

Abstract
Background and Purpose
Ceramide kinase (CerK) catalyzes the generation of ceramide-1-phosphate which may regulate various cellular functions, including inflammatory reactions and cell growth. Here, we studied the effect of a recently developed CerK inhibitor, NVP-231, on cancer cell proliferation and viability and investigated the role of cell cycle regulators implicated in these responses.
Experimental Approach
The breast and lung cancer cell lines MCF-7 and NCI-H358 were treated with increasing concentrations of NVP-231 and DNA synthesis, colony formation and cell death were determined. Flow cytometry was performed to analyse cell cycle distribution of cells and Western blot analysis was used to detect changes in cell cycle regulator expression and activation.
Key Results
In both cell lines, NVP-231 concentration-dependently reduced cell viability, DNA synthesis and colony formation. Moreover it induced apoptosis, as measured by increased DNA fragmentation and caspase-3 and caspase-9 cleavage. Cell cycle analysis revealed that NVP-231 decreased the number of cells in S phase and induced M phase arrest with an increased mitotic index, as determined by increased histone H3 phosphorylation. The effect on the cell cycle was even more pronounced when NVP-231 treatment was combined with staurosporine. Finally, overexpression of CerK protected, whereas down-regulation of CerK with siRNA sensitized, cells for staurosporine-induced apoptosis.
Conclusions and Implications
Our data demonstrate for the first time a crucial role for CerK in the M phase control in cancer cells and suggest its targeted inhibition, using drugs such as NVP-231, in combination with conventional pro-apoptotic chemotherapy.
Tables of Links
| TARGETS | |
| Caspase-3 | E3 ubiquitin protein ligase |
| Caspase-9 | Glucosylceramide synthase |
| CDK (cyclin-dependent kinase) | Glycogen synthase kinase-3β |
| CDK1 | p42-MAPK |
| CDK2 | p44-MAPK |
| CDK4 | PI3K |
| Ceramide | Sphingosine kinase 1 |
| CerK (ceramide kinase) | Wee1 kinase |
| Cyclin B1 (kalirin, RhoGEF kinase) |
| LiGANDS |
| NVP-231 |
| S1P (sphingosine 1-phosphate) |
| Staurosporine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Sphingolipids are essential structural components of cellular membranes but several subspecies were also shown to act as signalling molecules. Many studies have proven their key role in the regulation of cell viability and division. Especially, the role of ceramide and sphingosine 1-phosphate (S1P) in cell growth and death have been extensively studied over the last decades. Whereas ceramide exerts antiproliferative and pro-apoptotic effects, S1P seems to be a counter molecule to ceramide as in many cell types it exerts opposite effects, such as promoting cell proliferation and cell survival (Huwiler and Pfeilschifter, 2006; Huwiler and Zangemeister-Wittke, 2007; Pyne and Pyne, 2010; Van Brocklyn and Williams, 2012).
Recently, another phosphorylated sphingolipid species, ceramide 1-phosphate (C1P), has attracted attention as it was suggested to regulate various cellular functions, such as the release of synaptic vesicles (Shinghal et al., 1993), phagocytosis (Hinkovska-Galcheva et al., 1998), mast cell degranulation (Mitsutake et al., 2004), inflammatory reactions (Pettus et al., 2003), proliferation (Gomez-Muñoz et al., 1995; 1997,) and angiogenesis (Niwa et al., 2009) (reviewed in: Bornancin, 2011). Although C1P was suggested to trigger cell proliferation and migration (Gomez-Muñoz et al., 1995; 1997,), this effect was mainly seen when adding exogenous C1P to cell cultures. Regarding the migratory effect of exogenous C1P, it was proposed that it acts through a putative C1P receptor which was shown to be pertussis toxin sensitive (Granado et al., 2009). However, very high concentrations of C1P in the range of 30–50 μM were needed for this suggesting that if indeed a receptor is involved, interaction is of low affinity. So far, no high-affinity C1P receptor has been identified.
C1P is generated by the action of a ceramide kinase (CerK), which was first described in brain synaptic vesicles where it co-purified with synaptic vesicle markers (Bajjalieh et al., 1989). Meanwhile, CerK has been cloned from various species including mouse and human (Sugiura et al., 2002). CerK is a 537 amino acid protein showing homology to the DAG kinase and sphingosine kinase 1 but having strong substrate specificity for ceramides including the short-chain analogues C6-, C8- and C16-ceramide (Sugiura et al., 2002). CerK was shown to be localized in various subcellular compartments including Golgi, cytoplasm and nucleus (Carré et al., 2004; Rovina et al., 2009). The N-terminal sequence of CerK is thought to be myristoylated and to contain a pleckstrin homology domain, which is important for the Golgi localization (Carré et al., 2004). Furthermore, it was shown that the N-terminal region contains a nuclear import signal whereas the C-terminal region contains a nuclear export signal (Rovina et al., 2009). These findings led to the hypothesis that a nucleocytoplasmic shuttling of CerK represents a novel mechanism of regulation. In view of these reports, it is appealing that the primary site of C1P generation is inside the cell and consequently, the primary site of action may also be in the intracellular space, especially considering that so far no transporter of C1P has been identified.
In the present study, we investigated the effect of a recently developed catalytic inhibitor of CerK NVP-231 (Graf et al., 2008a) on the growth and survival of the breast and lung cancer cell lines MCF-7 and NCI-H358. We demonstrated that this inhibitor concentration-dependently reduces cell viability, DNA synthesis and colony formation, and that cells arrest in M phase of the cell cycle and subsequently undergo apoptosis. The death-promoting effect of NVP-231 was reduced when CerK was overexpressed in the cells. Furthermore, the combination of NVP-231 with staurosporine led to a synergistic effect on apoptosis induction. These findings demonstrate a crucial role of CerK in mitosis regulation and suggest that targeted CerK inhibition should be combined with pro-apoptotic anti-cancer agents for therapy.
Methods
Cell culturing and stimulation
The human lung cancer cell line NCI-H358 and the human breast cancer cell line MCF-7 were obtained from the American Type Culture Collection (Manassas, VA, USA). NCI-H358 cells were cultured as previously described (Huwiler et al., 2011). MCF-7 cells were cultured in DMEM supplemented with 10% (v·v−1) FBS, 10 mM HEPES pH 7.4, 6 μg·mL−1 insulin, 100 units·mL−1 penicillin, 100 μg·mL−1 streptomycin, and non-essential amino acids. Throughout all the experiments, cells were grown at 37°C in a humidified atmosphere containing 5% (v·v−1) CO2. Stimulated cells were homogenized in lysis buffer (Huwiler and Pfeilschifter, 2006; Huwiler et al., 2006) and centrifuged for 10 min at 13 000× g. The supernatant was taken for protein determination and 30 μg of protein was separated by SDS-PAGE, transferred to nitrocellulose membrane and subjected to Western blot analysis, as previously described (Huwiler and Pfeilschifter, 2006; Huwiler et al., 2006), using the antibodies indicated in the figure legends.
Cell transfection
For stable CerK overexpression, 4 × 105 MCF-7 cells per well were seeded in 6-well plates to be 50–70% confluent after 1 day. Cells were then incubated for 24 h in serum-free DMEM containing a mixture of 2 μg·mL−1 plasmid DNA (pcDNA3.1 plus human CerK cDNA) and 4 μL·mL−1 of TurboFectR transfection reagent (Fermentas GmbH, St. Leon-Rot, Germany). Control cells were transfected with the empty pcDNA3.1 vector plus TurboFect. Thereafter, transfected cells were maintained for at least 3 weeks in complete medium containing 1 mg·mL−1 G418 for selection before experiments were performed. For CerK down-regulation, cells were transfected with oligofectamine and 100 nM of a Smartpool siRNA of hCerK (Dharmacon RNAi, L-004061; Fisher Scientific, Wohlen, Switzerland) as recommended by the manufacturer. Forty-eight hours after transfection, cells were subcultured into 96-well plates for the DNA fragmentation assay. After 24 h, cells were treated as indicated in the figure legends.
