actin was used as the loading control (1:1000, C4, EMD Millipore),
Immunocytochemical analysis and image analysis
Briefly, (Patrick et al., 2011) coverslips or vibratome were incubated with rabbit polyclonal antibodies against total (T) CDK5 (1:1000, C-8, Santa Cruz Biotechnology) or p35/p25 (1:1000, C-19, Santa Cruz Biotechnology), or mouse monoclonal antibodies against p-tau (PHF1, 1:1000, generously provided by Dr Peter Davies, Albert Einstein College of Medicine), or a rabbit monoclonal antibody against total tau protein (1:1000, Sigma-Aldrich). All sections were processed under the same standardized conditions. Immunostained sections were imaged with a digital Olympus microscope BX41 (Olympus, Center Valley, PA, USA) and assessment of CDK, p35 and tau protein immunoreactivity was carried out with the Image-Pro Plus program (Media Cybernetics, Silver Spring, MD, USA). For each case, a total of three sections (10 digital images per section at 400×) were analysed in order to estimate the average number of immunolabelled cells per unit area (mm2) and the average intensity of the immunostaining (corrected optical density).
Analysis of neurodegeneration
Briefly, blind-coded, 40 μm thick vibratome sections were immunolabelled with the mouse monoclonal antibody against microtubule-associated protein-2 (MAP2, dendritic marker, 1:200, EMD Millipore) and the antibody against activated caspase-3 (Cell Signaling Technology, Inc., Beverly, MA, USA, 1:500). After overnight incubation, sections were treated with FITC-conjugated secondary antibodies (1:75, Vector Laboratories, Burlingame, CA) or Tyramide Red, (NEN Life Sciences, Boston, MA, USA) transferred to SuperFrost slides (Thermo Fisher Scientific), and mounted under glass coverslips with anti-fading media (Vector Laboratories). All sections were processed under the same standardized conditions. The immunolabelled blind-coded sections were serially imaged with a laser scanning confocal microscope (MRC1024, Bio-Rad) and analysed with the ImageJ program (NIH), as previously described (Toggas et al., 1994; Mucke et al., 1995). For each mouse, a total of three sections were analysed, and for each section, four fields in the frontal cortex and hippocampus were examined. Results were expressed as percentage of area of the neuropil occupied. An additional set of sections was immunostained with the mouse monoclonal antibodies against NeuN (neuronal marker, 1:500, EMD Millipore), GFAP (astroglial marker, 1:500, EMD Millipore) and Iba-1 (microglial marker, 1:5000, Wako Chemicals USA, Inc., Richmond, VA, USA). The GFAP- and NeuN-immunostained sections were imaged with a digital Olympus microscope and the Image-Pro Plus program (Media Cybernetics).
For detection of apoptosis the TUNEL detection method using the ApopTag In situ Apoptosis Detection Kit (Chemicon, Temecula, CA, USA) was used with modifications for free-floating sections as described previously (Biebl et al., 2000; 2005,; Cooper-Kuhn and Kuhn, 2002). Detection was performed with DAB and mounted under glass coverslips (Vector Laboratories) for digital brightfield microscopy analysis.
Double immunolabelling and confocal microscopy imaging
Double-immunocytochemical analysis was performed utilizing the Tyramide Signal Amplification™-Direct (Red) system (NEN Life Sciences) to detect CDK5. Specificity was tested by deleting each primary antibody. For this purpose, sections were double-labelled with the rabbit antibodies against CDK5 (1:20 000, EMD Millipore) detected with Tyramide Red, and p-tau (PHF-1) detected with FITC-conjugated secondary antibodies (1:75, Vector Laboratories). All sections were processed simultaneously under the same conditions, and experiments were performed twice for reproducibility. Sections were imaged on a laser scanning confocal microscope Bio-Rad Radiance 2000 (Hercules, CA) equipped with a Nikon E600FN Ellipse microscope (Tokyo, Japan) and using a Nikon Plan Apo 60× oil objective (NA 1.4; oil immersion).
Statistical analysis
All results are expressed as means ± SEM. All analyses were conducted on blind-coded samples. After the results were obtained, the code was broken and data were analysed with Graphpad Prism (San Diego, CA, USA). Comparisons among groups were performed with unpaired Student's t-test and chi-squared analysis or one-way anova with Dunnett's or Tukey-Kramer post hoc tests, as appropriate.
Materials
All 12 compounds tested for activity against CDK5 are listed in Table 1. Their sources were as follows: compounds 1,2,3 from WUXI, Shanghai, China; compounds 8,9 and 10 from Targegen, San Diego, California, USA; compounds 4,5,6,7,12 and 13 from Selleck Chemicals Houston TX, USA. Roscovitine was supplied by LC Laboratories, Woburn, MA, USA.
Table 1.
Kinase inhibitors tested for CDK5 activity
| Structure | Name | Target | Clinical use | MW (<400) | clogP (2-4) | cPSA (<100) | clogBB (>−0.5) | |
|---|---|---|---|---|---|---|---|---|
| 1 |  |
C07081504 | Various RTKs | 357.14 | 4.12 | 52.10 | −0.01 | |
| 2 | 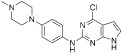 |
C07081505 | Various RTKs | 342.14 | 3.27 | 47.53 | −0.07 | |
| 3 |  |
C07081506 | Various RTKs | 338.10 | 3.31 | 52.89 | −0.14 | |
| 4 |  |
Lapatinib | VEGFR/Her2/neu | Breast cancer and solid tumours | 580.13 | 4.98 | 85.09 | −0.36 |
| 5 |  |
Dasatinib | c-Abl, c-kit, src | Prostate cancer | 487.16 | 3.18 | 83.53 | −0.61 |
| 6 | 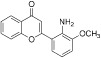 |
PD 98059 | MEK | 267.09 | 3.36 | 48.80 | −0.07 | |
| 7 |  |
Erlotinib | EGFR, v-src, v-abl | NSCLC, pancreatic cancer | 393.17 | 3.61 | 61.01 | −0.22 |
| 8 |  |
TG100572 | VEGFR2/, Src | 475.18 | 5.89 | 71.30 | −0.02 | |
| 9 | 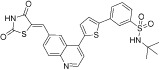 |
TG101910-A-2 | Src | 549.09 | 6.82 | 88.21 | −0.13 | |
| 10 |  |
TG-101525-A-2 | Various RTKs | 313.19 | 2.99 | 61.57 | −0.32 | |
| 11 |  |
TG100948 | Aurora A | 624.19 | 6.65 | 87.58 | −0.15 | |
| 12 |  |
Roscovitine | PLK/CDK | NSCLC | 353.23 | 2.46 | 75.77 | −0.61 |
| 13 |  |
Sunitinib | PDGF/VEGF | Renal cell carcinoma | 398.21 | 3.23 | 64.62 | −0.33 |
The values in parentheses are those preferred for CNS uptake.
logBB, blood–brain barrier permeation; logP, partition coefficient; logPS, permeation of neutral compounds from saline; MEK, MAPK kinase; MW, molecular weight; NSCLC, non-small cell lung carcinoma; PLK, serine/threonine protein kinase; Src, Rous sarcoma oncogene.
