

Abstract
Background and Purpose
GPR119 is a Gαs-coupled 7TM receptor activated by endogenous lipids such as oleoylethanolamide (OEA) and by the dietary triglyceride metabolite 2-monoacylglycerol. GPR119 stimulates enteroendocrine hormone and insulin secretion. But despite massive drug discovery efforts in the field, very little is known about the basic molecular pharmacology of GPR119.
Experimental Approach
GPR119 receptor signalling was studied in transfected cells. Mutational mapping (30 mutations in 23 positions) was performed on residues required for ligand-independent and agonist-induced GPR119 activation (AR231453 and OEA). Novel Rosetta-based receptor modelling was applied, using a composite template approach with segments from different X-ray structures and fully flexible ligand docking.
Key Results
The increased signalling induced by increasing the cell surface expression of GPR119 in the absence of agonist and the inhibitory effect of two synthetic inverse agonists demonstrated that GRP119 signals with a high degree of constitutive activity through the Gαs pathway. The mutational maps for AR231453 and OEA were very similar and, surprisingly, also similar to the mutational map for residues affecting the constitutive signalling – albeit with key differences. Surprisingly, almost all residues in extracellular loop-2b were important for the constitutive activity. The molecular modelling and docking demonstrated that AR231453 binds in a ‘vertical’ pocket in between mutational hits reaching from the centre of the receptor out to extracellular loop-2b.
Conclusions and Implications
The high constitutive activity of GPR119 should be taken into account in future drug discovery efforts, which can now be guided by the detailed knowledge of the physiochemical properties of the extended ligand-binding pocket.
Introduction
By use of bioinformatics, GPR119 was originally identified as an orphan, rhodopsin-like seven transmembrane (7TM) segment, GPCR, which is not closely related to any other 7TM receptor (Fredriksson et al., 2003). However, GPR119 had in fact been identified previously in the patent literature as an orphan receptor, that is highly expressed in pancreatic islets, under various names such as: SNORF25 (Bonini et al., 2001; Adham et al., 2003), RUP3, 19AJ, GPCR2, PFI-007 (Reilly, 2001; Ohishi et al., 2002), PS1, AXOR20, OSGPR116 (Griffin, 2006) and the glucose-dependent insulinotropic receptor (Jones, 2006). The ability of GPR119 to act as a pancreatic insulinotropic receptor was subsequently rediscovered after the receptor had obtained its official name, GPR119 (Soga et al., 2005; Sakamoto et al., 2006; Chu et al., 2008). Although the expression and function of GPR119 in beta cells has been repeatedly shown, it is still unclear to what extent GPR119 is also expressed in the other endocrine islet cells (Jones et al., 2009). However, GPR119 has been shown to be highly expressed in enteroendocrine cell lines such as GLUTag and STC-1 cells (Chu et al., 2008), as well as in natural enteroendocrine cells (Reimann et al., 2008; Parker et al., 2009).
GPR119 is activated by a number of different lipid metabolites, some of which are derived from dietary fat and some of which may be generated locally in the tissue (Hansen et al., 2012). Among the locally generated lipid metabolites, which presumably act in a paracrine or autocrine fashion, oleoylethanolamide (OEA) is particularly potent and could potentially be generated under physiological conditions in sufficient amounts to be a valid GPR119 agonist (Overton et al., 2006; Hansen et al., 2012). The same is true for 2-monoacylglycerols, which are generated in large amounts in the lumen of the gastrointestinal tract from dietary triglycerides by the action of pancreatic lipase and which consequently could act as luminal stimulators of enteroendocrine cells (Hansen et al., 2012). In contrast, N-oleoyldopamine and 5-hydroxy-eicosapentaenoic acid, which also have been reported to be GPR119 agonists (Chu et al., 2010; Kogure et al., 2011), are probably not generated in sufficient amounts under physiological conditions to be valid GPR119 agonists (Hansen et al., 2012). Importantly, a number of synthetic GPR119 agonists have been developed, which are highly selective and consequently have been crucial for the characterization of GPR119 function in vitro and in vivo (Jones et al., 2009).
GPR119 couples to Gαs (Chu et al., 2007) and is an efficient stimulator of cAMP production both in transfected cells (Soga et al., 2005; Overton et al., 2006; Chu et al., 2007) and in endocrine cell lines expressing GPR119 where the rise in cAMP levels is associated with hormone secretion. Thus, GPR119 activation results in the secretion of insulin from insulinoma cell lines (Soga et al., 2005; Chu et al., 2007; Ning et al., 2008) and GLP-1 from enteroendocrine cell lines (Chu et al., 2008; Lauffer et al., 2009) and primary intestinal cell cultures (Lan et al., 2012). The prototype GPR119 agonist, AR231453, has been shown to ameliorate glucose excursions in both normal and diabetic mice (Chu et al., 2007). Importantly, this effect was diminished when glucose was administered i.p. instead of p.o. GPR119 agonists induce both insulin and GLP-1 secretion in vivo and importantly, neither of these effects are observed in GPR119-deficient mice (Chu et al., 2007; 2008,). Thus, both a direct effect on the beta cells and an indirect incretin-based effect appear to contribute to the GPR119-mediated improvement in oral glucose tolerance. It has been suggested that GPR119 also plays a role in the control of food intake (Overton et al., 2006), but this is less certain than its well-established role in glucose control (Jones et al., 2009; Lan et al., 2009).
Early on, GPR119 became a major target for the development of novel anti-diabetic agents and several selective potent and efficacious GPR119 agonists have been in clinical trials (Overton et al., 2008; Shah, 2009; Dhayal and Morgan, 2010; Ohishi and Yoshida, 2012). But despite their apparent beneficial effects in various ex vivo and in vivo models, they have apparently not lived up to the high expectations, which originally surrounded GPR119. Several programmes have been terminated or have been given back to the biotech companies, from which they originated (Hansen et al., 2012). However, it is still unclear whether this apparent lack of clinical efficacy was caused by the broad pharmacology of GPR119 agonists stimulating not only incretin and insulin secretion but also counter-regulatory gut and pancreatic hormones such as somatostatin and glucagon, or whether the apparent lack of clinical efficacy of the present GPR119 agonists was related to difficulties in developing agonists for 7TM receptors with appropriate signalling properties.
Very little has been published about the basic molecular and cellular pharmacology of GPR119 despite the massive efforts invested in drug discovery processes targeting this receptor (Jones et al., 2009). In the present study, we focused on the apparent constitutive signalling of GPR119, and through extensive mutational mapping (Figure 1) combined with molecular modelling and computational chemistry, we characterized the molecular basis for constitutive versus agonist-induced signalling of GPR119. The broad conclusion is that both the prototype synthetic agonist AR231453 and the most potent endogenous agonist OEA bind in a similar vertical fashion to the main ligand-binding pocket of GPR119, and that overlapping – but not identical – residues in this pocket including key residues in extracellular loop-2b (ECL-2b) are also important for the constitutive signalling of the receptor.
Figure 1.
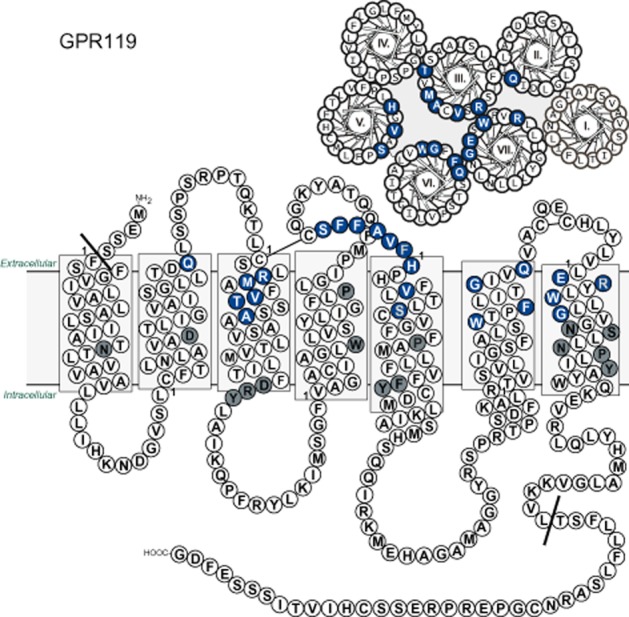
Helical wheel and serpentine model of the human GPR119. The sequence of GPR119 was depicted schematically from the membrane bilayer and from the extracellular side. The residues selected for the mutational analysis are marked with blue and white letters. Residues conserved in family A 7TM receptors and present in the GPR119 receptor are marked with grey. The Schwartz/Baldwin generic numbering system for 7TM receptors (Schwartz et al., 2006) is used to denote the residues in the transmembrane helices throughout the paper. The residues designated as the first of each helix are marked with 1. The mutations in ECL-2b are numbered according to their relative position from the conserved cysteine.