Cellular CerK activity assay
The cellular CerK activity assay was performed as described by Boath et al. (2008). The cellular IC50 value was calculated by using the Prism 5.03 software (GraphPad Software Inc., LaJolla, CA, USA) and by performing a non-linear curve fit.
DNA synthesis/proliferation assay
Cells were plated in a 96-well plate (1 × 104 cells per well) and maintained overnight in growth medium. The cells were then treated as indicated and for the last 24 h 5-bromo-2-deoxyuridine (BrdU) was added to culture medium. Incorporated BrdU was detected using the Cell Proliferation BrdU elisa kit according to the manufacturer's instructions (Roche Diagnostics GmbH, Mannheim, Germany).
AlamarBlue cell viability assay
Cell viability was measured using alamarBlueR reagent (resazurin) (Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA) (Larson et al., 1997). Cells were plated in a black 96-well plate (7.5 × 103 cells per well) and maintained overnight in complete medium. The cells were then treated as indicated and for the last 4 h, alamarBlue reagent was added. Intensity of fluorescence reflecting cell viability was measured at 544/590 nm excitation/emission wavelengths with a SpectraMax microplate reader (Molecular Devices, LLC, Sunnyvale, CA, USA). Alternatively, cell viability was detected by flow cytometry. For this, treated cells were trypsinized, washed once with PBS and resuspended in PBS containing 1% FCS and 10 μg·mL−1 propidium iodide. After 1 min of incubation at 25°C, cells were immediately analysed by flow cytometry. Propidium iodide (PI)-positive staining was detected in FL3 channel using a FACSCalibur (Becton & Dickinson, San Jose, CA, USA). At least 20 000 cells were counted in each sample.
Colony formation assay
Cells were subcultured in 60 mm diameter dishes at a density of 1000 cells per dish. After 24 h, cells were treated as indicated in growth medium and incubated for further 10 days (NCI-H358) or 14 days (MCF-7) to allow colony formation. Cells were then washed with PBS, air dried and stained for 30 min with 2% (w·v−1) crystal violet. The number of cell colonies was quantified by using a ColCount™ (Mammalian Cell Colony Counter, Oxford Optronix, Oxford, UK). Only colonies bigger than 50 cells were included in the counting.
Cell cycle analysis by flow cytometry
Stimulated cells were detached by trypsin/EDTA and fixed in 70% (v·v−1) ethanol for at least 1 h at −20°C. Cells were then incubated for 30 min in PBS containing 10 μg·mL−1 propidium iodide, and 100 μg·mL−1 RNase A and analysed by flow cytometry for DNA content using a FACSCalibur (Becton & Dickinson). To discriminate cells in M phase from cells in G2 phase, additional staining for the mitosis marker phospho-Ser10 histone H3 was performed before PI was added to the cell samples. The phospho-H3 positive cells were detected using an anti-rabbit Alexa-488 secondary antibody in FL1 channel. At least 20 000 cells were counted for each sample.
Quantification of C16-C1P and ceramides by LC-MS/MS
C1P and ceramides were extracted and quantified as recently described (Fayyaz et al., 2014). In brief, lipids were extracted from cells using C12-C1P and C17-ceramide as internal standards. Sample analysis was carried out by rapid-resolution liquid chromatography-MS/MS using a Q-TOF 6530 mass spectrometer (Agilent Technologies, Waldbronn, Germany) operating in the positive ESI mode for ceramides and in the negative ESI mode for C1P. The precursor ions of C12-C1P (m/z 560.408), C16-C1P (m/z 616.471) and ceramides (C16-ceramide (m/z 520.508), C17-ceramide (m/z 534.524), C18-ceramide (m/z 548.540), C18:1-ceramide (m/z 546.524), C20-ceramide (m/z 576.571), C22-ceramide (m/z 604.602), C24-ceramide (m/z 632.634), C24:1-ceramide (m/z 630.618) were cleaved into the fragment ions of m/z 78.960, m/z 78.960 and m/z 264.270 respectively. Quantification was performed using Mass Hunter Software (Agilent Technologies).
Statistical analysis
Statistical analysis of experimental data was performed using one-way anova followed by a Bonferroni's post hoc test for multiple comparisons or unpaired t-tests when only two groups were compared.
Chemicals
Secondary HRP-coupled IgGs, hyperfilm MPR and enhanced chemiluminescence reagents were from GE Health Care Systems (Glattbrugg, Switzerland). C6-ceramide was from Enzo Life Sciences AG (Lausen, Switzerland). C6–NBD-ceramide was from Cayman Chemicals Inc. (Ann Arbor, MI, USA) Antibodies against GAPDH, total histone H3, cleaved caspase-3, phospho-Ser133-cyclin B1, total cyclin B1, wee1, and cyclin-dependent kinase (CDK)4 were obtained from Cell Signaling Technology Europe B.V. (Leiden, the Netherlands); phospho-Ser10 histone H3 was from EMD Millipore Inc. (Darmstadt, Germany); cleaved caspase-9 and β-actin were from Santa Cruz Biotechnology, Inc. (Heidelberg, Germany); NVP-231 and N-[2-[(3,4-dimethoxybenzoyl)amino]-1,3-benzothiazol-6-yl]adamantane-1-carboxamide (NVP-995) were provided by Novartis Pharma Inc. (Basel, Switzerland); K1 was from Merck Chemicals Ltd (Nottingham, UK). All cell culture additives were from Invitrogen AG (Basel, Switzerland).
Results
NVP-231 reduces cell viability and DNA synthesis by triggering cell death of cancer cells
Previously, a novel CerK inhibitor NVP-231 was described by Graf et al. (2008a) that inhibited the catalytic activity of recombinant CerK in vitro with an IC50 of 12 nM. This inhibitor therefore represents an attractive tool to study the cellular functions of CerK.
Here, we investigated whether NVP-231 can inhibit CerK activity in intact cancer cells, and affects cancer cell responses. To this end, the breast cancer cell line MCF-7 was stably transfected with a cDNA construct containing human CerK. Cells were then incubated with a cell permeable fluorescently labelled C6-ceramide analog, NBD-ceramide, which acted as a CerK substrate to become phosphorylated. When cells were treated with increasing concentrations of NVP-231, cellular CerK activity, as measured by NBD-C1P formation, was gradually reduced (Figure 1A) demonstrating that NVP-231 active in transfected cells. The IC50 for CerK in the cellular system was calculated to be 59.70 ± 12 nM. Moreover, we tested an inactive compound, that is NVP-995, which shows the same chemical structure but additionally possesses two methoxy groups (Graf et al., 2009). We confirmed that NVP-995 had no inhibitory activity in the cellular CerK activity assay (Figure 1A). Furthermore, we quantified cellular C16-C1P and C16-ceramide levels in NVP-231-treated MCF-7 cells. C16-C1P (Figure 1B) declined concentration-dependently upon NVP-231 treatment whereas total ceramides levels increased (Figure 1C). The different ceramide subspecies were also analysed and showed highest levels of C16-, C24- and C24:1-ceramides. The subspecies C18-, C18:1-, C20- and C22-ceramides were hardly detectable (Figure 1C).
Figure 1.

Effect of NVP-231 and NVP-995 on CerK activity in transfected MCF-7 cells. (A) MCF-7 cells that were either left untransfected (white column, −) or transfected with a cDNA of human CerK (hCerK, +) were treated for 2 h with the indicated concentrations of NVP-231 (grey columns) or NVP-995 (black column) in the presence of NBD-C6-ceramide as described in the Methods section. Cells were harvested and taken for lipid extraction. Lipids were separated by TLC and analysed on a fluorescence imaging system. The density of spots corresponding to phosphorylated NBD-ceramide was measured and results expressed as % of CerK activity compared with Wt control cells. Data are means ± SD (n = 4). (B and C) MCF-7 cells were treated for 24 h with the indicated concentrations of NVP-231. Lipids were then extracted and taken for LC-MS/MS to quantify C16-C1P (B) and the various ceramide subspecies (C). Results are expressed as pmol lipids per 106 cells and are means ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 considered statistically significant when compared with the control samples; ###P < 0.001 statistically significant when compared with the hCerK overexpressed untreated samples.