Results
CDK5 inhibitory activity of sunitinib in a cell-free in vitro assay
ATP-mimetic TKIs have the potential to interact with the active ATP-binding site in other kinases. HIV-associated neurodegeneration is associated with aberrant signalling that includes the RTK and CDK5, thus compounds with dual activity might be useful as protection from HIV proteins toxicity. For this reason, we investigated potential activity of a small group of 12 RTK inhibitors for additional CDK5 inhibition in an in vitro cell-free kinase assay. To test known TKIs for activity against CDK5, a robust luminescence-based in vitro kinase assay was implemented using a CDC2 PK peptide as a substrate, and roscovitine was used as the positive control. Consistent with previous reports, roscovitine inhibited CDK5 with an IC50 of 1.6 μM (Figure 1A). Of the TKI compounds tested, which included anti-cancer TKIs lapatinib, dasatinib and erlotinib, only sunitinib inhibited CDK5 with an IC50 of <10 μM, at 4.2 μM (Figure 1B). In contrast, the other compounds showed only weak or no significant activity against CDK5 in the cell-free kinase assay (Figure 1C–H). As only sunitinib exhibited a favourable dose–response curve against CDK5 similar to roscovitine, we selected this compound for further study in cell-based and in vivo models of HIV neurotoxicity.
Figure 1.
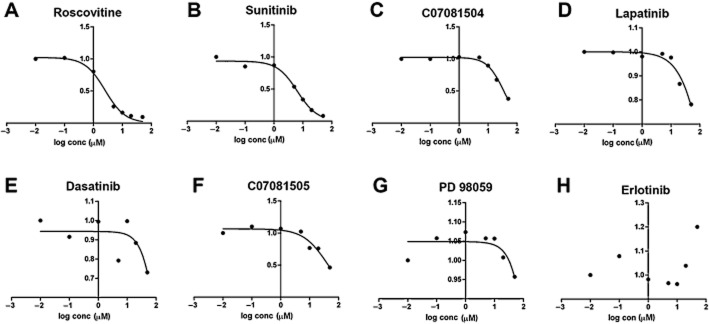
In vitro CDK5 activity assay with selected BBB-permeable RTK inhibitors. The activity of each compound was determined in a CDK5 ATP-based assay containing CDC2 PK peptide as substrate. (A–H) Roscovitine was used as a positive control showing an IC50 = 1.6 μM. Of the RTK inhibitors, only sunitinib showed CDK5 inhibitory activitywith IC50 = 4.2 μM; six other examples of RTK inhibitors did not show activity on CDK5 below 10 μM.
In silico studies of compounds fitting into the CDK5 pharmacophore
To confirm that compared with other TKIs, sunitinib can potentially interact with the CDK5 pocket, in silico molecular modelling and docking studies were performed. For this purpose, we generated a molecular surface model for the CDK5 ATP-binding site using the MOE2012.10 program. The pharmacophore was based on the residues of this pocket accessible to roscovitine (Figure 2A). The centres of the developed pharmacophore were placed on the circumferences of the circled pharmacophore pocket centres with the radii proportional to the open volume from a centre to the pocket wall. The F1 acceptor centre contacts the backbone nitrogen of Cys83. The residues Ile10 and Val18 were selected to contact three possible hydrophobic centres: F4, F5 and F10. Residue Lys33 contacts acceptor centre F6. Residue Asp86 contacts the donor centres F2 and F8. F6 donor/acceptor centre can contact Lys33 or Asn144. Residue Lys89 contacts the acceptor/donor centre F7, and the acceptor/aromatic centre F9. The Cys83 backbone oxygen contacts the donor centre F3 and backbone nitrogen the acceptor centre F1. The Gln130 backbone oxygen contacts the donor centre F2. After defining the three-dimensional positions of the key residues, we tested the preferred contact surfaces of roscovitine and sunitinib, taking in consideration the intra-pocket distribution of electrostatic field and hydrophobic surfaces along with the geometrical restriction of the pocket. A total of 10 possible pharmacophore centres in the pocket were identified (Figure 2B, C); however, given that it is difficult for a single compound to contact all 10, a partial pharmacophore with six out of the 10 contact centres was generated. Roscovitine (Figure 2D) and sunitinib (Figure 2E) were the only compounds that fit the six out of 10 contact centres in the partial pharmacophore. All other compounds fit five or fewer of 10 contact centres in the partial pharmacophores. When superimposed onto the pharmacophore, roscovitine and sunitinib displayed a similar fitting in the molecular pocket of CDK5 (Figure 2F). Both molecules fit four common pharmacophore centres: F9 acceptor/aromatic, F7 acceptor/donor centre, and F4, F5 hydrophobic centres. The additional two centres contacted are different for these compounds. Roscovitine fits F3 donor centre and F1 acceptor centre, sunitinib contacts F2 donor centre and the F10 hydrophobic centre.
Figure 2.

In silico comparison of the docking of roscovitine and sunitinib on a pharmacophore model for the ATP-binding pocket of CDK5. (A) The X-ray crystal structure of the human CDK5-p25/(R)-roscovitine complex (pdb ID 1UNL) was used to generate a pharmacophore-based model. (B, C) Representation of the CDK5 pharmacophore molecular surface generated using the program MOE2012.10 (Chemical Computing Group) showing the 10 centre features, with and without a mask. (D–F) Docking of roscovitine, sunitinib and both compounds in the 10 feature pharamacophore showing similar binding.
Next we compared the docking of sunitinib into both VEGFR-2 and CDK5 pockets. The results showed that sunitinib binds to both the RTK and CDK proteins in the ATP pocket (catalytic domain) in their hinge region below the β sheet scaffold and the C-helix, in a very similar mode with relative binding energies of −31 and −26 kcal·mol−1 respectively (Figure 3A, B).
Figure 3.

In silico comparison of sunitinib binding to the CDK5 and VEGFR-2 pharmacophores. Molecular modelling for CDK5 and VEGFR-2 was performed with Molsoft, ICM software using the crystal structure data from the protein databases. (A) Ribbon model structure of sunitinib docking to the CDK5 and (B) VEGFR-2 catalytic domain in their hinge region below the β sheet scaffold and the C-helix. Calculations of the binding parameters showed similar results for sunitinib binding to CDK5 and VEGFR-2.