Methods
Molecular biology
GPR119 receptor cDNA was cloned into pcDNA3.1(+) and FLAG tagged. A modified signal peptide from viral haemagglutinin was inserted in front of the FLAG tag to enhance surface expression (Guan et al., 1992). Mutations were introduced by site-directed mutagenesis using PCR. The resulting construct was transformed into competent Escherichia coli XL1 blue, cultured and purified with the maxi prep kit from Qiagen (Copenhagen, Denmark).
Cell culture and transfection
COS7 cells were grown in DMEM (low glucose) supplemented with 10% fetal calf serum, 2 mM glutamine and 0.01 mg·mL−1 pen-strep at 37°C, 10% CO2. Cells were transfected with 20 μg DNA/75 cm2 using calcium phosphate precipitation (Graham and van der Eb, 1973) with chloroquine addition. For the ‘gene dosing’ experiment, cells were transfected with 0, 5, 10, 20 or 40 μg DNA/75 cm2 GPR119 plasmid or 40 μg DNA/75 cm2 empty plasmid.
Cell surface expression measurements (elisa)
One day after transfection, cells were seeded in 96-well plates. The next day, cells were washed with Tris-buffered saline (TBS), fixed in 3.7% formaldehyde for 30 min, and washed and incubated with blocking buffer (2% BSA, 0.5 mM CaCl2 in TBS) for 1 h. Next, cells were incubated with a FLAG-M1 antibody (2 μg·mL−1) from mice for 2 h, washed, and incubated with a goat-anti-mouse antibody conjugated to HRP at 4°C overnight.
Then cells were washed and incubated with 150 μL tetramethylbenzidine for 5 min before the reaction was stopped with 0.2 M H2SO4. Absorbance at 450 nm was quantified using a multilabel counter (Wallac Victor2; Perkin Elmer, Broendby, Denmark). Determinations were made in quadruplicate.
cAMP accumulation assay
One day after transfection, cells were seeded in 12-well plates, 2 × 105 cells per well in 0.5 mL medium supplemented with 2 μL·mL−1 adenine-[2,8-3H] (PerkinElmer). The next day, cells were washed with HEPES-buffered saline and incubated with 1 mM IBMX in HEPES-buffered saline for 30 min at 37°C. Ligands were added, and cells were incubated for another 30 min at 37°C. Medium was removed, and cells were lysed with 5% trichloroacetic acid containing 0.1 mM ATP and 0.1 mM cAMP. The lysate was transferred to Dowex columns (50WX4; Sigma-Aldrich, Broendby, Denmark), washed with 2 mL H2O and eluted into Alumina columns (A9003; Sigma) with 10 mL H2O, from where cAMP was eluted with 6 mL 0.1 M imidazole. Then 15 mL of scintillation liquid (Gold Star; Meridian Biotechnology, Chessington, UK) was added to the eluate and radioactivity in the sample was measured on a scintillation counter. Determinations were made in duplicate.
Data analysis
Data were analysed using the Prism 5.0 software (GraphPad Software, San Diego, CA, USA). Surface expression of the mutant receptors was normalized to wild-type receptor surface expression (100%) and the signal from an empty vector (0%) tested in parallel. cAMP counts were normalized to the maximum AR231453-induced activation of the wild-type receptor tested in parallel (100%) and the mean value of cAMP accumulation from cells transfected with an empty vector (0%). EC50 values were determined by nonlinear regression. The statistical significance of the effects of the mutations on surface expression, basal activity, AR231453/OEA-induced activity and potency (logEC50) was assessed using ordinary one-way anova with Dunnett's post hoc test with correction for multiple comparisons.
GPR119 receptor modelling
A multiple alignment between the sequence of the human GPR119 receptor (accession code: Q8TDV5) and the sequence of 14 distinct template structures was generated using ICM version 3.7-2 (Molsoft LLC, San Diego, CA, USA). Two comparative GPR119 receptor models were generated based on a hybrid multiple template approach (Mobarec et al., 2009; Worth et al., 2011). The first model was based on: (i) adenosine A2A receptors (PDBID: 3QAK) to model the transmembrane helices TM-I to TM-VIII, including ICL1-3, ECL-1 and ECL-2b; (ii) the chemokine receptor CXCR4 (PDBID: 3ODU) to model ECL-2a; and (iii) dopamine D3 receptor (PDBID: 3PBL) to model ECL-3. The second model was based on: (i) A2A receptor template to model TM-I-III and TM-V-VIII, including ICL1-3, ECL-1 and ECL-2b; (ii) CXCR4 template to model TM-IV including ECL-2a; and (iii) dopamine D3 receptor to model ECL-3. The resulting models were optimized in 1000 steps of full-atom structure relaxation using Rosetta version 3.2.1 and the membrane force field (Barth et al., 2007; Leaver-Fay et al., 2011). Through the relaxation protocol, a disulphide bridge between CysIII:01 and Cys155 (ECL-2) was specified as a structural constraint. The top 300 (30%) best scored models (Rosetta energy) were clustered based on the root mean square deviation (RMSD) between main chain atoms of residue positions involved in direct ligand interactions in the various template structures using Rosetta cluster routine and a 1.0 Å cluster threshold. A total of 43 non-redundant energetically feasible GPR119 models (corresponding to the lowest energy structures from each cluster) were used as a receptor ensemble for docking.
Full flexible receptor-ligand docking of AR231453 into the GPR119-binding pocket
An ensemble of AR231453 containing nine conformations within 3 kcal·mol−1 of the global minimum was generated using the Merck Molecular Force Field as implemented in the ICM software package version 3.7-2 with a generalized born implicit solvation model. The conformational ensemble of AR231453 was docked 1000 times to each of the 43 possible binding pocket models using RosettaLigand with full receptor and ligand flexibility (Meiler and Baker, 2006; Davis and Baker, 2009; Fleishman et al., 2011; Lemmon and Meiler, 2012), producing a total of 43.000 receptor-ligand complexes. The top 10% of the best scored complexes with respect to ligand interaction energy were selected for further analysis, which previously have been demonstrated to enrich native-like ligand-binding conformations in a recent modelling and docking assessment study on a large set of distinct 7TM receptor X-ray structures (Nguyen et al., 2013). The RMSD between distinct ligand-binding modes was calculated for all complexes using an in-house script, and clustered with a hierarchical agglomerative clustering algorithm as implemented in BCL::Cluster (Alexander et al., 2011) using a clustering threshold of 2.5 Å. Low energy complexes from the 10 largest clusters were visually inspected and analysed in the context of the mutation analysis.
Materials
AR231453 was synthesized as previously described (Semple et al., 2008). The synthesis of AR436352 is described in Supporting Information Appendix S1. TM43718 was identified by virtual screening of worldwide vendor libraries of small molecule compounds based on ligand-based pharmacophore multiplet searches around the two lead structures AR246881 (Arena Pharmaceuticals, San Diego, CA, USA) and PSN632408 (Prosidion, Oxford, UK) using the UNITY modelling packages (Tripos, St. Louis, MO, USA). TM43718 was purchased as E897-0145 from ChemDiv (San Diego, CA, USA) and OEA from Sigma-Aldrich.
Results
GPR119 is constitutively active
Transfection experiments with increasing doses of expression plasmid demonstrated that GPR119 signals with a high degree of constitutive activity through the Gαs pathway. That is, increasing cell surface receptor expression as determined by elisa in the absence of agonist was associated with increasing stimulation of cAMP accumulation in transfected COS7 cells (Figure 2A and B). The degree of constitutive activity for GPR119 was 37 ± 0.8% of the Emax obtained with the prototype small molecule synthetic agonist AR231453 (Figure 2B and Table 1). The relative constitutive activity of GPR119 determined as percentage of Emax was quite independent of receptor expression (Figure 2B).
Figure 2.