Among the cellular functions that have been reported for C1P in the literature is stimulation of cell proliferation (Gomez-Muñoz et al., 1995; 1997,; Mitra et al., 2007; Gangoiti et al., 2012) although the detailed mechanisms have remained unclear. Additionally, Ruckhäberle et al. (2009) reported that in breast cancer patients, higher CerK expression is correlated with worse prognosis even suggesting CerK as a prognostic marker in breast cancer.
Here, we investigated whether cellular CerK plays a role in cancer cell viability and proliferation, and whether inhibition of CerK has an effect on cancer cell behaviour. When treating either MCF-7 or NCI-H358 cells with NVP-231 for 48 h, viability was concentration-dependently reduced with an IC50 of about 1 μM in MCF-7 cells (Figure 2A) and 500 nM in NCI-H358 cells (Figure 2B).
Figure 2.
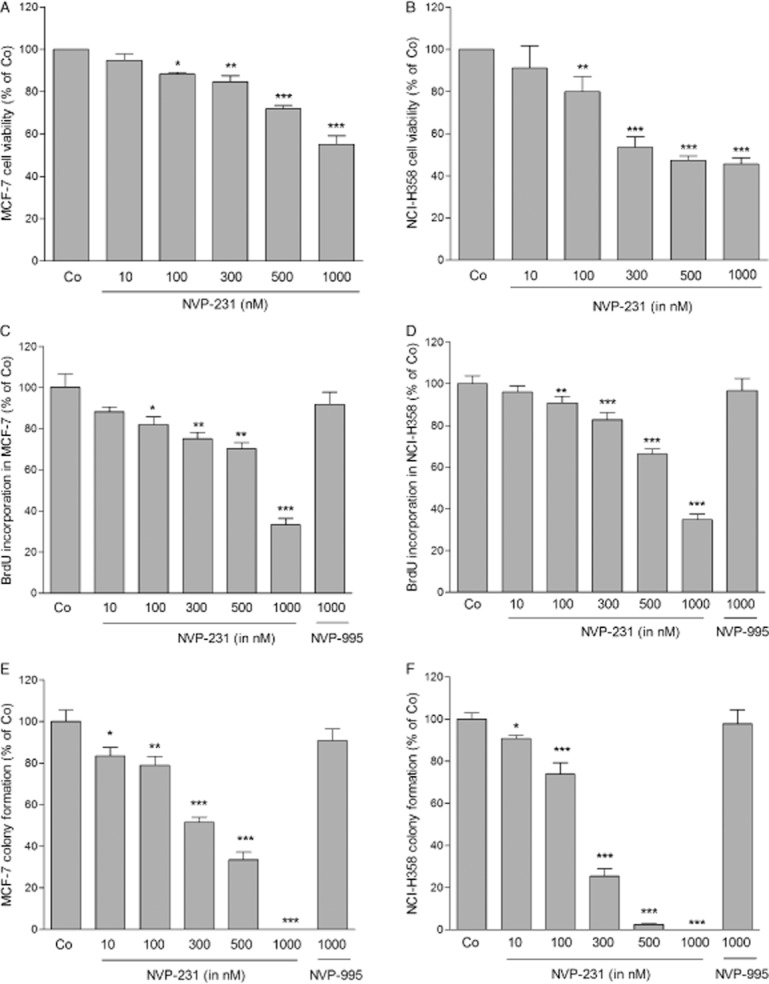
Effect of NVP-231 and NVP-995 on the viability of MCF-7 and NCI H358 cells. (A and B): MCF-7 (A) and NCI-H358 (B) cells were plated in a 96-well black plate (1 × 104 cells per well) and treated for 48 h with either vehicle (Co) or the indicated concentrations of NVP-231 (in nM). For the last 4 h of treatment, alamarBlueR was added and fluorescence was determined as described in the Methods section. Data are expressed as % of control and are means ± SD (n = 4). (C and D): MCF-7 (C) and NCI-H358 cells (D) were plated in a 96-well plate at a density of 1 × 104 cells per well and treated with the indicated concentrations NVP-231 or NVP-995 for 72 h. For the last 24 h, BrdU was added to the culture medium. Incorporated BrdU was measured by elisa using an anti-BrdU antibody according to the manufacturers' protocol. Data are expressed as % of BrdU incorporation compared with the control group and are means ± SD (n = 4). (E and F): MCF-7 cells (E) and NCI-H358 cells (F) were treated with the indicated concentrations of NVP-231 and NVP-995 in growth medium and incubated for further 10 days (NCI-H358 cells) or 14 days (MCF-7) to allow colony formation. Cells were stained with 2% (w·v−1) crystal violet and the numbers of colonies containing more than 50 cells were counted. Data are expressed as % of control and are means ± SD (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001 considered statistically significant when compared with the control groups.
We further measured the effect of NVP-231 on DNA synthesis by detecting the incorporation of BrdU into de novo synthesized DNA. NVP-231 treatment for 72 h reduced DNA synthesis in both cell lines. With 1 μM of NVP-231, the highest concentration tested, a 60–70% reduction after 72 h was detected in both cell lines (Figure 2C and D). In addition, the colony forming capacity of MCF-7 and NCI-H358 cells was monitored upon NVP-231 treatment during 10–14 days in a clonogenic assay. Both cell lines showed reduced colony formation upon NVP-231 treatment with a full inhibition obtained with 500 nM in NCI-H358 and with 1 μM in MCF-7 cells (Figure 2E and F).
To further investigate the reason for the reduced viability and DNA synthesis of both cell lines upon NVP-231 treatment, we analysed the amount of PI uptake as a measure of cell death. Upon treatment with 1 μM NVP-231, the number of PI-positive dead cells increased constantly reaching 20% in MCF-7 cells (Figure 3A) and more than 40% in NCI-H358 cells (Figure 3B) after a 72 h treatment, the latest time point analysed.
Figure 3.

Effect of NVP-231 on cell death of MCF-7 and NCI-H358 cells. MCF-7 (A and C) and NCI-H358 (B and D) cells were treated for 24–72 h with either vehicle (Co, 0) or 1 μM NVP-231. Cells were collected and stained with propidium iodide and analysed by flow cytometry to detect PI-positive cells (A and B). Data are expressed as % of cells with positive PI staining and are means ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 considered statistically significant when compared with the control groups. In parallel, cell lysates were separated by SDS-PAGE, transferred to nitrocellulose and subjected to Western blot analysis (C and D) using antibodies against cleaved caspase-3 (upper panels), cleaved caspase-9 (middle panels) and GAPDH (lower panels) at dilutions of 1:1000 each. Data show duplicate samples and are representative of three independent experiments giving similar results. (E and F) Graphs show the densitometric evaluation of Western blot data of MCF-7 (E) and NCI-H358 cells (F). Results are expressed as % of control and are means SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 considered statistically significant when compared with the control groups.
To see whether the increased cell death was due to programmed cell death, we analysed cell lysates for markers of apoptosis such as cleaved caspase-3 and caspase-9. Notably, it was previously reported that MCF-7 cells are deficient of caspase 3 (Kurokawa et al., 1999). However, the MCF-7 cell line used in this study clearly expresses caspase-3 (Figure 3C). Both cell lines responded to NVP-231 with enhanced caspase-3 (Figure 3C and E) and caspase-9 (Figure 3D and F) cleavage. However, the time course was different in the two cell lines. In MCF-7 cells, highest caspase-3 and caspase-9 cleavage and thus activation occurred at 24 h and thereafter decreased again, suggesting that at the later time points necrosis-like changes occurred which account for the high amount of PI-positive cells at 48 h and 72 h (Figure 3C and E). In NCI-H358 cells, caspase-3 and caspase-9 cleavage occurred continuously over 72 h (Figure 3D and F).
NVP-231 treatment leads to M phase arrest
We further studied the effect of NVP-231 on cell cycle progression. Notably, the normal constitutive distribution of growing cells in the cell cycle was different between the two cell lines. MCF-7 cells showed strikingly more polyploid cells (>4N DNA content) than NCI-H358 cells but less cells in S phase and less cells in the sub-G1 phase, which comprises cells with fragmented DNA typical for apoptotic/necrotic cells. Treatment of MCF-7 (Figure 4A and C upper panels) or NCI-H358 cells (Figure 4B and C lower panels) for 24 h with increasing concentrations of NVP-231 decreased the number of cells in S phase and resulted in an accumulation of cells in G2/M (4N DNA content). In parallel, more cells containing fragmented DNA were detected in the sub-G1 fraction. The amount of polyploid cells (>4N DNA content) was not affected by NVP-231 in neither of the two cell lines. As expected, the inactive compound NVP-995 did not affect cell cycle distribution.