Sunitinib treatment reduces CDK5 phosphorylation and protects neuronal cells from gp120-mediated neurite injury
In addition to RTK inhibition, sunitinib displayed CDK5 inhibitory activity comparable with roscovitine in cell-free kinase assays, cell-based assays were performed to determine the capacity of this compound to protect NPCs from the toxic effects of gp120. In these neural lineage cells, gp120 toxicity is manifested as a reduction in the length of neurites immunostained with the MAP2 antibody (Figure 4A, B). Sunitinib treatment protected neuronal progeny from gp120 toxicity in a dose-dependent manner (Figure 4B). Gp120 treatment alone increased levels of pCDK5 immunoreactivity, and this effect was reversed by sunitinib treatment in a dose-dependent manner (Figure 4C). To confirm the immunocytochemical results by an independent method, Western blot analysis was performed. Neuronal cells treated with gp120 exhibited increased pCDK5 and p35 fragmentation to p25 (Figure 4D–F), consistent with aberrant activation of CDK5 observed in the brains of patients with HIV-associated neurodegeneration (Patrick et al., 2011). Treatment with sunitinib reduced pCDK5 expression and p25 generation to levels comparable with vehicle-treated controls (Figure 4D–F).
Figure 4.

In vitro neuroprotective effects of sunitinib in a neuronal cell model challenged with HIV1-gp120. Neuronal cells derived from adult rat hippocampal NPCs were treated with increasing concentrations (0.1, 1, 10 μM) of sunitinib followed by gp120 (HIV-1SF162 gp120) at a 200 μg·mL−1. (A) Immunocytochemical analysis with an antibody against the neurite marker MAP2 and pCDK5. In saline-treated cells, there were abundant and elongated neuritic processes (arrowheads). NPCs challenged with gp120 displayed reduced MAP2 and increased pCDK5 immunostaining. (B) Image analysis for neurite length and (C) pCDK5 immunoreactivity showing that sunitinib protects NPCs against gp120 and reduces pCDK5 levels. (D) Western blot analysis with antibodies against T-CDK5, pCDK5 and p35. NPCs challenged with gp120 display increased pCDK5 and p25 immunoreactive bands. (E) Image analysis for neurite length and (F) pCDK5 immunoreactivity showing that sunitinib protects NPCs against gp120 and reduces pCDK5 levels. Bar = 10 μm. *P < 0.05, significantly different from vehicle controls, one-way ANOVA with Dunnett's test; # P < 0.05, significantly different from gp120 alone, one way ANOVA with Tukey-Kramer test.
Sunitinib treatment reduces CDK5 hyperactivation and tau protein phosphorylation in gp120 tg mice
Next we investigated whether treatment with sunitinib could reverse the neuropathology in gp120 tg mice, a model that reproduces some neurodegenerative and behavioural changes characteristic of patients with HIV neuropathology. For this purpose, non-tg and gp120 tg mice (12 months old, n = 10 per group) were treated with vehicle alone or sunitinib (10 mg·kg−1) for 4 weeks. Pharmacokinetic analysis showed that, at this dose, sunitinib crossed the BBB into the CNS at high levels and with a favourable brain to plasma ratio (B/P area under the curve = 1.96). The t1/2 in brain was 4.57 h and in plasma 1.16 h and the plasma stability after 1 h was 90%.
Immunoblot analysis demonstrated that compared with vehicle-treated non-tg mice, gp120 tg mice showed increased CDK5 phosphorylation and p35 cleavage to p25 (Figure 5A, B). Treatment of gp120 tg mice with sunitinib reduced levels of pCDK5 and p25 to those comparable with non-tg controls (Figure 5A, B). We have previously shown that in HIV patients as well as in the gp120 tg mice there is increased phosphorylation of CDK5 substrates such as CRMP2 and tau protein (Crews et al., 2011; Patrick et al., 2011). Immunoblot results confirmed increased levels of pCRMP2 in the brains of vehicle-treated gp120 mice, while gp120 tg mice treated with sunitinib displayed pCRMP2 levels comparable with non-tg controls (Figure 5A, C). Next we investigated the levels of p-tau with antibodies that detect epitopes phosphorylated by CDK5 (Figure 6). Levels of total tau protein were similar between non-tg and gp120 tg groups (Figure 6A). In contrast, vehicle-treated gp120 tg mice displayed increased tau protein phosphorylation with the AT270, CP13, AT8 and PHF-1 antibodies (Figure 6A–E). Treatment with sunitinib normalized the levels of tau protein phosphorylation in the gp120 tg mice (Figure 6A–E).
Figure 5.

Immunoblot analysis of sunitinib's effects on CDK5 in gp120 tg mice. Non-tg and gp120 tg mice were treated with vehicle or sunitinib for 4 weeks and analysed by immunoblot. (A) Representative Western blot reacted with antibodies against T-CDK5, pCDK5, p35/25, pCRMP2, Tuj-1 and actin, as a loading control. (B, C) Image-Quant analysis of the ratio of pCDK5/T-CDK5, p25/35, pCRMP2 and Tuj1. In gp120 tg mice, levels of pCDK5/T-CDK5, p25/35 and pCRMP2 were higher than in non-tg mice. *P < 0.05; one-way anova with Dunnett's test. Sunitinib treatment resulted in a reduction of these levels compared with vehicle controls. #P < 0.05; one-way anova with Tukey–Kramer test. Mice were 12 months old, n = 10 per group.
Figure 6.

Immunoblot analysis of sunitinib's effects on tau protein in gp120 tg mice. Non-tg and gp120 tg mice were treated with vehicle or sunitinib for 4 weeks and analysed by immunoblot. (A) Representative Western blot treated with antibodies against total tau protein, p-tau (AT270, CP13, AT8 and PHF1) and actin, as a loading control. (B–E) Image-Quant analysis of AT270, CP13, AT8 and PHF1. In gp120 tg mice levels of p-tau were higher than in non-tg mice. *P < 0.05; one-way anova with Dunnett's test. Sunitinib treatment resulted in a reduction of p-tau levels compared with vehicle controls. #P < 0.05; one-way anova with Tukey–Kramer test. Mice were 12 months old, n = 10 per group.
Immunocytochemical analysis was performed to confirm these results by an independent method and to investigate the cellular patterns of CDK5 and tau protein phosphorylation in gp120 tg mice treated with sunitinib (Figure 7A). Compared with the non-tg vehicle-treated and sunitinib-treated mice, the vehicle-treated gp120 tg mice showed increased pCDK5 and p-tau immunoreactivity in pyramidal cells in the neocortex and hippocampus (Figure 7A–C). Consistent with the immunoblot results, treatment with sunitinib reduced pCDK5 and p-tau immunostaining in the gp120 tg mice to levels comparable to the non-tg controls (Figure 7A–C). In agreement with the immunoperoxidase results, double-labelling and confocal microscopy analysis showed that compared with non-tg mice, in vehicle-treated gp120 tg mice, there was greater co-localization of pCDK5 with p-tau immunoreactive neurones (Figure 7D). Treatment with sunitinib reduced co-labelling of pCDK5 and p-tau in the gp120 tg mice to levels equivalent to the non-tg controls (Figure 7D).
Figure 7.