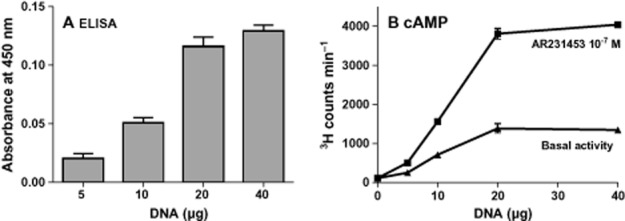
Constitutive activity of GPR119. (A) Surface expression of FLAG-tagged GPR119 resulting from transfections with increasing concentrations of a GPR119 construct and measured with elisa. (B) Basal level and AR231453-induced cAMP accumulation from cells transfected with increasing concentrations of the GPR119 construct – 20 μg of DNA was chosen as the optimal amount for further studies.
Tables of Links
| TARGETS | |
|---|---|
| β2-adrenoceptor | D3 receptor |
| κ-opioid receptor | Ghrelin receptor |
| A2A receptor | GPR119 |
| Chemokine receptors | H1 receptor |
| CXCR4 | M2 receptor |
Table 1.
Surface expression and basal activity of wild-type GPR119 and receptor mutants along with efficacy and potency of AR231453 and the potency fold change between wild type and mutants (Fmut)
| Mutant | Surface expression | Basal activity | Emax | AR231453 EC50 | Fmut | n | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (%) | SEM | (%) | SEM | (%) | SEM | Log | SEM | nM | |||
| Wild type | 100 | 2.1 | 37.0 | 0.7 | 96 | 0.9 | −8.73 | 0.03 | 1.9 | 26 | |
| Binding pocket | |||||||||||
| GlnII:24Ala (65) | 122 | 9.4 | 31.0 | 5.3 | 99 | 7.1 | −8.9 | 0.24 | 1.2 | 0.6 | 4 |
| ArgIII:04Ala (81) | 51* | 2.4 | 1.9* | 1.5 | 41* | 2.5 | −7.94* | 0.15 | 11.6 | 6.1 | 4 |
| MetIII:05Ala (82) | 78 | 5.8 | 8.3* | 1.4 | 24* | 1.9 | −8.37 | 0.19 | 4.3 | 2.3 | 4 |
| ValIII:08Ala (85) | 115 | 9.2 | 29.0 | 6.0 | 88 | 7.2 | −8.80 | 0.28 | 1.6 | 0.8 | 4 |
| ThrIII:09Ala (86) | 55* | 12.0 | 13.0* | 2.1 | 39* | 4.5 | −7.80* | 0.36 | 15.8 | 8.3 | 4 |
| ThrIII:09Val (86) | 71 | 14.0 | 32.0 | 7.2 | 61* | 6.2 | −8.90 | 0.63 | 1.3 | 0.7 | 4 |
| AlaIII:12Val (89) | 231* | 43.0 | 27.0 | 6.0 | 90 | 32.0 | −6.99* | 0.81 | 103.0 | 54.0 | 4 |
| HisV:01Ala (162) | 58* | 4.3 | 20.0* | 1.9 | 73 | 2.4 | −8.75 | 0.08 | 1.8 | 0.9 | 3 |
| ValV:05Ala (166) | 53* | 9.1 | 35.0 | 6.6 | 70* | 8.5 | −8.03* | 0.61 | 9.3 | 4.9 | 3 |
| SerV:09Ala (170) | 157* | 17.0 | 57.0* | 7.5 | 108 | 7.4 | −8.64 | 0.38 | 2.3 | 1.2 | 4 |
| SerV:09Val (170) | 164* | 30.0 | 12.0* | 3.5 | 77 | 5.0 | −8.02* | 0.16 | 9.7 | 5.1 | 4 |
| TrpVI:13Ala (238) | 40* | 2.6 | – | – | – | – | – | – | – | – | 4 |
| PheVI:16Ala (241) | 72 | 4.7 | 11.0* | 1.8 | 27* | 4.4 | −7.47* | 0.50 | 34.0 | 18.0 | 3 |
| GlyVI:20Ala (245) | 136* | 3.7 | 48.0* | 3.1 | 106 | 4.1 | −8.78 | 0.16 | 1.7 | 0.9 | 5 |
| GlyVI:20Ser (245) | 116 | 5.6 | 57.0* | 3.4 | 111 | 4.1 | −8.75 | 0.15 | 1.8 | 0.9 | 4 |
| GlyVI:20Val (245) | 71 | 4.5 | 30.0 | 1.9 | 61* | 2.4 | −8.82 | 0.16 | 1.5 | 0.8 | 5 |
| GlnVI:23Ala (248) | 111 | 18.0 | 31.0 | 7.9 | 87 | 11.0 | −8.07 | 0.41 | 8.6 | 4.5 | 3 |
| GluVII:02Ala (261) | 54* | 5.1 | 13.0* | 1.3 | 54* | 3.3 | −7.75* | 0.16 | 17.7 | 9.3 | 4 |
| ArgVII:03Ala (262) | 50* | 4.1 | −0.1* | 0.7 | 18* | 1.2 | −7.94* | 0.13 | 11.6 | 6.1 | 4 |
| TrpVII:06Ala (265) | 40* | 2.8 | 34.0 | 2.7 | – | – | – | – | – | – | 5 |
| GlyVII:09Ala (268) | 148* | 6.1 | 19.0* | 2.5 | 78 | 3.8 | −8.46 | 0.13 | 3.4 | 1.8 | 4 |
| GlyVII:09Val (268) | 206* | 7.0 | 20.0* | 2.8 | – | – | – | – | – | – | 4 |
| GlyVII:09Phe (268) | 155* | 16.0 | 20.0* | 1.6 | 57* | 8.4 | −7.10* | 0.38 | 80.0 | 42.0 | 3 |
| ECL-2b | |||||||||||
| SerC+1Ala (156) | 71 | 3.5 | 6.9* | 1.3 | 48* | 2.2 | −8.44 | 0.12 | 3.7 | 1.9 | 3 |
| PheC+2Ala (157) | 69 | 4.4 | 6.4* | 0.5 | 23* | 1.7 | −6.83* | 0.18 | 147.0 | 77.0 | 5 |
| PheC+3Ala (158) | 73 | 3.6 | 3.9* | 0.6 | 24* | 1.2 | −7.35* | 0.13 | 45.0 | 24.0 | 5 |
| AlaC+4Gly (159) | 87 | 3.8 | 18.0* | 3.3 | 75 | 4.6 | −8.60 | 0.17 | 2.5 | 1.3 | 3 |
| ValC+5Ala (160) | 66 | 3.4 | 5.7* | 0.9 | 34* | 1.4 | −8.13* | 0.11 | 7.4 | 3.9 | 3 |
| PheC+6Ala (161) | 56* | 2.3 | 5.6* | 1.0 | 21* | 1.2 | −8.65 | 0.15 | 2.2 | 1.2 | 3 |
Statistically significant changes from wild type are marked with an asterisk
Residue positions are given in parentheses after the Schwartz/Baldwin name.
During a ligand-based drug discovery process for GPR119 agonists, it was discovered that one of the compounds – TM43718 (Figure 3A) – surprisingly instead functioned as a full-inverse agonist for GPR119, as it inhibited the spontaneous ligand-independent signalling in a dose-dependent manner (EC50 = 1.6 μM), that is, down to the level observed in cells transfected with empty vector (Figure 3B and C). TM43718 also functioned as an antagonist of the AR231453-induced Gαs signalling (Figure 3C).
Figure 3.

Constitutive activity of GPR119 – effect of combined small molecule antagonists and inverse agonists. (A, D) Structure of TM43718 and AR436352. (B, E) Normalized concentration–response curves for TM43718 and AR436352 measured by cAMP accumulation in GPR119-transfected cells and for AR231453. (C, F, G) Normalized concentration–response curves for AR231453 or OEA measured by cAMP accumulation in GPR119-transfected cells treated with TM43718 or AR436352 before the AR231453 or OEA treatment.
In the original screening hit in the drug discovery process for AR231453, it was described as being an inverse agonist, although it was not further characterized as such at that time (Semple et al., 2008). However, as shown in Figure 3D and E, a distantly related second-generation tri-substituted pyrimidine, AR436352, functioned as a 20 nM potent partial inverse agonist. Like TM43718, AR436352 also acted as an antagonist against AR231453 (Figure 3E). AR436352 also functioned as an antagonist against OEA, but in contrast to TM43718, AR436352 seemed to be a non-competitive antagonist (Figure 3G).
Thus, both ‘gene-dosing’ experiments and experiments with two different inverse agonists indicate that GPR119 signals with high constitutive activity through Gαs.