Figure 4.
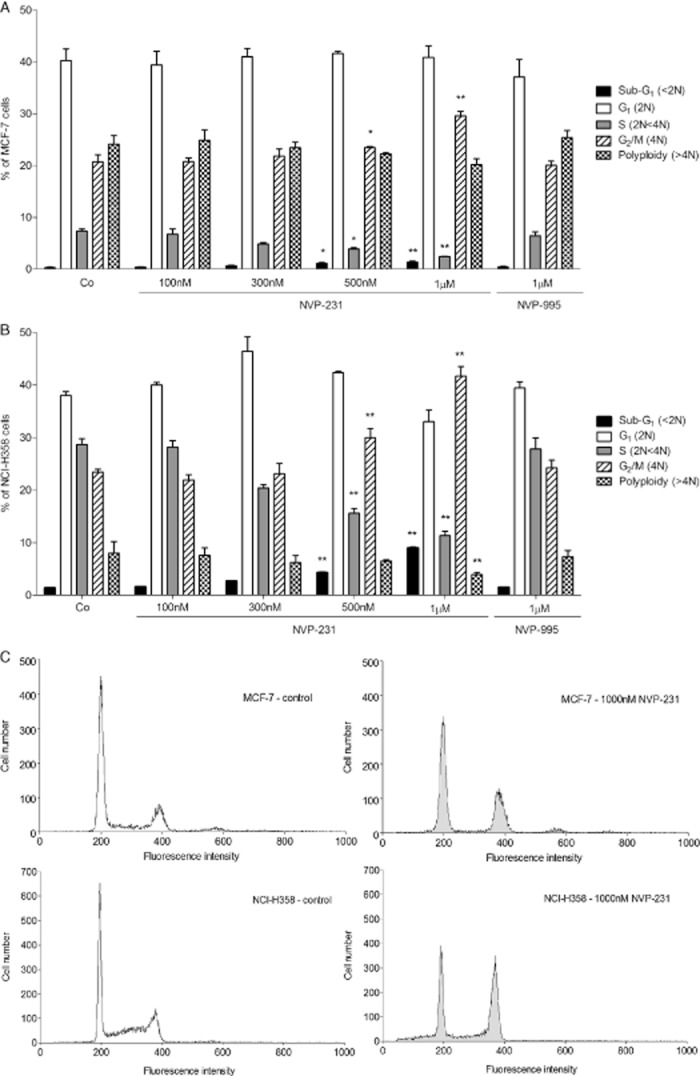
Effect of NVP-231 and NVP-995 on cell cycle progression of MCF-7 and NCI-H358 cells. MCF-7 (A) and NCI-H358 (B) cells were treated for 24 h with either vehicle (Co) or the indicated concentrations of NVP-231 and NVP-995 (in nM). Cells were collected, stained with propidium iodide and analysed by flow cytometry for DNA content. Data are expressed as % of cells in the corresponding cell cycle phases and are means ± SD (n = 6). *P < 0.05, **P < 0.01 considered statistically significant when compared with the respective control groups. (C) Graphs show representative samples of MCF-7 control cells (upper left panel), MCF-7 cells treated with 1000 nM NVP-231 (upper right panel), NCI-H358 control cells (lower left panel) and NCI-H358 cells treated with 1000 nM NVP-231 (lower right panel).
To further analyse the observed accumulation of cells in G2/M, we investigated whether cells arrested at the G2/M boundary or in the M phase upon CerK inhibition. To this end, phosphorylated histone H3 was analysed as a well-accepted marker for mitosis (Colman et al., 2006). NVP-231 treatment of both MCF-7 cells (Figure 5A) and NCI-H358 cells (Figure 5B) led to a concentration-dependent increase of the mitotic index of the cells detected by flow cytometry. In parallel, Western blot analysis of phospho- Ser10 histone H3 in cellular lysates was performed which also showed enhanced staining upon NVP-231 treatment (data not shown). These data demonstrate that NVP-231-treated cells committed M phase arrest rather than arrest at the G2/M boundary.
Figure 5.
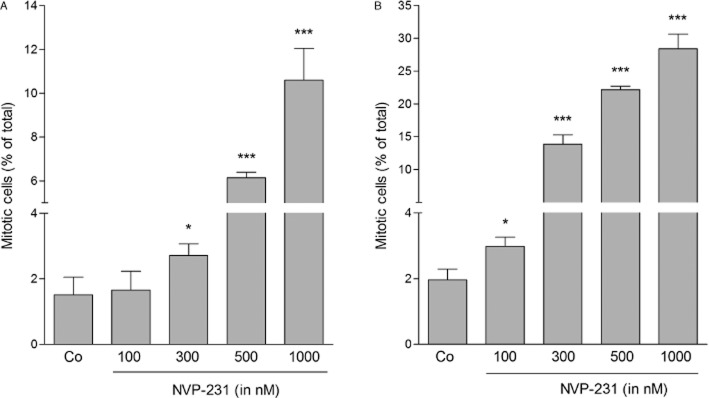
Effect of NVP-231 on the mitotic index of MCF-7 and NCI-H358 cells. MCF-7 (A) and NCI-H358 (B) cells were treated for 24 h with either vehicle (−) or the indicated concentrations of NVP-231 (in nM). Cells were collected and stained with an anti-phospho-Ser10 histone H3 antibody and analysed by flow cytometry. Data are expressed as % mitotic cells and are means ± SD (n = 6). *P < 0.05, ***P < 0.001 considered statistically significant when compared with the control groups.
We next investigated additional molecular factors known to be involved in cell cycle regulation, especially in G2/M transition. This transition is known to be regulated by the so called mitosis-promoting factor, which represents a heterodimeric protein composed of cyclin B and cdc2 (CDK1). In the two cell lines studied here, NVP-231 treatment caused a concentration-dependent up-regulation of cyclin B1 phosphorylation at Ser133 and a reduction of CDK1 phosphorylation at Tyr15 (Figure 6). Both events are required for G2/M transition and further support the finding that cells were not arrested in G2 but rather enter the M phase. However, total CDK1 expression also declined upon CerK inhibition (Figure 6). Moreover, we found that wee1 known as one of the kinases regulating CDK1 activity was down-regulated by NVP-231 (Figure 6). In addition, we also detected a down-regulation of CDK4, a kinase important for G1/S transition (Sheppard and McArthur, 2013). The reduction of CDK4 protein expression occurred in a time-dependent manner over 72 h and was more pronounced in NCI-H358 cells (Figure 7B and C) than in MCF-7 cells (Figure 7A and C), thus reflecting the fact that NCI-H358 cells have a shorter doubling time than MCF-7 cells. This down-regulation of CDK4 may explain the reduced number of cells in S phase.
Figure 6.

Effect of NVP-231 and NVP-995 on cyclin B1, CDK1 and wee1 expressions in MCF-7 and NCI-H358 cells. MCF-7 (A) and NCI-H358 (B) cells were treated for 24 h with either vehicle (Co) or the indicated concentrations of the NVP-231 and NVP-995 (in nM). Cell lysates were separated by SDS-PAGE, transferred to nitrocellulose and subjected to Western blot analysis using antibodies against phospho-Ser133 cyclin B1, total cyclin B1, phospho-Tyr15 CDK1, total CDK1, wee1 and GAPDH. Data show duplicate samples and are representative of at least three independent experiments giving similar results. (C and D) Graphs show the densitometric evaluation of Western blot data of MCF-7 (C) and NCI-H358 cells (D). Results are expressed as % of control and are means SD (n = 4–6). *P < 0.05, **P < 0.01, ***P < 0.001 considered statistically significant when compared with the control groups.
Figure 7.