Immunocytochemical analysis of sunitinib's effects on pCDK5 and p-tau in gp120 tg mice. Non-tg and gp120 tg mice were treated with vehicle or sunitinib for 4 weeks and immunostained serial vibratome sections were analysed by digital brightfield or confocal microscopy. (A) Representative brightfield microscopic images of sections incubated with antibodies against pCDK5 and p-tau (PHF1) showing increased immunoreactivity in cortical neurones in vehicle-treated gp120 tg mice. Outlined images to the right of each low-power micrograph are higher magnifications of the inset. (B, C) Computer-aided ImageJ analysis of pCDK5 and PHF1. In vehicle-treated gp120 tg mice, levels of p-tau were higher than in non-tg mice. *P < 0.05; one-way anova with Dunnett's test. Sunitinib treatment resulted in a reduction of pCDK5 and p-tau levels compared with vehicle controls. #P < 0.05; one-way anova with Tukey–Kramer test.. Mice were 12 months old, n = 10 per group. (D) Immunocytochemical analysis and confocal microscopy of sections double-labelled with antibodies against PHF1 (FITC channel) and pCDK5 (red channel). Co-localization (arrows) was observed in sections from vehicle-treated gp120 tg mice compared with controls or sunitinib treatment. Mice were 12 months old, n = 10 per group. Scale bar = 10 μm.
Sunitinib treatment ameliorates the neurodegenerative pathology in gp120 tg mice
Next we analysed the effects of sunitinib on the neuro-inflammatory and neurodegenerative pathology in gp120 tg mice. Compared with non-tg controls, vehicle-treated gp120 tg mice displayed increased microgliosis (Iba1) (Figure 8A, B) and astrogliosis (GFAP) (Figure 8C, D) in the neocortex and hippocampus. Sunitinib treatment partially reduced the levels of microgliosis in the neocortex and hippocampus (Figure 8A, B), and to some extent also decreased the levels of astrocytosis (Figure 8C, D). As previously described, compared with non-tg controls, vehicle-treated gp120 tg mice displayed reduced neuronal density as shown by NeuN staining (Figure 9A, B) and levels of MAP2-immunoreactive dendrites in the neocortex (Figure 9C, D). Sunitinib-treated gp120 tg mice displayed levels of NeuN (Figure 9A, B) and MAP2 similar to saline-treated non-tg controls (Figure 9C, D). Next, sections were analysed with an antibody against active caspase-3. Compared with non-tg controls, vehicle-treated gp120 tg mice displayed increased immunostaining in the neocortex and hippocampus (Figure 9E, F). Sunitinib-treated gp120 tg mice displayed levels of active caspase-3 immunostaining (Figure 9E, F) similar to saline-treated non-tg controls (Figure 9E, F). Additional analysis of neurodegeneration was performed with the TUNEL assay. The vehicle-treated gp120 tg mice displayed increased numbers of TUNEL-positive cells in the neocortex and hippocampus (Figure 9G, H) compared with non-tg controls. In contrast, sunitinib-treated gp120 tg mice displayed low levels of TUNEL (Figure 9G, H) comparable with saline-treated non-tg controls (Figure 9G, H).
Figure 8.

Immunocytochemical analysis of the effects of sunitinib in markers of neuro-inflammation in gp120 tg mice. Non-tg and gp120 tg mice were treated with vehicle or sunitinib for 4 weeks and immunostained serial vibratome sections were analysed by digital brightfield microscopy. (A) Representative brightfield microscopic images of sections incubated with an antibody against the microglial marker Iba1 showing increased immunoreactivity in the neocortex and hippocampus in vehicle-treated gp120 tg mice. Outlined images below each low-power micrograph are higher magnifications of the inset in the neocortex. (B) Computer-aided ImageJ analysis of Iba1. In vehicle-treated gp120 tg mice levels of Iba1 were higher than in non-tg mice. *P < 0.05; one-way anova with Dunnett's test. Sunitinib treatment resulted in a reduction of Iba1 levels compared with vehicle controls. #P < 0.05; one-way anova with Tukey–Kramer test. (C) Representative brightfield microscopic images of sections incubated with an antibody against the astroglial marker GFAP showing increased immunoreactivity in the neocortex and hippocampus in vehicle-treated gp120 tg mice. Outlined images below each low-power micrograph are higher magnifications of the inset in the neocortex. (D) Computer-aided ImageJ analysis of Iba1. In vehicle-treated gp120 tg mice levels of GFAP were higher than in non-tg mice. *P < 0.05; one-way anova with Dunnett's test. Sunitinib treatment resulted in a reduction of GFAP levels compared with vehicle controls. #P < 0.05; one-way anova with Tukey–Kramer test. Mice were 12 months old, n = 10 per group. Bar = 250 and 10 μm.
Figure 9.

Effects of sunitinib on markers of neurodegeneration in gp120 tg mice. Non-tg and gp120 tg mice were treated with vehicle or sunitinib for 4 weeks and immunostained serial vibratome sections were analysed by digital brightfield microscopy and confocal microscopy. (A) Representative brightfield microscopic images of sections incubated with an antibody against the neuronal marker NeuN showing decreased immunoreactivity in the neocortex in vehicle-treated gp120 tg mice. Outlined images below each low-power micrograph are higher magnifications of the inset in the neocortex. (B) Computer-aided Image-J analysis of the numbers of positive NeuN cells. In vehicle-treated gp120 tg mice, there were fewer than in non-tg mice. *P < 0.05; one-way anova with Dunnett's test. Sunitinib treatment ameliorated the neuronal changes, compared with vehicle-treated gp120 tg mice. #P < 0.05; one-way anova with Tukey–Kramer test. (C) Immunocytochemical analysis and confocal microscopy of sections labelled with an antibody against the dendritic marker MAP2 (FITC channel). (D) Computer-aided ImageJ analysis of the percentage of area of the neuropil that is MAP2 immunostained. In vehicle-treated gp120 tg mice there was less MAP2 immunoreactivity than in non-tg mice. *P < 0.05; one-way anova with Dunnett's test. Sunitinib treatment ameliorated the dendritic alterations compared with vehicle-treated gp120 tg mice. #P < 0.05; one-way anova with Tukey–Kramer test. Bar = 250 and 10 μm. (E) Representative confocal microscopy images of sections incubated with an antibody against active caspase-3 showing increased immunoreactivity in the neocortex in vehicle-treated gp120 tg mice. (F) Computer-aided Image-J analysis of pixel intensity. In vehicle-treated gp120 tg mice, there was more capsase-3 staining than in non-tg mice. *P < 0.05; one-way anova with Dunnett's test. Sunitinib treatment reduced caspase-3 in gp120 tg mice. #P < 0.05; one-way anova with Tukey–Kramer test. (G) Representative digital microscopy images of sections stained with the TUNEL assay showing increased positive cells in the neocortex in vehicle-treated gp120 tg mice. (H) Computer-aided Image-J analysis of positive cells. In vehicle-treated gp120 tg mice there was more staining than in non-tg mice. *P < 0.05; one-way anova with Dunnett's test. Sunitinib treatment reduced TUNEL staining in gp120 tg mice. #P < 0.05; one-way anova with Tukey–Kramer test. Mice were 12 months old, n = 10 per group.