Mutational analysis of structural basis for the constitutive activity of GPR119
To examine the structural basis for the constitutive activity and to map ligand-receptor interactions in GPR119, alanine substitutions as well as steric hindrance mutagenesis were performed. The residues selected for the mutational analysis were located in the main ligand-binding pocket distributed on TM-II, TM-III, TM-V, TM-VI and TM-VII (Figure 1). In addition, residues in ECL-2b were mutated, as this loop has been shown to be a generally important structural element involved in ligand binding at the entrance to the main ligand-binding pocket (Nygaard et al., 2009). The effect of the mutations on the constitutive and ligand-induced receptor activation was examined by cAMP accumulation assays in transfected COS7 cells. In the following, we first describe the effects of the mutations on the ligand-independent, constitutive activity of GPR119 and then the effects upon agonist-induced receptor activation.
ECL-2b is broadly important for the constitutive activity of GPR1119
Surprisingly, Ala substitutions of each of the residues in ECL-2b strongly reduced the constitutive activity of GPR119, that is, from nearly 40% to 5–7.5% of Emax, except for the AlaC+4 (AlaC+4 was mutated to Gly) and the HisV:01 mutant at the end of ECL-2b in which the constitutive activity was reduced only down to 18 and 20% of Emax respectively (Figure 4 and Table 1). The surface expression of the ECL-2b-mutated receptors was between 56 and 87% of wild type, indicating that the decrease in constitutive activity was not resulting (solely) from a decrease in surface expression.
Figure 4.
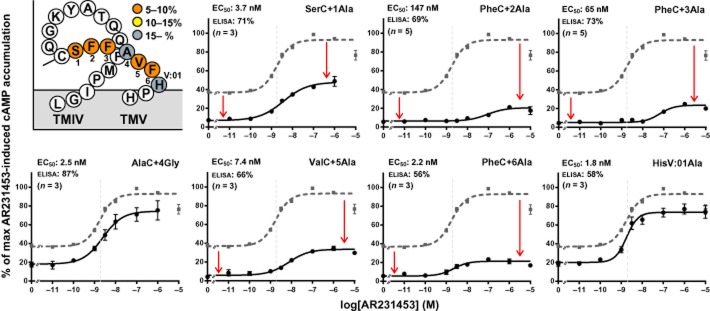
Structural basis for the constitutive activity – ECL-2b. The graphs show normalized concentration–response curves for AR231453 on GPR119 receptors mutated in ECL-2b (black). On each graph, the wild-type curve is shown in stippled grey for comparison. The EC50 values are indicated in grey stippled lines and can also be found in the upper left corner of each graph together with the surface expression level of the mutant and number of repetitions. The figure on the upper left shows the ECL-2 of GPR119, colour coded for the level of constitutive activity obtained with the receptor mutants. The constitutive activity was calculated by setting the Emax for AR231453-induced activation of the wild-type receptor to 100 and the mean value from cells transfected with an empty vector to 0.
The strong decrease in constitutive activity resulting from Ala substitution at multiple positions suggests that ECL-2b functions as a ‘tethered agonist’ in GPR119, and is important for stabilization of an active conformation of the receptor.
Key residues in the main ligand binding pocket are also important for constitutive activity of GPR119
The mutational analysis revealed three positions in the main ligand binding pocket where Ala substitution completely abolished the constitutive activity of GPR119. These included not only the highly conserved TrpVI:13 located in the middle of the receptor, which previously has been implicated as a ‘micro-switch’ in the general 7TM activation mechanism (Nygaard et al., 2009; Holst et al., 2010), but also rather surprisingly two Arg residues located at the extracellular ends of TM-III and TM-VII – ArgIII:04 and ArgVII:03 respectively (Figure 5 and Table 1). Ala substitution of the third charged residue of the main ligand-binding pocket, GluVII:02, located at the extracellular end of TM-VII in close spatial proximity to the two Arg residues, also decreased the constitutive activity albeit only down to 13% (Table 1). Similarly, Ala substitution of the neighbouring residue to ArgIII:04, MetIII:05, reduced the constitutive activity to 8.3%. Ala substitution of PheVI:16 and ThrIII:09, located deeper in the binding pocket, reduced the constitutive activity of the receptor to 11 and 13% of Emax respectively. In the case of SerV:09, Ala substitution in fact increased the constitutive signalling of the receptor whereas substitution with Val decreased the activity to 12% (Figure 5 and Table 1).
Figure 5.

Structural basis for the constitutive activity – main ligand-binding pocket. The graphs show normalized concentration–response curves for AR231453 on selected receptors with mutations in the main ligand-binding pocket (black). On each graph, the wild-type curve is shown in stippled grey for comparison. The EC50 values are indicated in grey stippled lines and can also be found in the upper left corner of each graph together with the surface expression level of the mutant and number of repetitions, except for graphs showing more than one mutant receptor – see Table 1. The helical wheel in the middle shows the location of the mutated residues, and a colour code denotes the level of constitutive activity obtained with the GPR119 receptor mutants. The colours are based on alanine substitutions. The constitutive activity was calculated by setting the Emax for AR231453-induced activation of the wild-type receptor to 100 and the mean value from cells transfected with an empty vector to 0.
In conclusion, a cluster of residues located at the upper part of the main ligand-binding pocket in between TM-III, TM-VI and TM-VII are highly important for maintaining the constitutive activity of GPR119.
Mutational mapping of the ligand interactions in the main ligand-binding pocket of GPR119
Although there was a large degree of overlap, the mutational map for residues involved in agonist-induced receptor activation differed at key positions from the mutational map of residues being important for the constitutive activity of GPR119 (Figure 6, Table 1). Three mutations in the main ligand-binding pocket completely abolished AR231453-induced receptor activation: TrpVI:13Ala, TrpVII:06Ala and GlyVII:09Val. As described above, TrpVI:13 is involved in the general 7TM activation mechanism, but could also be interacting directly with the ligand from its position in the middle of the receptor. The indole side chain of TrpVII:06 is located in TM-VII two helical turns above TrpVI:13 and faces into the main ligand-binding pocket, thus TrpVII:06 likely interacts directly with the AR231453 agonist. In the case of GlyVII:09, introduction of Ala or Phe resulted in an 18- and 50-fold decrease in AR231453 potency, respectively, while the β-branched side chain of Val completely abolished AR231453-induced receptor activation.
Figure 6.
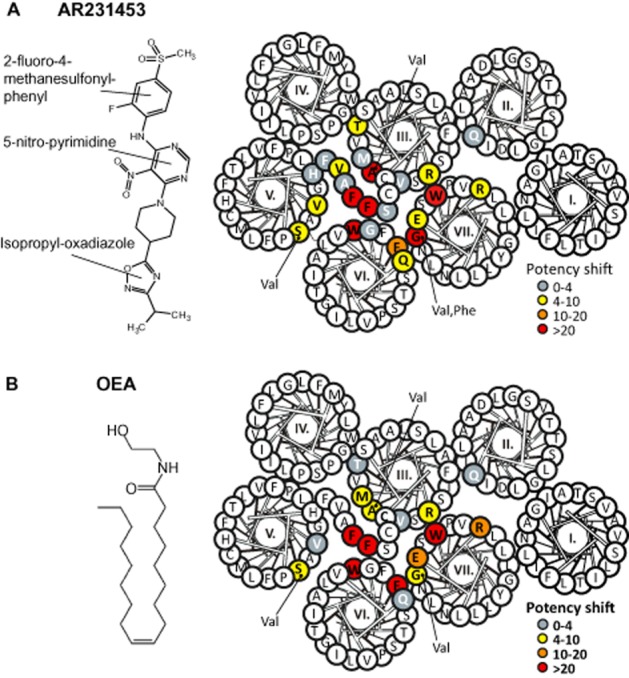
Structural basis for agonist-induced activation. The structures of AR231453 or OEA are shown to the left. In the helical wheel shown together with ECL-2b, the mutated residues are coloured corresponding to their effect on the potency of (A) AR231453 or (B) OEA in the activation of GPR119. The colours are based on alanine substitutions, except for AlaIII:12, SerV:09 and GlyVII:09, for which the valine substitution determines the colour.