Time-dependent effect of NVP-231 on CDK4 protein expression in MCF-7 and NCI-H358 cells. MCF-7 (A) and NCI-H358 (B) cells were treated for 24 h with either vehicle (Co) or for the indicated time periods with 1 μM of NVP-231. Cell lysates were separated by SDS-PAGE, transferred to nitrocellulose and subjected to Western blot analysis using antibodies against CDK4 and β-actin. Data show duplicate samples and are representative of three independent experiments giving similar results. (C) Graphs show the densitometric evaluation of Western blot data of MCF-7 and NCI-H358 cells. Results are expressed as % of control and are means ± SD (n = 6). **P < 0.01, ***P < 0.001 considered statistically significant when compared with the control groups. ###P < 0.001 compared with the NCI-H358 control group.
CerK inhibition sensitizes cells to drug-induced apoptosis
To test whether CerK inhibition sensitizes cancer cells to drug-induced apoptosis, we co-treated cells with NVP-231 and staurosporine. Staurosporine was chosen as a non-specific but potent inducer of apoptosis and used in a low concentration to better assess the effect of NVP-231. NCI-H358 cells treated with 20 nM of staurosporine showed a twofold increase of DNA fragmented cells compared with control cells (Figure 8A). When cells were pretreated with increasing concentrations of NVP-231, staurosporine stimulated a synergistic increase of DNA fragmentation (Figure 8A). Interestingly, MCF-7 cells reacted in a different manner to staurosporine than NCI-H358 cells. Using the same concentration of 20 nM staurosporine, only a minor increase of DNA fragmentation occurred (data not shown). Therefore, we measured staurosporine-induced G2/M arrest and found that pretreatment of cells with NVP-231 indeed enhanced staurosporine-induced G2/M arrest (Figure 8B). Next, we used an alternative approach to block CerK action, that is by down-regulating the enzyme using RNA interference. Under conditions where CerK mRNA expression was reduced to 30–40% of control transfected cells (data not shown), staurosporine-induced DNA fragmentation was significantly up-regulated in NCI-H358 (Figure 8C) and MCF-7 cells (Figure 8D).
Figure 8.

Effect of NVP-231 and CerK down-regulation with siRNA on staurosporine-induced cell death. NCI-H358 (A) and MCF-7 cells (B) were pretreated for 4 h with either vehicle (DMSO, 0) or the indicated concentrations of the NVP-231 and NVP-995 (in nM) before the addition of 20 nM staurosporine (+) for an additional 20 h. Cells were collected and analysed for DNA fragmentation and G2/M arrest. Data are means ± SD (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 considered statistically significant when compared with the untreated control group; ###P < 0.001 compared with the staurosporine-treated control group; §§§P < 0.001 compared with the corresponding NVP-231-treated groups. NCI-H358 (C) and MCF-7 cells (D) were transfected with a control siRNA (ctrl-siRNA) or siRNA of CerK (siCerK) and then treated for 20 h with the indicated concentrations of staurosporine (in nM). Cells were collected and analysed for DNA fragmentation. Data are means ± SD (n = 3). *P < 0.05, ***P < 0.001 considered statistically significant when compared with the untreated control group; ##P < 0.01, ###P < 0.001 compared with the untreated siCerK group; §P < 0.05, §§P < 0.01, §§§P < 0.001 compared with the corresponding staurosporine-treated groups.
Finally, we tested whether overexpression of CerK in MCF-7 cells protects from drug-induced apoptosis. To this end, cells were transiently transfected with a human CerK cDNA construct prior to treatment with staurosporine. As shown in Figure 9A, staurosporine reduced MCF-7 cell viability in a concentration-dependent manner. When CerK was overexpressed, the proapoptotic effect of staurosporine was reversed (Figure 9A), suggesting that endogenously produced C1P may promote cell viability. Moreover, overexpression of CerK reduced the NVP-231-triggered increase of phospho-histone H3 (Figure 9B).
Figure 9.

Effect of CerK overexpression on staurosporine-stimulated apoptosis and NVP-231-mediated histone H3 phosphorylation. (A) Cells transfected with the empty vector (pcDNA3) or with a cDNA construct of hCerK (pcDNA3-hCerK) were plated in a 96-well black plate (1 × 104 cells per well) and treated for 48 h with either vehicle (Co) or the indicated concentrations of staurosporine (in nM). For the last 4 h, alamarBlueR was added and fluorescence was determined as described in the Methods section. Data are expressed as % of control and are means ± SD (n = 5), **P < 0.01, ***P < 0.001 considered statistically significant when compared with the corresponding value of pcDNA3 transfected samples. (B) NCI-H358 cells transfected with the empty vector (pcDNA3) or with a cDNA construct of hCerK (pcDNA3-hCerK) were treated for 24 h with either vehicle (Co) or the indicated concentrations of NVP-231. Cells were then taken for phospho-histone H3 analysis by flow cytometry. Results are expressed as % of phospho-H3 positive cells and are means ± SD (n = 3). ***P < 0.001 when compared with the pcDNA3 transfected control samples; §P < 0.05, §§§P < 0.001 when compared with the corresponding NVP-231-treated pcDNA3 transfected samples.
Discussion
Previous studies suggested a role for CerK and its product C1P in the regulation of cell proliferation and apoptosis. However, the exact mechanisms have remained largely unclear. It was first shown that CerK is an important player in the control of plant cell death. By mutating the plant CerK homologue accelerated cell death 5 (acd5) in Arabidopsis, the plants not only accumulated ceramides, but also showed excessive cell death upon pathogen infection (Liang et al., 2003). Later on, in an in vitro system with human lung cancer cells, down-regulation of CerK by siRNA transfection, also resulted in reduced DNA synthesis and increased apoptosis (Mitra et al., 2007).
To unveil the cellular function of the lipid C1P, many studies used exogenous C1P added to cell cultures, either as liposomes or micelles in rather high concentrations or diluted in solvents containing dodecane. Using these approaches, C1P seems to trigger, in a cell type-specific manner, either proliferation or apoptosis (Gomez-Muñoz et al., 1997; Tauzin et al., 2007). Recently, Gangoiti et al. (2012) showed that in C2C12 myoblasts, exogenous C1P induced cell proliferation by stimulating the phosphorylation of glycogen synthase kinase-3β, increasing retinoblastoma (Rb) phosphorylation and enhancing cyclin D1 expression. Clearly, C1P is a cell-impermeable lipid which cannot enter the cell without a transporter or, as may be the case for dodecane, by triggering pore formation. Alternatively, exogenous C1P, like S1P, may act via a cell surface receptor. However, so far, neither a specific C1P export system like a transporter, nor a specific high-affinity cell surface receptor for C1P has been identified. Therefore, it remains unclear whether extracellular C1P-triggered cellular effects are indeed mediated by a specific action on a putative receptor or result, at least to some extent, from membrane disturbances like those triggered by detergents. Nevertheless, C1P is primarily generated inside the cell and therefore, its preferential site of action is most likely intracellular.
In the present study, we used a recently developed potent catalytic inhibitor of CerK, that is NVP-231 (Graf et al., 2008a), which inhibits CerK in the low nM range in a cell-free in vitro but also in transfected cells (Figure 1). We report that in two cancer cell lines of distinct histotypes, that is the breast cancer cell line MCF-7 and the lung cancer cell line NCI-H358, treatment with NVP-231 concentration-dependently reduced cell viability, DNA synthesis, as well as colony formation. The cells showed abnormalities in cell cycle progression causing a striking accumulation of cells in the M phase and, additionally, a reduced number of cells in S phase. Later on, cell death increased. Certainly, in pharmacology, the specificity of an inhibitor is crucial. As NVP-231 was identified as a ceramide-competitive CerK inhibitor, it is possible that, if at all, the inhibitor interferes with other enzymes with affinity for lipids, particularly to ceramide. The specificity of NVP-231 for CerK was addressed by Graf et al. (2008a). Various other lipid kinases and other enzymes with affinity for ceramide were tested. The most sensitive enzyme responding to NVP-231 was DAGKα which was inhibited with an IC50 of 5 μM whereas all other tested enzymes were inhibited with IC50 values between 10 and 100 μM.