Taken together, these results support the notion that sunitinib might exert neuroprotective effects in models of HIV gp120 toxicity, and that the dual ability of this compound at blocking RTK and CDK5 signalling might contribute to the therapeutic effects.
Discussion and conclusions
Sunitinib is an orally effective RTK inhibitor utilized for the treatment of RCC that targets both angiogenic pathways (i.e. those from VEGF or PDGF receptors) and direct pro-oncogenic pathways, such as stem cell factor receptor and feline McDonough sarcoma-like tyrosine kinase-3. The present study showed that in addition to the effects on RTKs, sunitinib is also capable of blocking CDK5, at levels comparable with those of roscovitine. In cellular models of HIV-gp120 toxicity, sunitinib reduced CDK5 activation and tau protein hyperphosphorylation, and promoted neurite outgrowth. Likewise, in GFAP-gp120 tg mice, sunitinib reduced CDK5 and tau protein hyperphosphorylation and ameliorated the neuro-inflammatory and neurodegenerative pathology.
A common consequence of targeted cancer therapeutics in rapidly dividing bulk tumour cells is the ability of a compound to arrest cell division, usually leading to either differentiation or apoptosis. Interestingly, in the case of neurodegenerative pathways associated with CDK5 activation, there is often transient reactivation of cell cycle machinery directly preceding neuronal cell death (Cicero and Herrup, 2005). Therefore, anti-cancer compounds that modulate the cell cycle may be active even in typically non-dividing cells of the brain, and furthermore, the differentiation-promoting effects may support production of mature neuronal progeny from endogenous regions of neurogenesis.
The dual effects of sunitinib at inhibiting RTKs and CDK5 is consistent with previous studies that have shown that other RTK inhibitors such as erlotinib that primarily targets VEGF receptors can also modulate CDK2/4 (Ling et al., 2007) and macrocyclic aminopyrimidines that block VEGF receptors can also inhibit CDK2 (Lucking et al., 2007). Earlier studies had indicated that sunitinib exhibited some inhibitory activity against CDK1 (IC50 = 2.6 μM) (Mendel et al., 2003), and a more recent study showed that sunitinib binds to CDK2 through hydrogen bonding interactions with the hinge region of the ATP site, namely the backbone of residues Glu81 and Leu83 (Martin et al., 2012). This is consistent with studies showing that sunitinib treatment causes cell cycle arrest in the G0/G1 phase by increasing p27Kip1, pRb1 and p130/Rb2, and decreasing the expression of cyclin D1, cyclin D3, and CDK2 (Teng et al., 2013). In the CNS, while increased p27 promotes cell survival and is neuroprotective, decreasing p27 reduces neuronal cell viability. Therefore, the neuroprotective effects of sunitinib might involve several mechanisms including direct binding to the catalytic pocket and down-regulation of hyperactive RTKs and CDK5 and increased expression of p27 that can also regulate CDK5. Moreover, another intriguing possibility is that sunitinib's effects on CDK5 might be indirect via regulation of the phosphorylation of CDK5 at Tyr15. Previous studies have shown that phosphorylation at this residue might regulate the state of CDK5 activation In this regard, we found that in the gp120 model of toxicity phosphorylation at Tyr15 of CDK5 is increased, while sunitinib treatment reduced Tyr15 phosphorylation of CDK5 to baseline levels.
Abnormal activation of the CDK5 (Wang et al., 2007; Crews et al., 2009), GSK3β (Maggirwar et al., 1999; Dewhurst et al., 2007; Crews et al., 2009; Schifitto et al., 2009)and RTKs such as insulin-like receptor (Gerena et al., 2012), ephrin receptors (Yuferov et al., 2013), STAT3 (Peng et al., 2011), PDGF (Potula et al., 2004; Eggert et al., 2009) and insulin-like growth factor (Ying Wang et al., 2003) signalling pathways have been implicated in HIV-associated neurocognitive impairment and in gp120 tg models of HIV-1 neurotoxicity. As discussed earlier, all of these are pathways that are potentially affected by sunitinib and might explain the mechanisms of neuroprotection in models of gp120 toxicity. Moreover, the results of the current study are in agreement with previous studies showing that roscovitine, a CDK inhibitor that inhibits CDK5 (as well as some other CDKs) in the low micromolar range, protected primary neurones from the neurotoxic effects of HIV (Wang et al., 2007), and ameliorated the neurotoxic and neurobehavioural alteration of the HIV-1 protein gp120 in tg animals (Patrick et al., 2011). Likewise, lithium chloride, which blocks GSK3β activity, and to some extent CDK5 hyperactivation, also reduces the behavioural deficits in patients with HIV encephalitis (HIVE) and in gp120 tg mice (Everall et al., 2002). Therefore, compounds that block CDK5 hyperactivation such as roscovitine are of potential importance in the management of neurodegenerative disorders; for this reason, we investigated the potential of sunitinib. However, it is worth mentioning that novel CDK5 inhibitors with greater selectivity and bioavailability are currently being developed. For example, a series of new purine-based fluoroaryl triazoles were developed and shown to be neuroprotective against Aβ-induced neurotoxicity. Roscovitine and the triazole 7 bind to the ATP-binding site of CDK5/p25 with comparable binding energies (Nair et al., 2011).
As previous studies have shown that in patients with HIVE, the abnormal activation of CDK5 and RTKs might be associated with behavioural deficits and neurotoxicity, we postulate that sunitinib's neuroprotective effects in HIV models might be related to the dual effects targeting the CDK5 and RTK signalling pathways. For this study, we focused on the effects of sunitinib via CDK5. However, future studies will need to evaluate the effects of this compound at modulating RTKs and other signalling pathways that are dysregulated during the progression of HIV neurotoxicity.
In addition to the dual effects on RTKs and CDK5, inhibition with sunitinib may have better pharmacokinetic characteristics than compounds such as roscovitine, which exhibits a brain/plasma (B/P) concentration ratio of 0.15–0.30. (Vita et al., 2005). Our results demonstrate that the B/P ratio for sunitinib was 1.96 with a favourable pharmacological half-life in the CNS of treated mice. However, it is worth taking into consideration that brain accumulation of sunitinib in mice is restricted by the drug transporters Abcb1 (P-glycoprotein) and Abcg2 (Tang et al., 2012). Complete inhibition of both transporters significantly increases brain accumulation of sunitinib, and it has been suggested that the co-administration of elacridar and sunitinib might be beneficial (Tang et al., 2012; Oberoi et al., 2013).