In addition to TrpVII:06, Ala substitution of three charged residues, ArgIII:04, GluVII:02 and ArgVII:03, located at the interface between TM-VII and TM-III and the interface between TM-VII and TM-VI each decreased the potency of AR231453 6- to 10-fold (Figure 6, Table 1). Similarly, mutations of a group of residues located fairly deep in the main ligand-binding pocket at the interface between TM-III and TM-V also decreased AR231453 potency 4- to 10-fold, that is, ThrIII:09, ValV:05 and SerV:09, and in the case of AlaIII:12 to Val even 54-fold (Figure 6, Table 1).
With regard to OEA, the mutational map closely resembled that of AR231453 although subtle differences were observed (Figure 6, Table 2). The most noticeable was that the AlaIII:12 to Val and the GlyVII:09 to Val mutations had a greater effect on AR231453-induced activation than on OEA-induced activation, whereas the PheVI:16 to Ala mutation, which decreased the potency of AR231453 18-fold, resulted in a complete lack of activation by OEA.
| LIGANDS | |
|---|---|
| Adenine | GLP-1 |
| AR231453 | Insulin |
| ATP | OEA |
| cAMP |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Table 2.
Efficacy and potency of OEA on wild-type GPR119 and receptor mutants as well as the potency fold change between wild type and mutants (Fmut)
| Mutant | Emax | OEA EC50 | Fmut | n | |||
|---|---|---|---|---|---|---|---|
| (%) | SEM | Log | SEM | nM | |||
| Wild type | 97 | 2.0 | −6.54 | 0.13 | 290 | 7 | |
| Binding pocket | |||||||
| GlnII:24Ala (65) | 101 | 5.6 | −6.23 | 0.26 | 596 | 3.5 | 3 |
| ArgIII:04Ala (81) | 34* | 2.4 | −5.83 | 0.12 | 1470 | 8.6 | 3 |
| MetIII:05Ala (82) | 26* | 1.5 | −6.01 | 0.08 | 975 | 5.7 | 3 |
| ValIII:08Ala (85) | 97 | 3.4 | −6.18 | 0.12 | 663 | 3.9 | 3 |
| ThrIII:09Ala (86) | 37* | 2.1 | −6.11 | 0.20 | 784 | 2.0 | 4 |
| ThrIII:09Val (86) | 63* | 8.7 | −6.59 | 0.77 | 257 | 0.67 | 4 |
| AlaIII:12Val (89) | 116 | 19.0 | −5.80 | 0.42 | 1600 | 4.1 | 4 |
| ValV:05Ala (166) | 69 | 13.0 | −6.07 | 0.88 | 861 | 2.2 | 3 |
| SerV:09Ala (170) | 120 | 16.0 | −6.73 | 0.65 | 185 | 0.5 | 4 |
| SerV:09Val (170) | 47* | 17.0 | −5.76 | 0.70 | 1760 | 4.6 | 4 |
| TrpVI:13Ala (238) | – | – | – | – | – | – | 3 |
| PheVI:16Ala (241) | – | – | – | – | – | – | 3 |
| GlyVI:20Val (245) | 80 | 2.6 | −6.58 | 0.28 | 262 | 1.5 | 3 |
| GlnVI:23Ala (248) | 73 | 8.4 | −5.90 | 0.39 | 1270 | 3.3 | 3 |
| GluVII:02Ala (261) | 71 | 9.2 | −5.69 | 0.28 | 2040 | 12.0 | 3 |
| ArgVII:03Ala (262) | 20* | 2.7 | −5.54 | 0.17 | 2860 | 17.0 | 3 |
| TrpVII:06Ala (265) | – | – | – | – | – | – | 4 |
| GlyVII:09Val (268) | 43* | 2.5 | −5.79 | 0.30 | 1630 | 9.6 | 3 |
| ECL-2b | |||||||
| PheC+2Ala (157) | – | – | – | – | – | – | 3 |
| PheC+3Ala (158) | 19* | 2.9 | –5.12 | 0.51 | 7816 | 21.0 | 4 |
Statistically significant changes from wild type are marked with an asterisk
Residue positions are given in parentheses after the Schwartz/Baldwin name.
Two phenylalanines in ECL-2b of GPR119 are important for ligand potency
In contrast to the constitutive signalling of GPR119, where nearly all substitutions in ECL-2b had major effects, only Ala substitutions of the two neighbouring phenylalanines, PheC+2 and PheC+3, seriously affected the agonist-induced signalling, that is, the potency of AR231453 was impaired 77- and 34-fold, respectively, and OEA had no effect in the PheC+2 to Ala mutant and was impaired 22-fold by the PheC+3 mutant. Ala substitutions of SerC+1, AlaC+4, PheC+6 and HisV:01 had no effect on agonist potency, whereas Ala substitution of ValC+5 decreased the AR231453 potency 3.9-fold (Figure 6, Table 1).
It should be noted, however, that nearly all of the substitutions in ECL-2b, which decreased the constitutive activity, also decreased the efficacy of AR231453, that is, by around 40% (Figure 4), indicating that ECL-2b is generally important for the stabilization of the active conformation of GPR119, although many of the mutations did not affect agonist potency.
Molecular models of predicted binding modes of AR231453 in GPR119
Initially, we compared the molecular models built over single GPCR X-ray structures with two models, Combi1 and Combi2, based on a composed multiple template approach in which structural fragments from three different GPCR X-ray structures were joined: A2A, CXC4 and dopamine D3 receptors. For example, the structure of ECL-3 from the D3 receptor was selected because it has an internal disulphide bridge similar to the one in GPR119 (Figure 1). The models were optimized using full-atom structure relaxation with Rosetta, and the 30% best scoring models were subjected to cluster analysis and 43 non-redundant energetically feasible models corresponding to the lowest energy structures from each cluster were used as a receptor ensemble for ligand docking (see Methods for details). The energy of the models based on multiple templates was approximately 10 Rosetta energy units (REU) lower compared with the best models developed from single-template structures (Figure 7A). The Combi2 model was used for further analysis.
Figure 7.

Molecular model of proposed binding mode for AR231453 in GPR119. (A) Rosetta conformational energies of relaxed GPR119 homology models based on the single-template structures of adenosine A2A receptors (PDBID:3EML), CXCR4 (PDBID:3ODU), dopamine D3 receptors (PDBID:3PBL), histamine H1 receptors (PDBID:3RZE) and muscarinic M2 receptors (PDBID:3UON), and two GPR119 models (Combi1 and Combi2) based on hybrid template structures assembled by fragments from the adenosine A2A, CXCR4 and the dopamine D3 receptors. (B) Density of computed distances between the isopropyl moiety of the ligand and a point in the extracellular loop region and computed distances between the sulfonyl moiety and a point in the bottom of the main binding site for the top 10% complexes with the best scored docking pose. (C) Ligplot illustrating predicted receptor-ligand interactions in good agreement with the mutational data. (D) Extracellular and membrane view for (i) the most populated AR231453 binding conformation based on clustering; (ii) the best scored SO2-up; and (iii) the best scored SO2-down binding mode of AR231453 among the 10 most populated clusters. AR231453 is shown as sphere with green carbons. Mutated residues are shown in sticks and colour coded as described in previous figures.
A conformational ensemble of low-energy structures of AR231453 in Combi2 was generated and docked 1000 times to each of the 43 selected binding pocket ensembles using RosettaLigand with full receptor and ligand flexibility. To elucidate possible binding modes of AR231453 in the GPR119 receptor, we analysed the top 10% best scoring complexes (4300) with respect to ligand interaction energy.
Two overall ligand-binding possibilities were identified in which AR231453 binds in the main ligand-binding pocket in an extended conformation perpendicular to the plane of the membrane between TM-III, -V, -VI and -VII, where the sulfonyl moiety of the ligand is either pointing to the ECL region, which we call SO2-up, or towards the bottom of the main ligand-binding pocket, SO2-down. The SO2-up binding pose was observed in approximately 80% of the top 10% best scored models energy-wise from the docking simulation (Figure 7B). The analysis of the 10 most populated binding modes identified by clustering based on RMSD between distinct ligand-binding modes showed that six clusters represented the SO2-up pose and four represented the SO2-down pose. Model scores and ligand interaction energies associated with models in the most populated clusters were surprisingly similar, although a slightly better score (−36.6 REU) was observed for the best SO2-up docking pose (Figure 7D.2) compared with the best (−35.3 REU) SO2-down (Figure 7D.3). The largest, most populated cluster was with AR231453 in a SO2-up docking pose of which the best scored model is shown in Figure 7D.1.