Regulation of cell cycle progression in mammalian cells is generally associated with expression and activity of four CDKs including CDK1 (Cdc2), CDK2, CDK4 and CDK6, and their regulatory cyclin partners comprising cyclin A, B, D and E (Leake, 1996; Lim and Kaldis, 2013). CDK1 represents an absolutely essential kinase and its dysfunction is fatal for cell cycle progression, cell division and viability. CDK1 is tightly involved in the processes of DNA replication, chromosome cohesion and condensation, assembly, positioning and stability of the mitotic spindle, chromosomes attachment, alignment and segregation (Enserink and Kolodner, 2010). All of these events may be disturbed when CDK1 activity is compromised. Recently, Que et al. (2013) reported that silencing of CDK1 in osteosarcoma cells resulted, similar to our study, in a reduced proliferation and accumulation of cells in G2/M and a reduced number of cells in S phase. Additionally, Rani et al. (1995) showed that treatment of fibroblasts with the glucosylceramide synthase inhibitor 1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) not only led to a reduced level of glucosylceramide and an enhanced level of cellular ceramide, but also induced accumulation of cells in G2/M and a reduction in S phase. Concomitantly, they observed a decrease of CDK1 activity upon PDMP treatment. The authors suggested that especially ceramide was responsible for G2/M arrest, whereas the loss of glucosylceramide contributed to the decrease of S-phase cells (Rani et al., 1995). Moreover, Drobnik et al. (1999) showed that in fibroblasts from Tangier disease patients, which are characterized by a disturbed lipid metabolism due to a mutated ABC1 transporter (Bodzioch et al., 1999), cellular ceramide concentrations were also increased, and in response to different mitogens, cells accumulated in G2/M and disappeared from S phase (Drobnik et al., 1999).
As inhibition or knockout of CerK also results in the accumulation of ceramide (Graf et al., 2008b and Figure 1C in this study), it is well possible that similar mechanisms are triggered by these treatment strategies which all lead to the same cellular phenotype. However, whether this phenotype is mediated by ceramide remains uncertain due to controversial data about the role of ceramide in cell cycle regulation. Ceramide was also shown to arrest cells, including MCF-7 cells, in G1 (Jayadev et al., 1995; Struckhoff et al., 2010), which seems to involve dephosphorylation of the Rb protein (Dbaibo et al., 1995) and up-regulation of the CDK inhibitory factor p21 (Oh et al., 1998; Lee et al., 2000). In another study, it was shown that inhibition of de novo sphingolipid biosynthesis by myriocin, which results in a reduction of various sphingolipids including ceramide, reduced DNA synthesis and proliferation of melanoma cells, and cells accumulated in G2/M and disappeared from S phase. On a molecular level, myriocin was shown to induce a significant loss of CDK1 protein, which could be completely prevented by addition of exogenous C8-ceramide, leading the authors to conclude that ceramide or one of its catabolites is essential for CDK1 expression (Lee et al., 2011). Certainly, the ceramide effect may be cell type specific and depends on the cell's equipment with ceramide-converting enzymes, such as ceramidases and sphingosine kinases, which are known as crucial determinants of ceramide's actions (Huwiler and Pfeilschifter, 2006; Huwiler and Zangemeister-Wittke, 2007).
In an attempt to further trace the signal upstream, we found that wee1 was down-regulated by NVP-231 in both NCI-H358 and MCF-7 cells. Wee1 kinase is a critical regulator of CDK1 activity by mediating Tyr15 phosphorylation and thus CDK1 inactivation, and it has been reported that wee1 must be degraded by an E3 ubiquitin ligase activity to ensure entry into mitosis (Smith et al., 2007). It is thus tempting to speculate that CerK inhibition leads to enhanced E3 ubiquitin protein ligase activity which then promotes wee1 degradation and the onset of mitosis.
Our data also demonstrate that upon NVP-231 treatment of cancer cells, CDK4 protein was concentration- and time-dependently down-regulated. This effect was even more pronounced in synchronized cells (data not shown). CDK4 is a central player of the p16/cyclin D/CDK4 and 6/retinoblastoma pathway promoting G1 to S cell cycle transition (Sheppard and McArthur, 2013). Furthermore, inhibition or down-regulation of CDK4 not only induces G1 arrest but also increases cell death (Sheppard and McArthur, 2013).
The observed down-regulation of CDK4 by CerK inhibition may explain the observed decline of cells from S phase and increased apoptosis. However, the mechanism underlying these effects is still unclear and further studies are needed to address this issue. Especially important is the question which lipid subspecies is directly responsible for the effects: is it the accumulated ceramide or the loss of C1P or another metabolite? Also it is worth noting in this context, that in intestinal polyps of SphK1-deficient mice CDK4 expression was down-regulated correlating with reduced epithelial cell proliferation (Kohno et al., 2006). In that study, in vitro experiments with rat intestinal epithelial cells revealed that exogenous sphingosine, but not ceramide, could mimic the effect of reduced CDK4 expression and proliferation (Kohno et al., 2006). On the contrary, Kim et al. (2000) showed that in HL-60 cells, ceramide inhibited CDK4 activity and thereby contributed to G1 arrest and subsequent apoptosis. Thus further studies are needed to clarify the involvement of the different sphingolipid species in the cell cycle machinery.
Cell cycle-regulating processes are not only restricted to the nucleus but may also occur in the cytoplasm. In view of this, unravelling the subcellular localization of CerK in the dynamic range of the cell cycle will be crucial to identify possible direct targets of C1P. The fact that CerK contains both nuclear import and export signals in its sequence makes a nuclear function very likely although this is still unproven. By transfecting COS-1 cells with a GFP-coupled CerK construct, Rovina et al. (2009) demonstrated the localization of the wild-type CerK in the Golgi, the cytoplasm and in the nucleus. These data make it tempting to suggest that CerK exerts a function in the nucleus.
Furthermore, Chen et al. (2010) previously showed that CerK contains various phosphorylation sites, including Ser340 and Ser408. These two residues represent classical phosphorylation sites for proline-directed PKs including MAPKs and CDKs. Because both of these families of PKs are crucially involved in cell proliferation and cell cycle regulation, coupling of CerK to the cell cycle in one or the other way is very likely.
A keystone to successful anti-cancer therapy is to sensitize cancer cells to proapoptotic chemotherapy. Our data indicate that CerK inhibition per se is sufficient to induce cancer cell death, and in addition, sensitizes cells to death-promoting agents such as the unspecific kinase inhibitor staurosporine. This sensitizing effect is indeed due to CerK inhibition and not due to an unspecific effect, as CerK overexpression rendered cells less susceptible to death induction by staurosporine. A role for C1P in cell survival was also proposed by Gomez-Muñoz et al. (2005). These authors showed that in mouse macrophages exogenous, C1P promoted cell survival by activating PI3K and Akt/PKB and by up-regulating the anti-apoptotic factor Bcl-xL. However, in our cell systems, the overexpression of CerK did not cause enhanced Akt (PKB) or p42/p44-MAPK phosphorylation (data not shown) thus excluding a role of C1P in early survival signalling and rather supporting our hypothesis that C1P acts within the nucleus to promote M phase transition. Further studies are needed to clarify which combinations of NVP-231 and conventional anti-cancer agents are most promising to optimize the synergistic anti-cancer effect not only in vitro in cancer cell lines but finally also in vivo in patients.
This crucial function of CerK in proper mitosis transition and cell proliferation proposed by our data also suggests a therapeutic use of NVP-231 in other proliferation-associated diseases. In this regard, we have recently shown that in renal mesangial cells, CerK knockout or NVP-231 treatment reduces cell proliferation and therefore, the use of NVP-231 to treat mesangioproliferative glomerulonephritis represents an attractive novel strategy (Pastukhov et al., 2014). However, to date, an enhanced expression of CerK has only been shown for breast cancer but not for other diseases. Thus, Ruckhäberle et al. (2009) reported that high CerK expression is correlated with a worse prognosis of breast cancer patients even suggesting that CerK may be a useful prognostic marker in breast cancer.
In summary, our findings demonstrate for the first time a crucial role for CerK in M-phase regulation and propose that combining targeted CerK inhibition with proapoptotic anti-cancer agents has potential for more effective cancer therapy.