Further supporting a potential neuroprotective role for sunitinib, this anti-cancer compound has been under consideration for the treatment of glioblastoma, and a recent study showed that sunitinib increases neuronal survival and neurotrophic effects via NF-κB (Sanchez et al., 2013). The NF-κB pathway is also involved in the progression of HIV infection of the CNS (Alvarez et al., 2007), thus providing another target for the neuroprotective effects of sunitinib in HIV neurotoxicity. Moreover, the CDK5 and RTK signalling pathways are also affected in other neurodegenerative disorders such as AD, Parkinson's disease and Huntington's disease (Patrick et al., 1999; Lee et al., 2000; Cuny, 2009; Czapski et al., 2013). Therefore, utilizing an anti-cancer compound such as sunitinib with oral bioavailability and inhibitory activity against both RTKs and CDKs might offer a unique opportunity for drug repositioning in an effort to develop new treatments for neurodegenerative disorders.
Acknowledgments
This study was supported by the National Institutes of Health (MH45294, MH5974, MH58164) and the California NeuroAIDS Tissue Network (U01 MH83506). The HIV Neurobehavioral Research Center is supported by a Center Award (MH62512) from the National Institute of Mental Health.
Glossary
- AD
Alzheimer's disease
- ART
antiretroviral therapy
- BBB
blood–brain barrier
- CRMP2
collapsin response mediator protein-2
- GFAP
glial fibrillary acidic protein
- Grb2
growth factor receptor-bound protein 2
- GSK3β
glycogen synthase kinase-3β
- HAND
HIV-associated neurocognitive disorders
- HIVE
HIV encephalitis
- MAP2
microtubule-associated protein-2
- MEF2
myocyte enhancer factor-2
- RTKs
receptor tyrosine kinases
- tg
transgenic
- TKIs
tyrosine kinase inhibitors
Author contributions
W. W., E. S., I. F. T. and V. L. K. performed in silico modelling. D. P. and A. P. performed in vitro kinase assays. T. G. performed cell-based assays. L. A. C. and C. R. O. drafted and edited the manuscript. C. P. performed Western blot and immunocytochemistry. E. R. performed in vivo studies. E. M. performed neuropathology, image analysis, manuscript preparation, statistical analysis and idea conception.
Conflict of interest
The authors declare no conflicts of interest.
References
- Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, et al. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci U S A. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol. 2013a;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013c;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alirezaei M, Watry DD, Flynn CF, Kiosses WB, Masliah E, Williams BR, et al. Human immunodeficiency virus-1/surface glycoprotein 120 induces apoptosis through RNA-activated protein kinase signaling in neurons. J Neurosci. 2007;27:11047–11055. doi: 10.1523/JNEUROSCI.2733-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alirezaei M, Kiosses WB, Fox HS. Decreased neuronal autophagy in HIV dementia: a mechanism of indirect neurotoxicity. Autophagy. 2008;4:963–966. doi: 10.4161/auto.6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez S, Serramia MJ, Fresno M, Munoz-Fernandez MA. HIV-1 envelope glycoprotein 120 induces cyclooxygenase-2 expression in astrocytoma cells through a nuclear factor-kappaB-dependent mechanism. Neuromolecular Med. 2007;9:179–193. doi: 10.1007/BF02685891. [DOI] [PubMed] [Google Scholar]
- Bell JE. An update on the neuropathology of HIV in the HAART era. Histopathology. 2004;45:549–559. doi: 10.1111/j.1365-2559.2004.02004.x. [DOI] [PubMed] [Google Scholar]
- Bethel-Brown C, Yao H, Hu G, Buch S. Platelet-derived growth factor (PDGF)-BB-mediated induction of monocyte chemoattractant protein 1 in human astrocytes: implications for HIV-associated neuroinflammation. J Neuroinflammation. 2012;9:262. doi: 10.1186/1742-2094-9-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biebl M, Cooper CM, Winkler J, Kuhn HG. Analysis of neurogenesis and programmed cell death reveals a self-renewing capacity in the adult rat brain. Neurosci Lett. 2000;291:17–20. doi: 10.1016/s0304-3940(00)01368-9. [DOI] [PubMed] [Google Scholar]
- Biebl M, Winner B, Winkler J. Caspase inhibition decreases cell death in regions of adult neurogenesis. Neuroreport. 2005;16:1147–1150. doi: 10.1097/00001756-200508010-00003. [DOI] [PubMed] [Google Scholar]
- Cary DC, Clements JE, Henderson AJ. RON receptor tyrosine kinase, a negative regulator of inflammation, is decreased during simian immunodeficiency virus-associated central nervous system disease. J Immunol. 2013;191:4280–4287. doi: 10.4049/jimmunol.1300797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC. 2005. pp. 1–63. HIV/AIDS surveillance report.
- CDC. 2007. pp. 1–54. HIV/AIDS surveillance report.
- Chambers LA, Wilson MG, Rueda S, Gogolishvili D, Shi MQ, Rourke SB, et al. Evidence informing the intersection of HIV, aging and health: a scoping review. AIDS Behav. 2014;18:661–675. doi: 10.1007/s10461-013-0627-5. [DOI] [PubMed] [Google Scholar]
- Cicero S, Herrup K. Cyclin-dependent kinase 5 is essential for neuronal cell cycle arrest and differentiation. J Neurosci. 2005;25:9658–9668. doi: 10.1523/JNEUROSCI.1773-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Kuhn CM, Kuhn HG. Is it all DNA repair? Methodological considerations for detecting neurogenesis in the adult brain. Brain Res Dev Brain Res. 2002;134:13–21. doi: 10.1016/s0165-3806(01)00243-7. [DOI] [PubMed] [Google Scholar]
- Crews L, Patrick C, Achim CL, Everall IP, Masliah E. Molecular pathology of neuro-AIDS (CNS-HIV) Int J Mol Sci. 2009;10:1045–1063. doi: 10.3390/ijms10031045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, et al. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS ONE. 2010;5:e9313. doi: 10.1371/journal.pone.0009313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Crews L, Ruf R, Patrick C, Dumaop W, Trejo-Morales M, Achim CL, et al. Phosphorylation of collapsin response mediator protein-2 disrupts neuronal maturation in a model of adult neurogenesis: implications for neurodegenerative disorders. Mol Neurodegener. 2011;6:67. doi: 10.1186/1750-1326-6-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuny GD. Kinase inhibitors as potential therapeutics for acute and chronic neurodegenerative conditions. Curr Pharm Des. 2009;15:3919–3939. doi: 10.2174/138161209789649330. [DOI] [PubMed] [Google Scholar]