The major AR231453 binding mode – ‘SO2-up’
Although the detailed receptor-ligand interaction varied among the best scoring clusters and models, the overall SO2-up binding mode represented by the largest clusters was similar, as illustrated in the ‘lig-plot’ (Figure 7C) and shown in the two models in Figure 7D.1–2. In this binding pose, the isopropyl-oxadiazole moiety is located deep in the main binding pocket involved in hydrophobic/aromatic interactions with AlaIII:12 and the highly conserved TrpVI:13 – and is making hydrogen bond interactions with ThrIII:09 and SerV:09 in most models. In contrast, analysis of the top scored models suggests that the effect of the GlyVII:09 mutations is most likely related to indirect effects.
The 5-nitro-pyrimidine scaffold of AR231453 is located centrally in the ligand-binding pocket and predicted to be involved in substantial aromatic/hydrophobic interactions with the key residues PheVI:16, TrpVII:06 and PheC+2 (ECL-2b) across the clusters (Figure 7D.1–3), supporting the importance of these residues in the mutational analysis. In the models associated with the cluster including the best scored SO2-up binding pose (Figure 7D.2), the nitro group is involved in polar interactions with GlnVI:23, while either of the pyridyl nitrogens (in the pyrimidine scaffold) make hydrogen bond interactions with the amine group of the indole side chain of TrpVII:06. Accordingly, GluVII:02 (located opposite to GlnVI:23) is involved in hydrogen bond interactions with the adjacent aniline linker. In models associated with the most populated binding conformation (Figure 7D.1), AR231453 is basically rotated 180° around its ‘vertical’ isopropyl-sulphonyl axis. In these models, GlnVI:23 is involved in the hydrogen bond interaction with GluVII:02 and/or either of the pyridyl nitrogens. Both of these SO2-up possibilities are supported by the effect of mutations of key residues, PheVI:16, TrpVII:06 and PheC+2, and by the moderate effect of the GlnVI:23 and GluVII:02 mutations. Importantly, no obvious polar contact seems to be formed between these residues and AR231453 in the models associated with the best scored reversed SO2-down binding mode (Figure 7D.3). In these SO2-down models, GlnVI:23 and GluVII:02 were often found to interact with each other instead of with AR231453.
With regard to ECL-2, the terminal 2-fluoro-4-methanesulfonyl-phenyl moiety of AR231453 is in the SO2-up pose surrounded by PheC+2 and PheC+3 as well as hydrophobic residues at the extracellular end of TM-VII and ECL-3, indicating that the pronounced effects of Ala substitutions of PheC+2 and PheC+3 in ECL-2b are due to the disruption of direct interactions with the agonist. In contrast, the modest effects of mutations of ValC+5 ArgIII:04 and ArgVII:03 on the potency of AR231453 are likely to be indirect as these residues mainly make interactions with other residues in the receptor.
The structural basis for the mutations which did not affect the potency of AR231453 (e.g. GlnII:24, MetIII:05, ValIII:08, HisV:01 and GlyVI:20) could in most cases be explained by their limited interaction or remote positions to AR231453 (Figure 7C and D.1–3).
In conclusion, although both SO2-down and SO2-up poses are energetically allowed and found in surprisingly comparable numbers of clusters, the best scoring models and the most populated clusters of models for AR231453 in its ‘vertical’ binding pocket in GPR119 correspond to SO2-up poses, where it, for example, makes strong polar interactions with key residues in the middle of the pocket.
Discussion and conclusions
In the present study, the structural basis for both the ligand-induced and constitutive signalling of the enteroendocrine lipid sensor GPR119 was characterized by mutational analysis and molecular modelling. Although there are key differences, a surprisingly large degree of overlap was found between residues located at the extracellular ends of TM-III, TM-V, TM-VI and TM-VII, and in ECL-2b being important for both ligand-independent and ligand-induced signalling. For the prototype synthetic GPR119 ligand AR231453, a picture is emerging, where it activates the receptor by binding in a tight vertical pocket, most likely with its isopropyl-oxadiazole moiety penetrating deep into the receptor interacting with key micro-switch residues, and with the 2-fluoro-4-methanesulfonyl-phenyl moiety at the opposite end of the molecule pointing towards the extracellular space interacting with residues in ECL-2b.
Structural basis for GPR119 constitutive activity
Although it was initially reported that one of the earliest compounds selective for GPR119 was an inverse agonist (Semple et al., 2008), very little focus has since been placed on the fact that GPR119 is highly constitutively active, at least when expressed in transfected cells. As GPR119 is activated by a range of different fatty acid derivatives (Hansen et al., 2012), the observed apparent constitutive activity might in fact instead be mediated by some unknown lipid ligand produced by the cells. However, we did observe key differences between residues being important for constitutive activity and residues important for OEA-induced activation. This is most clear for the TrpVII:06 to Ala substitution, which did not affect the high constitutive activity of the receptor, but eliminated the activity of OEA. This suggests that GPR119 is truly constitutively active. But it is still possible that some other cellular-derived, unidentified ligand binding slightly different from OEA is constitutively activating the receptor. To elucidate this, an inverse agonist without antagonistic properties would be useful, so that inhibition of constitutive activity could be distinguished from that induced by blocking endogenously produced GPR119 ligands, as demonstrated in the ghrelin system by Holst and co-workers (Holst et al., 2003; Petersen et al., 2009).
The structural basis for the constitutive activity of GPR119 is particularly dependent upon two arginine residues in the TM region – ArgIII:04 and ArgVII:03 – and surprisingly almost the entire ECL-2b. Because ECL-2b displays great aromaticity, it could be speculated that cationic π-interactions between the arginine residues in the upper part of the main ligand-binding pocket and the phenylalanines in ECL-2b could be stabilizing the ligand-independent active receptor conformation. However, our current models of GPR119 in complex with agonist ligands do not support that notion.
In the ghrelin receptor, it is a particular aromatic cluster of residues – PheVI:16, PheVII:06 and PheVII:09 – located relatively deep in the main ligand-binding pocket at the interface between TM-VI and TM-VII that are important for the constitutive activity (Holst et al., 2003). Two of these residues are also found in GPR119; however, even though the PheVI:16 to Ala mutation in GPR119 also decreases the constitutive activity, the receptor still displays a constitutive activity of 11% and the TrpVII:06 to Ala mutation does not affect the ligand-independent signalling at all. Position VII:09 is a glycine in GPR119, and introduction of a phenylalanine here results in a decrease in constitutive activity. Thus, although being structurally similar, the interface of TM-VI and VII does not serve the same function in GPR119 as in the ghrelin receptor.
Structural basis for agonist activation of GPR119
The mutational mapping identified residues located all the way from the bottom of the main ligand-binding pocket, [i.e. TrpVI:13 (6.50)] to the ECL structures (i.e. PheC+2 and PheC+3 in ECL-2b) to be important for agonist-induced activation of GPR119. Between these sites, in particular AlaIII:12, PheVI:16, TrpVII:06 and GlyVII:09 were important for AR231453-induced activation of GPR119. These are all residues facing the main ligand-binding pocket, which recently have been confirmed to be involved in ligand binding in a large series of X-ray structures of receptor-ligand complexes (Nygaard et al., 2009; Katritch et al., 2013). In fact, the elongated overall shape of AR231453 and its proposed vertical binding mode in the main ligand-binding pocket is somewhat similar to the antagonist ZM241385 and its binding in the A2A receptors (Jaakola et al., 2008; Nygaard et al., 2009). Interestingly, the mutational map for the endogenous high-potency agonist OEA and for the prototype synthetic agonist AR231453 was similar – including the two Phes in ECL-2b. This is rather surprising because the structures of the two ligands are very different and because OEA is a highly flexible ligand (Figure 6). In relation to agonist binding, it should be noted that activating metal ion sites have been built between positions VI:16 (6.45) and III:08 (3.32) in the β2-adrenoceptor and the κ-opioid receptor (Thirstrup et al., 1996; Elling et al., 2006), that is, right in the middle of the proposed binding site for AR231453 in GPR119. An interesting key residue involved in agonist binding in GPR119 is VII:06. This position is well known, particularly in chemokine receptors, which nearly all have a glutamic acid residue at this position serving as an anchor point for a centrally located positively charged nitrogen of the different types of ligands (Rosenkilde and Schwartz, 2006; Jensen et al., 2007; Thiele et al., 2012). GPR119 is unique in having a Trp residue at position VII:06, which is not found in any other 7TM receptors (Mirzadegan et al., 2003).