Acknowledgments
We thank Isolde Römer and Svetlana Kokin for excellent technical assistance. This work was supported by the German Research Foundation (SPP1267/2, SFB1039, HU842/5-1) and the Swiss National Science Foundation (310030_135619).
Glossary
- BrdU
5-bromo-2-deoxyuridine
- C1P
ceramide-1-phosphate
- CerK
ceramide kinase
- CDK
cyclin-dependent kinase
- NVP-231
N-[2-benzoylamino-1,3-benzothiazol-6-yl]adamantane-1-carboxamide
- NVP-995
N-[2-[(3,4-dimethoxybenzoyl)amino]-1,3-benzothiazol-6-yl]adamantane-1-carboxamide
- S1P
sphingosine-1-phosphate
Author contributions
O. P., S. S. and L. J. performed the experiments. U. Z.-W., J. P. and A. H. designed the research study. D. F. and F. B. contributed essential reagents and tools. O. P., S. S., L. J., U. Z.-W., B. K., J. P. and A. H. analysed the data. U. Z.-W., J. P. and A. H. wrote the paper.
Conflict of interest
None.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajjalieh SM, Martin TF, Floor E. Synaptic vesicle ceramide kinase. A calcium-stimulated lipid kinase that co-purifies with brain synaptic vesicles. J Biol Chem. 1989;264:14354–14360. [PubMed] [Google Scholar]
- Boath A, Graf C, Lidome E, Ullrich T, Nussbaumer P, Bornancin F. Regulation and traffic of ceramide 1-phosphate produced by ceramide kinase: comparative analysis to glucosylceramide and sphingomyelin. J Biol Chem. 2008;283:8517–8526. doi: 10.1074/jbc.M707107200. [DOI] [PubMed] [Google Scholar]
- Bodzioch M, Orsó E, Klucken J, Langmann T, Böttcher A, Diederich W, et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet. 1999;22:347–351. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- Bornancin F. Ceramide kinase: the first decade. Cell Signal. 2011;23:999–1008. doi: 10.1016/j.cellsig.2010.11.012. [DOI] [PubMed] [Google Scholar]
- Carré A, Graf C, Stora S, Mechtcheriakova D, Csonga R, Urtz N, et al. Ceramide kinase targeting and activity determined by its N-terminal pleckstrin homology domain. Biochem Biophys Res Commun. 2004;324:1215–1219. doi: 10.1016/j.bbrc.2004.09.181. [DOI] [PubMed] [Google Scholar]
- Chen WQ, Graf C, Zimmel D, Rovina P, Krapfenbauer K, Jaritz M, et al. Ceramide kinase profiling by mass spectrometry reveals a conserved phosphorylation pattern downstream of the catalytic site. J Proteome Res. 2010;9:420–429. doi: 10.1021/pr900763z. [DOI] [PubMed] [Google Scholar]
- Colman H, Giannini C, Huang L, Gonzalez J, Hess K, Bruner J, et al. Assessment and prognostic significance of mitotic index using the mitosis marker phospho-histone H3 in low and intermediate-grade infiltrating astrocytomas. Am J Surg Pathol. 2006;30:657–664. doi: 10.1097/01.pas.0000202048.28203.25. [DOI] [PubMed] [Google Scholar]
- Dbaibo GS, Pushkareva MY, Jayadev S, Schwarz JK, Horowitz JM, Obeid LM, et al. Retinoblastoma gene product as a downstream target for a ceramide-dependent pathway of growth arrest. Proc Natl Acad Sci U S A. 1995;92:1347–1351. doi: 10.1073/pnas.92.5.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobnik W, Liebisch G, Biederer C, Trümbach B, Rogler G, Müller P, et al. Growth and cell cycle abnormalities of fibroblasts from Tangier disease patients. Arterioscler Thromb Vasc Biol. 1999;19:28–38. doi: 10.1161/01.atv.19.1.28. [DOI] [PubMed] [Google Scholar]
- Enserink JM, Kolodner RD. An overview of Cdk1-controlled targets and processes. Cell Div. 2010;5:11. doi: 10.1186/1747-1028-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayyaz S, Henkel J, Japtok L, Krämer S, Damm G, Seehofer D, et al. Involvement of sphingosine 1-phosphate in palmitate-induced insulin resistance of hepatocytes via the S1P2 receptor subtype. Diabetologia. 2014;57:373–382. doi: 10.1007/s00125-013-3123-6. [DOI] [PubMed] [Google Scholar]
- Gangoiti P, Bernacchioni C, Donati C, Cencetti F, Ouro A, Gómez-Muñoz A, et al. Ceramide 1-phosphate stimulates proliferation of C2C12 myoblasts. Biochimie. 2012;94:597–607. doi: 10.1016/j.biochi.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Muñoz A, Duffy PA, Martin A, O'Brien L, Byun HS, Bittman R, et al. Short-chain ceramide-1-phosphates are novel stimulators of DNA synthesis and cell division: antagonism by cell-permeable ceramides. Mol Pharmacol. 1995;47:833–839. [PubMed] [Google Scholar]
- Gomez-Muñoz A, Frago LM, Alvarez L, Varela-Nieto I. Stimulation of DNA synthesis by natural ceramide 1-phosphate. Biochem J. 1997;325:435–440. doi: 10.1042/bj3250435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Muñoz A, Kong JY, Parhar K, Wang SW, Gangoiti P, Gonzalez M, et al. Ceramide-1-phosphate promotes cell survival through activation of the phosphatidylinositol 3-kinase/protein kinase B pathway. FEBS Lett. 2005;579:3744–3750. doi: 10.1016/j.febslet.2005.05.067. [DOI] [PubMed] [Google Scholar]
- Graf C, Klumpp M, Habig M, Rovina P, Billich A, Baumruker T, et al. Targeting ceramide metabolism with a potent and specific ceramide kinase inhibitor. Mol Pharmacol. 2008a;74:925–932. doi: 10.1124/mol.108.048652. [DOI] [PubMed] [Google Scholar]
- Graf C, Zemann B, Rovina P, Urtz N, Schanzer A, Reuschel R, et al. Neutropenia with impaired immune response to Streptococcus pneumoniae in ceramide kinase-deficient mice. J Immunol. 2008b;180:3457–3466. doi: 10.4049/jimmunol.180.5.3457. [DOI] [PubMed] [Google Scholar]
- Graf C, Rovina P, Bornancin F. A secondary assay for ceramide kinase inhibitors based on cell growth inhibition by short-chain ceramides. Anal Biochem. 2009;384:166–169. doi: 10.1016/j.ab.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Granado MH, Gangoiti P, Ouro A, Arana L, González M, Trueba M, et al. Ceramide 1-phosphate (C1P) promotes cell migration Involvement of a specific C1P receptor. Cell Signal. 2009;21:405–412. doi: 10.1016/j.cellsig.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Hinkovska-Galcheva VT, Boxer LA, Mansfield PJ, Harsh D, Blackwood A, Shayman JA. The formation of ceramide-1-phosphate during neutrophil phagocytosis and its role in liposome fusion. J Biol Chem. 1998;273:33203–33209. doi: 10.1074/jbc.273.50.33203. [DOI] [PubMed] [Google Scholar]
- Huwiler A, Pfeilschifter J. Altering the sphingosine-1-phosphate/ceramide balance: a promising approach for tumor therapy. Curr Pharm Des. 2006;12:4625–4635. doi: 10.2174/138161206779010422. [DOI] [PubMed] [Google Scholar]