- Czapski GA, Gassowska M, Wilkaniec A, Cieslik M, Adamczyk A. Extracellular alpha-synuclein induces calpain-dependent overactivation of cyclin-dependent kinase 5 in vitro. FEBS Lett. 2013;587:3135–3141. doi: 10.1016/j.febslet.2013.07.053. [DOI] [PubMed] [Google Scholar]
- Dewhurst S, Maggirwar SB, Schifitto G, Gendelman HE, Gelbard HA. Glycogen synthase kinase 3 beta (GSK-3 beta) as a therapeutic target in neuroAIDS. J Neuroimmune Pharmacol. 2007;2:93–96. doi: 10.1007/s11481-006-9051-1. [DOI] [PubMed] [Google Scholar]
- Diesing TS, Swindells S, Gelbard H, Gendelman HE. HIV-1-associated dementia: a basic science and clinical perspective. AIDS Read. 2002;12:358–368. [PubMed] [Google Scholar]
- Eggert D, Dash PK, Serradji N, Dong CZ, Clayette P, Heymans F, et al. Development of a platelet-activating factor antagonist for HIV-1 associated neurocognitive disorders. J Neuroimmunol. 2009;213:47–59. doi: 10.1016/j.jneuroim.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8:33–44. doi: 10.1038/nrn2040. [DOI] [PubMed] [Google Scholar]
- Everall IP, Bell C, Mallory M, Langford D, Adame A, Rockestein E, et al. Lithium ameliorates HIV-gp120-mediated neurotoxicity. Mol Cell Neurosci. 2002;21:493–501. doi: 10.1006/mcne.2002.1196. [DOI] [PubMed] [Google Scholar]
- Fields J, Dumaop W, Adame A, Ellis RJ, Letendre S, Grant I, et al. Alterations in the levels of vesicular trafficking proteins involved in HIV replication in the brains and CSF of patients with HIV-associated neurocognitive disorders. J Neuroimmune Pharmacol. 2013;8:1197–1209. doi: 10.1007/s11481-013-9511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, D'Emilia DM, et al. Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J Neurosci. 2002;22:4015–4024. doi: 10.1523/JNEUROSCI.22-10-04015.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerena Y, Skolasky RL, Velez JM, Toro-Nieves D, Mayo R, Nath A, et al. Soluble and cell-associated insulin receptor dysfunction correlates with severity of HAND in HIV-infected women. PLoS ONE. 2012;7:e37358. doi: 10.1371/journal.pone.0037358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Grant I, Heaton RK, Atkinson JH. Neurocognitive disorders in HIV-1 infection. HNRC Group. HIV Neurobehavioral Research Center. Curr Top Microbiol Immunol. 1995;202:11–32. [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Mechanisms of neuronal injury and death in HIV-1 associated dementia. Curr HIV Res. 2006;4:307–318. doi: 10.2174/157016206777709384. [DOI] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Wang T, Jang MH, Steiner J, Haughey N, Ming GL, et al. Rescue of adult hippocampal neurogenesis in a mouse model of HIV neurologic disease. Neurobiol Dis. 2011;41:678–687. doi: 10.1016/j.nbd.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- Lindl KA, Marks DR, Kolson DL, Jordan-Sciutto KL. HIV-associated neurocognitive disorder: pathogenesis and therapeutic opportunities. J Neuroimmune Pharmacol. 2010;5:294–309. doi: 10.1007/s11481-010-9205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling YH, Li T, Yuan Z, Haigentz M, Jr, Weber TK, Perez-Soler R. Erlotinib, an effective epidermal growth factor receptor tyrosine kinase inhibitor, induces p27KIP1 up-regulation and nuclear translocation in association with cell growth inhibition and G1/S phase arrest in human non-small-cell lung cancer cell lines. Mol Pharmacol. 2007;72:248–258. doi: 10.1124/mol.107.034827. [DOI] [PubMed] [Google Scholar]
- Lipton S, Rosenberg P. Mechanisms of disease: excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Lopes JP, Oliveira CR, Agostinho P. Neurodegeneration in an Abeta-induced model of Alzheimer's disease: the role of Cdk5. Aging Cell. 2010;9:64–77. doi: 10.1111/j.1474-9726.2009.00536.x. [DOI] [PubMed] [Google Scholar]
- Lucking U, Siemeister G, Schafer M, Briem H, Kruger M, Lienau P, et al. Macrocyclic aminopyrimidines as multitarget CDK and VEGF-R inhibitors with potent antiproliferative activities. ChemMedChem. 2007;2:63–77. doi: 10.1002/cmdc.200600199. [DOI] [PubMed] [Google Scholar]
- Maggirwar SB, Tong N, Ramirez S, Gelbard HA, Dewhurst S. HIV-1 Tat-mediated activation of glycogen synthase kinase-3beta contributes to Tat-mediated neurotoxicity. J Neurochem. 1999;73:578–586. doi: 10.1046/j.1471-4159.1999.0730578.x. [DOI] [PubMed] [Google Scholar]
- Mapelli M, Massimiliano L, Crovace C, Seeliger MA, Tsai LH, Meijer L, et al. Mechanism of CDK5/p25 binding by CDK inhibitors. J Med Chem. 2005;48:671–679. doi: 10.1021/jm049323m. [DOI] [PubMed] [Google Scholar]
- Martin MP, Alam R, Betzi S, Ingles DJ, Zhu JY, Schonbrunn E. A novel approach to the discovery of small-molecule ligands of CDK2. Chembiochem. 2012;13:2128–2136. doi: 10.1002/cbic.201200316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Achim C, Ge N, DeTeresa R, Terry R, Wiley C. Spectrum of human immunodeficiency virus-associated neocortical damage. Ann Neurol. 1992;32:321–329. doi: 10.1002/ana.410320304. [DOI] [PubMed] [Google Scholar]
- Masliah E, Roberts ES, Langford D, Everall I, Crews L, Adame A, et al. Patterns of gene dysregulation in the frontal cortex of patients with HIV encephalitis [Erratum appeared in J Neuroimmunol 2005, 162:197] J Neuroimmunol. 2004;157:163–175. doi: 10.1016/j.jneuroim.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Haughey NJ, Nath A. Cell death in HIV dementia. Cell Death Differ. 2005;12(Suppl. 1):893–904. doi: 10.1038/sj.cdd.4401577. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Haughey N, Gartner S, Conant K, Pardo C, Nath A, et al. Human immunodeficiency virus-associated dementia: an evolving disease. J Neurovirol. 2003;9:205–221. doi: 10.1080/13550280390194109. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–337. [PubMed] [Google Scholar]
- Menn B, Bach S, Blevins TL, Campbell M, Meijer L, Timsit S. Delayed treatment with systemic (S)-roscovitine provides neuroprotection and inhibits in vivo CDK5 activity increase in animal stroke models. PLoS ONE. 2010;5:e12117. doi: 10.1371/journal.pone.0012117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Abraham CR, Ruppe MD, Rockenstein EM, Toggas SM, Mallory M, et al. Protection against HIV-1 gp120-induced brain damage by neuronal expression of human amyloid precursor protein. J Exp Med. 1995;181:1551–1556. doi: 10.1084/jem.181.4.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair N, Kudo W, Smith MA, Abrol R, Goddard WA, 3rd, Reddy VP. Novel purine-based fluoroaryl-1,2,3-triazoles as neuroprotecting agents: synthesis, neuronal cell culture investigations, and CDK5 docking studies. Bioorg Med Chem Lett. 2011;21:3957–3961. doi: 10.1016/j.bmcl.2011.05.019. [DOI] [PubMed] [Google Scholar]