The many high-resolution X-ray structures of different GPCRs have revealed that ECL-2b is a very important structural element, as it not only borders the entrance to the main ligand-binding pocket, but importantly, residues in this loop are involved in direct ligand interactions in most receptors that have, at present, been characterized (Nygaard et al., 2009; Peeters et al., 2014). For GPR119, it is interesting that key residues in ECL-2b, that is, PheC+2, PheC+3 and ValC+5, are conserved between not only the human and mouse homologues but even in zebrafish GPR119, despite the fact that human and zebrafish GPR119 only share 35% identity. Normally, residues in the extracellular domains of 7TM receptors vary the most. The surprising conservation of these residues in ECL-2b supports the notion that this loop has a major functional role in GPR119 either as a ‘tethered agonist’ – leading to the apparent high ligand-independent constitutive activity receptor – or is involved in binding some unknown autocrine or paracrine lipid agonist. In the ghrelin receptor, a naturally occurring mutation in ECL-2b has been shown to completely eliminate the constitutive activity (Holst and Schwartz, 2006; Mokrosinski et al., 2012).
Molecular modelling of GPR119 agonist complex
In general, molecular models are built over the X-ray structure of the receptor which is most closely related to the target of interest. However, here we found that molecular models with apparent lower energy can be generated when different segments are sampled from different receptors based on local and not overall homology, that is, using a composed template as proposed by Worth et al. (2011). Docking of ligands in molecular models is far from trivial, particularly when no experimental data are available (Kufareva et al., 2011). However, as demonstrated in a recent benchmark study where we ‘re-docked’ ligands into known X-ray structures as if they were molecular models using full flexible receptor-ligand simulations, the results are much improved if experimental data are available concerning ligand-receptor interactions (Nguyen et al., 2013). Consequently we believe that the preferred docking pose for AR4231453 presented in the present study (Figure 7C and D.1) is probably quite close to the real binding mode for this prototype GPR119 agonist.
In conclusion, the present study demonstrates that GPR119 signals with high constitutive activity through Gαs which should be taken into account in the drug discovery process. Moreover, the mutational mapping and novel insight into the receptor binding mode of agonists will improve structure-based drug discovery efforts for GPR119 to control gut hormone and insulin secretion.
Acknowledgments
The Novo Nordisk Foundation Center for Basic Metabolic Research (http://www.metabol.ku.dk) is supported by an unconditional grant from the Novo Nordisk Foundation to University of Copenhagen. M. S. E. and M. H. P. were supported by a PhD scholarship from the Faculty of Health and Medical Sciences, University of Copenhagen.
Glossary
- 7TM
seven transmembrane
- ECL
extracellular loop
- OEA
oleoylethanolamide
- REU
Rosetta energy units
- RMSD
root mean square deviation
Author contributions
M. S. E., C. N., M. H. P., N. D. H. and T. M. F. performed the research. M. S. E., C. N., N. D. H., T. M. F. and T. W. S. designed the research study. L. E., J. L., R. M. J., C. N. and T. M. F. contributed essential reagents or tools. M. S. E., C. N., N. D. H., T. M. F. and T. W. S. analysed the data. M. S. E., C. N., T. M. F. and T. W. S. wrote the paper.
Conflict of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12877
Supplementary methods.
References
- Adham N, Bonini J, Borowsky B, Boyle N, Thompson T. 2003. DNA encoding SNORF25 receptor. U.S. Pat. Appl. Publ. US20030125539 A1.
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander N, Woetzel N, Meiler J. 2011. pp. 13–18. BCL::Cluster: a method for clustering biological molecules coupled with visualization in the Pymol Molecular Graphics System. Computational Advances in Bio and Medical Sciences (ICCABS), IEEE 1st International Conference. [DOI] [PMC free article] [PubMed]
- Barth P, Schonbrun J, Baker D. Toward high-resolution prediction and design of transmembrane helical protein structures. Proc Natl Acad Sci U S A. 2007;104:15682–15687. doi: 10.1073/pnas.0702515104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini JA, Borowsky BE, Montclair NA, Ridgewood NB, Cliffside P, Thompson TO. 2001. DNA encoding SNORF25 receptor. United States Patent US 6,221,660 B1.
- Chu ZL, Jones RM, He H, Carroll C, Gutierrez V, Lucman A, et al. A role for beta-cell-expressed G protein-coupled receptor 119 in glycemic control by enhancing glucose-dependent insulin release. Endocrinology. 2007;148:2601–2609. doi: 10.1210/en.2006-1608. [DOI] [PubMed] [Google Scholar]
- Chu ZL, Carroll C, Alfonso J, Gutierrez V, He H, Lucman A, et al. A role for intestinal endocrine cell-expressed g protein-coupled receptor 119 in glycemic control by enhancing glucagon-like Peptide-1 and glucose-dependent insulinotropic peptide release. Endocrinology. 2008;149:2038–2047. doi: 10.1210/en.2007-0966. [DOI] [PubMed] [Google Scholar]
- Chu ZL, Carroll C, Chen R, Alfonso J, Gutierrez V, He H, et al. N-oleoyldopamine enhances glucose homeostasis through the activation of GPR119. Mol Endocrinol. 2010;24:161–170. doi: 10.1210/me.2009-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis IW, Baker D. RosettaLigand docking with full ligand and receptor flexibility. J Mol Biol. 2009;385:381–392. doi: 10.1016/j.jmb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Dhayal S, Morgan NG. The significance of GPR119 agonists as a future treatment for type 2 diabetes. Drug News Perspect. 2010;23:418–424. doi: 10.1358/dnp.2010.23.7.1468395. [DOI] [PubMed] [Google Scholar]
- Elling CE, Frimurer TM, Gerlach LO, Jorgensen R, Holst B, Schwartz TW. Metal ion site engineering indicates a global toggle switch model for seven-transmembrane receptor activation. J Biol Chem. 2006;281:17337–17346. doi: 10.1074/jbc.M512510200. [DOI] [PubMed] [Google Scholar]
- Fleishman SJ, Leaver-Fay A, Corn JE, Strauch EM, Khare SD, Koga N, et al. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS ONE. 2011;6:e20161. doi: 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson R, Hoglund PJ, Gloriam DE, Lagerstrom MC, Schioth HB. Seven evolutionarily conserved human rhodopsin G protein-coupled receptors lacking close relatives. FEBS Lett. 2003;554:381–388. doi: 10.1016/s0014-5793(03)01196-7. [DOI] [PubMed] [Google Scholar]
- Graham FL, van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- Griffin G. 2006. Methods for identification of modulators of OSGPR116 activity. United States Patent US 7083933 B1.
- Guan XM, Kobilka TS, Kobilka BK. Enhancement of membrane insertion and function in a type IIIb membrane protein following introduction of a cleavable signal peptide. J Biol Chem. 1992;267:21995–21998. [PubMed] [Google Scholar]
- Hansen HS, Rosenkilde MM, Holst JJ, Schwartz TW. GPR119 as a fat sensor. Trends Pharmacol Sci. 2012;33:374–381. doi: 10.1016/j.tips.2012.03.014. [DOI] [PubMed] [Google Scholar]
- Holst B, Schwartz TW. Ghrelin receptor mutations – too little height and too much hunger. J Clin Invest. 2006;116:637–641. doi: 10.1172/JCI27999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst B, Cygankiewicz A, Jensen TH, Ankersen M, Schwartz TW. High constitutive signaling of the ghrelin receptor – identification of a potent inverse agonist. Mol Endocrinol. 2003;17:2201–2210. doi: 10.1210/me.2003-0069. [DOI] [PubMed] [Google Scholar]
- Holst B, Nygaard R, Valentin-Hansen L, Bach A, Engelstoft MS, Petersen PS, et al. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J Biol Chem. 2010;285:3973–3985. doi: 10.1074/jbc.M109.064725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PC, Nygaard R, Thiele S, Elder A, Zhu G, Kolbeck R, et al. Molecular interaction of a potent nonpeptide agonist with the chemokine receptor CCR8. Mol Pharmacol. 2007;72:327–340. doi: 10.1124/mol.106.035543. [DOI] [PubMed] [Google Scholar]
- Jones RM. 2006. Discovery of agonists of the glucose dependent insulinotropic receptor, GPR119, a pancreatic beta-cell oGPCR, for the treatment of NIDDM. 232nd ACS National Meeting, San Francisco, CA, United States, MEDI-275.