- Nath A. Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J Infect Dis. 2002;186(Suppl. 2):S193–S198. doi: 10.1086/344528. [DOI] [PubMed] [Google Scholar]
- Norman JP, Perry SW, Reynolds HM, Kiebala M, De Mesy Bentley KL, Trejo M, et al. HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PLoS ONE. 2008;3:e3731. doi: 10.1371/journal.pone.0003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberoi RK, Mittapalli RK, Elmquist WF. Pharmacokinetic assessment of efflux transport in sunitinib distribution to the brain. J Pharmacol Exp Ther. 2013;347:755–764. doi: 10.1124/jpet.113.208959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto S, Kang Y-J, Brechtel CW, Siviglia E, Russo R, Clemente A, et al. HIV/gp120 decreases adult neural progenitor cell proliferation via checkpoint kinase-mediated cell-cycle withdrawal and G1 arrest. Cell Stem Cell. 2007;1:230–236. doi: 10.1016/j.stem.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Ozdener H. Molecular mechanisms of HIV-1 associated neurodegeneration. J Biosci. 2005;30:391–405. doi: 10.1007/BF02703676. [DOI] [PubMed] [Google Scholar]
- Patrick C, Crews L, Desplats P, Dumaop W, Rockenstein E, Achim CL, et al. Increased CDK5 expression in HIV encephalitis contributes to neurodegeneration via tau phosphorylation and is reversed with Roscovitine. Am J Pathol. 2011;178:1646–1661. doi: 10.1016/j.ajpath.2010.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Sun L, Jia B, Lan X, Zhu B, Wu Y, et al. HIV-1-infected and immune-activated macrophages induce astrocytic differentiation of human cortical neural progenitor cells via the STAT3 pathway. PLoS ONE. 2011;6:e19439. doi: 10.1371/journal.pone.0019439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potula R, Dhillion N, Sui Y, Zien CA, Funa K, Pinson D, et al. Association of platelet-derived growth factor-B chain with simian human immunodeficiency virus encephalitis. Am J Pathol. 2004;165:815–824. doi: 10.1016/S0002-9440(10)63344-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasheed S, Yan JS, Hussain A, Lai B. Proteomic characterization of HIV-modulated membrane receptors, kinases and signaling proteins involved in novel angiogenic pathways. J Transl Med. 2009;7:75. doi: 10.1186/1479-5876-7-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rom S, Pacifici M, Passiatore G, Aprea S, Waligorska A, Del Valle L, et al. HIV-1 Tat binds to SH3 domains: cellular and viral outcome of Tat/Grb2 interaction. Biochim Biophys Acta. 2011;1813:1836–1844. doi: 10.1016/j.bbamcr.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, et al. HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirol. 2002;8:136–142. doi: 10.1080/13550280290049615. [DOI] [PubMed] [Google Scholar]
- Sanchez A, Tripathy D, Yin X, Luo J, Martinez JM, Grammas P. Sunitinib enhances neuronal survival in vitro via NF-kappaB-mediated signaling and expression of cyclooxygenase-2 and inducible nitric oxide synthase. J Neuroinflammation. 2013;10:93. doi: 10.1186/1742-2094-10-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schifitto G, Zhong J, Gill D, Peterson DR, Gaugh MD, Zhu T, et al. Lithium therapy for human immunodeficiency virus type 1-associated neurocognitive impairment. J Neurovirol. 2009;15:176–186. doi: 10.1080/13550280902758973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JC, Woods SP, Carey CL, Weber E, Bondi MW, Grant I. Neurocognitive consequences of HIV infection in older adults: an evaluation of the ‘cortical’ hypothesis. AIDS Behav. 2011;15:1187–1196. doi: 10.1007/s10461-010-9815-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G. Statement of Senator Gordon H Smith. Aging Hearing: HIV Over Fifty, Exploring the New Threat. Washington, DC: Senate Committee on Aging; 2005. [Google Scholar]
- Tang L, Wang Y, Strom A, Gustafsson JA, Guan X. Lapatinib induces p27(Kip1)-dependent G(1) arrest through both transcriptional and post-translational mechanisms. Cell Cycle. 2013;12:2665–2674. doi: 10.4161/cc.25728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SC, Lagas JS, Lankheet NA, Poller B, Hillebrand MJ, Rosing H, et al. Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib coadministration. Int J Cancer. 2012;130:223–233. doi: 10.1002/ijc.26000. [DOI] [PubMed] [Google Scholar]
- Teng CL, Yu CT, Hwang WL, Tsai JR, Liu HC, Hwang GY, et al. Effector mechanisms of sunitinib-induced G1 cell cycle arrest, differentiation, and apoptosis in human acute myeloid leukaemia HL60 and KG-1 cells. Ann Hematol. 2013;92:301–313. doi: 10.1007/s00277-012-1627-7. [DOI] [PubMed] [Google Scholar]
- Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- Vita M, Abdel-Rehim M, Olofsson S, Hassan Z, Meurling L, Siden A, et al. Tissue distribution, pharmacokinetics and identification of roscovitine metabolites in rat. Eur J Pharm Sci. 2005;25:91–103. doi: 10.1016/j.ejps.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Wang Y, White MG, Akay C, Chodroff RA, Robinson J, Lindl KA, et al. Activation of cyclin-dependent kinase 5 by calpains contributes to human immunodeficiency virus-induced neurotoxicity. J Neurochem. 2007;103:439–455. doi: 10.1111/j.1471-4159.2007.04746.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Gao J, Zhang D, Zhang J, Ma J, Jiang H. New insights into the antifibrotic effects of sorafenib on hepatic stellate cells and liver fibrosis. J Hepatol. 2010;53:132–144. doi: 10.1016/j.jhep.2010.02.027. [DOI] [PubMed] [Google Scholar]
- Ying Wang J, Peruzzi F, Lassak A, Del Valle L, Radhakrishnan S, Rappaport J, et al. Neuroprotective effects of IGF-I against TNFalpha-induced neuronal damage in HIV-associated dementia. Virology. 2003;305:66–76. doi: 10.1006/viro.2002.1690. [DOI] [PubMed] [Google Scholar]
- Yuferov V, Ho A, Morgello S, Yang Y, Ott J, Kreek MJ. Expression of ephrin receptors and ligands in postmortem brains of HIV-infected subjects with and without cognitive impairment. J Neuroimmune Pharmacol. 2013;8:333–344. doi: 10.1007/s11481-012-9429-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapata-Torres G, Opazo F, Salgado C, Munoz JP, Krautwurst H, Mascayano C, et al. Effects of natural flavones and flavonols on the kinase activity of Cdk5. J Nat Prod. 2004;67:416–420. doi: 10.1021/np034011s. [DOI] [PubMed] [Google Scholar]
- Zhou D, Masliah E, Spector SA. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J Infect Dis. 2011;203:1647–1657. doi: 10.1093/infdis/jir163. [DOI] [PMC free article] [PubMed] [Google Scholar]