- Jones RM, Leonard JN, Buzard DJ, Lehmann J. GPR119 agonists for the treatment of type 2 diabetes. Expert Opin Ther Pat. 2009;19:1339–1359. doi: 10.1517/13543770903153878. [DOI] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogure R, Toyama K, Hiyamuta S, Kojima I, Takeda S. 5-Hydroxy-eicosapentaenoic acid is an endogenous GPR119 agonist and enhances glucose-dependent insulin secretion. Biochem Biophys Res Commun. 2011;416:58–63. doi: 10.1016/j.bbrc.2011.10.141. [DOI] [PubMed] [Google Scholar]
- Kufareva I, Rueda M, Katritch V, Stevens RC, Abagyan R, Participants GD. Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure. 2011;19:1108–1126. doi: 10.1016/j.str.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan H, Vassileva G, Corona A, Liu L, Baker H, Golovko A, et al. GPR119 is required for physiological regulation of glucagon-like peptide-1 secretion but not for metabolic homeostasis. J Endocrinol. 2009;201:219–230. doi: 10.1677/JOE-08-0453. [DOI] [PubMed] [Google Scholar]
- Lan H, Lin HV, Wang CF, Wright MJ, Xu S, Kang L, et al. Agonists at GPR119 mediate secretion of GLP-1 from mouse enteroendocrine cells through glucose-independent pathways. Br J Pharmacol. 2012;165:2799–2807. doi: 10.1111/j.1476-5381.2011.01754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffer LM, Iakoubov R, Brubaker PL. GPR119 is essential for oleoylethanolamide-induced glucagon-like peptide-1 secretion from the intestinal enteroendocrine L-cell. Diabetes. 2009;58:1058–1066. doi: 10.2337/db08-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon G, Meiler J. Rosetta Ligand docking with flexible XML protocols. Methods Mol Biol. 2012;819:143–155. doi: 10.1007/978-1-61779-465-0_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiler J, Baker D. ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins. 2006;65:538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- Mirzadegan T, Benko G, Filipek S, Palczewski K. Sequence analyses of G-protein-coupled receptors: similarities to rhodopsin. Biochemistry. 2003;42:2759–2767. doi: 10.1021/bi027224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobarec JC, Sanchez R, Filizola M. Modern homology modeling of G-protein coupled receptors: which structural template to use? J Med Chem. 2009;52:5207–5216. doi: 10.1021/jm9005252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokrosinski J, Frimurer TM, Sivertsen B, Schwartz TW, Holst B. Modulation of constitutive activity and signaling bias of the ghrelin receptor by conformational constraint in the second extracellular loop. J Biol Chem. 2012;287:33488–33502. doi: 10.1074/jbc.M112.383240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen ED, Norn C, Frimurer TM, Meiler J. Assessment and challenges of ligand docking into comparative models of G-protein coupled receptors. PLoS ONE. 2013;8:e67302. doi: 10.1371/journal.pone.0067302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Y, O'Neill K, Lan H, Pang L, Shan LX, Hawes BE, et al. Endogenous and synthetic agonists of GPR119 differ in signalling pathways and their effects on insulin secretion in MIN6c4 insulinoma cells. Br J Pharmacol. 2008;155:1056–1065. doi: 10.1038/bjp.2008.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R, Frimurer TM, Holst B, Rosenkilde MM, Schwartz TW. Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol Sci. 2009;30:249–259. doi: 10.1016/j.tips.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Ohishi T, Yoshida S. The therapeutic potential of GPR119 agonists for type 2 diabetes. Expert Opin Investig Drugs. 2012;21:321–328. doi: 10.1517/13543784.2012.657797. [DOI] [PubMed] [Google Scholar]
- Ohishi T, Takasaki J, Matsumoto M, Saito T, Kamohara M, Soga T, et al. Method of screening remedy for diabetes. PCT International Application. 2002 WO 2002044362 A1. [Google Scholar]
- Overton HA, Babbs AJ, Doel SM, Fyfe MC, Gardner LS, Griffin G, et al. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006;3:167–175. doi: 10.1016/j.cmet.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Overton HA, Fyfe MC, Reynet C. GPR119, a novel G protein-coupled receptor target for the treatment of type 2 diabetes and obesity. Br J Pharmacol. 2008;153(Suppl. 1):S76–S81. doi: 10.1038/sj.bjp.0707529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker HE, Habib AM, Rogers GJ, Gribble FM, Reimann F. Nutrient-dependent secretion of glucose-dependent insulinotropic polypeptide from primary murine K cells. Diabetologia. 2009;52:289–298. doi: 10.1007/s00125-008-1202-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters MC, van Westen GJ, Li Q, Ap IJ. Importance of the extracellular loops in G protein-coupled receptors for ligand recognition and receptor activation. Trends Pharmacol Sci. 2011;32:35–42. doi: 10.1016/j.tips.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Petersen PS, Woldbye DP, Madsen AN, Egerod KL, Jin C, Lang M, et al. In vivo characterization of high Basal signaling from the ghrelin receptor. Endocrinology. 2009;150:4920–4930. doi: 10.1210/en.2008-1638. [DOI] [PubMed] [Google Scholar]
- Reilly MA. G-protein receptor. 2001. Eur. Pat. Appl. EPO Patent 1092727-A2.
- Reimann F, Habib AM, Tolhurst G, Parker HE, Rogers GJ, Gribble FM. Glucose sensing in L cells: a primary cell study. Cell Metab. 2008;8:532–539. doi: 10.1016/j.cmet.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkilde MM, Schwartz TW. GluVII:06 – a highly conserved and selective anchor point for non-peptide ligands in chemokine receptors. Curr Top Med Chem. 2006;6:1319–1333. doi: 10.2174/15680266106061319. [DOI] [PubMed] [Google Scholar]
- Sakamoto Y, Inoue H, Kawakami S, Miyawaki K, Miyamoto T, Mizuta K, et al. Expression and distribution of Gpr119 in the pancreatic islets of mice and rats: predominant localization in pancreatic polypeptide-secreting PP-cells. Biochem Biophys Res Commun. 2006;351:474–480. doi: 10.1016/j.bbrc.2006.10.076. [DOI] [PubMed] [Google Scholar]
- Schwartz TW, Frimurer TM, Holst B, Rosenkilde MM, Elling CE. Molecular mechanism of 7TM receptor activation – a global toggle switch model. Annu Rev Pharmacol Toxicol. 2006;46:481–519. doi: 10.1146/annurev.pharmtox.46.120604.141218. [DOI] [PubMed] [Google Scholar]
- Semple G, Fioravanti B, Pereira G, Calderon I, Uy J, Choi K, et al. Discovery of the first potent and orally efficacious agonist of the orphan G-protein coupled receptor 119. J Med Chem. 2008;51:5172–5175. doi: 10.1021/jm8006867. [DOI] [PubMed] [Google Scholar]
- Shah U. GPR119 agonists: a promising new approach for the treatment of type 2 diabetes and related metabolic disorders. Curr Opin Drug Discov Devel. 2009;12:519–532. [PubMed] [Google Scholar]
- Soga T, Ohishi T, Matsui T, Saito T, Matsumoto M, Takasaki J, et al. Lysophosphatidylcholine enhances glucose-dependent insulin secretion via an orphan G-protein-coupled receptor. Biochem Biophys Res Commun. 2005;326:744–751. doi: 10.1016/j.bbrc.2004.11.120. [DOI] [PubMed] [Google Scholar]
- Thiele S, Malmgaard-Clausen M, Engel-Andreasen J, Steen A, Rummel PC, Nielsen MC, et al. Modulation in selectivity and allosteric properties of small-molecule ligands for CC-chemokine receptors. J Med Chem. 2012;55:8164–8177. doi: 10.1021/jm301121j. [DOI] [PubMed] [Google Scholar]
- Thirstrup K, Elling CE, Hjorth SA, Schwartz TW. Construction of a high affinity zinc switch in the kappa-opioid receptor. J Biol Chem. 1996;271:7875–7878. doi: 10.1074/jbc.271.14.7875. [DOI] [PubMed] [Google Scholar]
- Worth CL, Kreuchwig A, Kleinau G, Krause G. GPCR-SSFE: a comprehensive database of G-protein-coupled receptor template predictions and homology models. BMC Bioinformatics. 2011;12:185. doi: 10.1186/1471-2105-12-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary methods.