- Huwiler A, Zangemeister-Wittke U. Targeting the conversion of ceramide to sphingosine 1-phosphate as a novel strategy for cancer therapy. Crit Rev Oncol Hematol. 2007;63:150–159. doi: 10.1016/j.critrevonc.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Huwiler A, Döll F, Ren S, Klawitter S, Greening A, Römer I, et al. Histamine increases sphingosine kinase-1 expression and activity in the human arterial endothelial cell line EA.hy 926 by a PKC-a-dependent mechanism. Biochim Biophys Acta. 2006;1761:367–376. doi: 10.1016/j.bbalip.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Huwiler A, Kotelevets N, Xin C, Pastukhov O, Pfeilschifter J, Zangemeister-Wittke U. Loss of sphingosine kinase-1 in carcinoma cells increases formation of reactive oxygen species and sensitivity to doxorubicin-induced DNA damage. Br J Pharmacol. 2011;162:532–543. doi: 10.1111/j.1476-5381.2010.01053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayadev S, Liu B, Bielawska AE, Lee JY, Nazaire F, Pushkareva M, et al. Role for ceramide in cell cycle arrest. J Biol Chem. 1995;270:2047–2052. doi: 10.1074/jbc.270.5.2047. [DOI] [PubMed] [Google Scholar]
- Kim WH, Ghil KC, Lee JH, Yeo SH, Chun YJ, Choi KH, et al. Involvement of p27(kip1) in ceramide-mediated apoptosis in HL-60 cells. Cancer Lett. 2000;151:39–48. doi: 10.1016/s0304-3835(99)00402-4. [DOI] [PubMed] [Google Scholar]
- Kohno M, Momoi M, Oo ML, Paik JH, Lee YM, Venkataraman K, et al. Intracellular role for sphingosine kinase 1 in intestinal adenoma cell proliferation. Mol Cell Biol. 2006;26:7211–7223. doi: 10.1128/MCB.02341-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa H, Nishio K, Fukumoto H, Tomonari A, Suzuki T, Saijo N. Alteration of Caspase-3 (CPP32/Yama/apopain) in wild-type MCF-7, breast cancer cells. Oncol Rep. 1999;6:33–37. doi: 10.3892/or.6.1.33. [DOI] [PubMed] [Google Scholar]
- Larson EM, Doughman DJ, Gregerson DS, Obritsch WF. A new, simple, nonradioactive, nontoxic in vitro assay to monitor corneal endothelial cell viability. Invest Ophthalmol Vis Sci. 1997;38:1929–1933. [PubMed] [Google Scholar]
- Leake R. The cell cycle and regulation of cancer cell growth. Ann N Y Acad Sci. 1996;784:252–262. doi: 10.1111/j.1749-6632.1996.tb16240.x. [DOI] [PubMed] [Google Scholar]
- Lee JY, Bielawska AE, Obeid LM. Regulation of cyclin-dependent kinase 2 activity by ceramide. Exp Cell Res. 2000;26:303–311. doi: 10.1006/excr.2000.5028. [DOI] [PubMed] [Google Scholar]
- Lee YS, Choi KM, Choi MH, Ji SY, Lee S, Sin DM, et al. Serine palmitoyltransferase inhibitor myriocin induces growth inhibition of B16F10 melanoma cells through G(2)/M phase arrest. Cell Prolif. 2011;44:320–329. doi: 10.1111/j.1365-2184.2011.00761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Yao N, Song JT, Luo S, Lu H, Greenberg JT. Ceramides modulate programmed cell death in plants. Genes Dev. 2003;17:2636–26341. doi: 10.1101/gad.1140503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140:3079–3093. doi: 10.1242/dev.091744. [DOI] [PubMed] [Google Scholar]
- Mitra P, Maceyka M, Payne SG, Lamour N, Milstien S, Chalfant CE, et al. Ceramide kinase regulates growth and survival of A549 human lung adenocarcinoma cells. FEBS Lett. 2007;58:735–740. doi: 10.1016/j.febslet.2007.01.041. [DOI] [PubMed] [Google Scholar]
- Mitsutake S, Kim TJ, Inagaki Y, Kato M, Yamashita T, Igarashi Y. Ceramide kinase is a mediator of calcium-dependent degranulation in mast cells. J Biol Chem. 2004;279:17570–17577. doi: 10.1074/jbc.M312885200. [DOI] [PubMed] [Google Scholar]
- Niwa S, Graf C, Bornancin F. Ceramide kinase deficiency impairs microendothelial cell angiogenesis in vitro. Microvasc Res. 2009;77:389–393. doi: 10.1016/j.mvr.2009.01.006. [DOI] [PubMed] [Google Scholar]
- Oh WJ, Kim WH, Kang KH, Kim TY, Kim MY, Choi KH. Induction of p21 during ceramide-mediated apoptosis in human hepatocarcinoma cells. Cancer Lett. 1998;129:215–222. doi: 10.1016/s0304-3835(98)00105-0. [DOI] [PubMed] [Google Scholar]
- Pastukhov O, Schwalm S, Römer I, Zangemeister-Wittke U, Pfeilschifter J, Huwiler A. Ceramide kinase contributes to proliferation but not to prostaglandin E2 formation in renal mesangial cells and fibroblasts. Cell Physiol Biochem. 2014;34:119–133. doi: 10.1159/000362989. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettus BJ, Bielawska A, Spiegel S, Roddy P, Hannun YA, Chalfant CE. Ceramide kinase mediates cytokine- and calcium ionophore-induced arachidonic acid release. J Biol Chem. 2003;278:38206–38213. doi: 10.1074/jbc.M304816200. [DOI] [PubMed] [Google Scholar]
- Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- Que XY, Li Y, Han Y, Li XZ. Effects of siRNA–mediated Cdc2 silencing on MG63 cell proliferation and apoptosis. Mol Med Rep. 2013;7:466–470. doi: 10.3892/mmr.2012.1209. [DOI] [PubMed] [Google Scholar]
- Rani CS, Abe A, Chang Y, Rosenzweig N, Saltiel AR, Radin NS, et al. Cell cycle arrest induced by an inhibitor of glucosylceramide synthase. Correlation with cyclin-dependent kinases. J Biol Chem. 1995;270:2859–2867. doi: 10.1074/jbc.270.6.2859. [DOI] [PubMed] [Google Scholar]
- Rovina P, Schanzer A, Graf C, Mechtcheriakova D, Jaritz M, Bornancin F. Subcellular localization of ceramide kinase and ceramide kinase-like protein requires interplay of their Pleckstrin Homology domain-containing N-terminal regions together with C-terminal domains. Biochim Biophys Acta. 2009;1791:1023–1030. doi: 10.1016/j.bbalip.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Ruckhäberle E, Karn T, Rody A, Hanker L, Gätje R, Metzler D, et al. Gene expression of ceramide kinase, galactosyl ceramide synthase and ganglioside GD3 synthase is associated with prognosis in breast cancer. J Cancer Res Clin Oncol. 2009;135:1005–1013. doi: 10.1007/s00432-008-0536-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard KE, McArthur GA. The cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clin Cancer Res. 2013;19:5320–5328. doi: 10.1158/1078-0432.CCR-13-0259. [DOI] [PubMed] [Google Scholar]
- Shinghal R, Scheller RH, Bajialieh SM. Ceramide 1-phosphate phosphatase activity in brain. J Neurochem. 1993;61:2279–2285. doi: 10.1111/j.1471-4159.1993.tb07470.x. [DOI] [PubMed] [Google Scholar]
- Smith A, Simanski S, Fallahi M, Ayad NG. Redundant ubiquitin ligase activities regulate wee1 degradation and mitotic entry. Cell Cycle. 2007;6:2795–2799. doi: 10.4161/cc.6.22.4919. [DOI] [PubMed] [Google Scholar]
- Struckhoff AP, Patel B, Beckman BS. Inhibition of p53 sensitizes MCF-7 cells to ceramide treatment. Int J Oncol. 2010;37:21–30. doi: 10.3892/ijo_00000649. [DOI] [PubMed] [Google Scholar]
- Sugiura M, Kono K, Liu H, Shimizugawa T, Minekura H, Spiegel S, et al. Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. J Biol Chem. 2002;277:23294–23300. doi: 10.1074/jbc.M201535200. [DOI] [PubMed] [Google Scholar]
- Tauzin L, Graf C, Sun M, Rovina P, Bouveyron N, Jaritz M, et al. Effects of ceramide-1-phosphate on cultured cells: dependence on dodecane in the vehicle. J Lipid Res. 2007;48:66–76. doi: 10.1194/jlr.M600399-JLR200. [DOI] [PubMed] [Google Scholar]
- Van Brocklyn JR, Williams JB. The control of the balance between ceramide and sphingosine-1-phosphate by sphingosine kinase: oxidative stress and the seesaw of cell survival and death. Comp Biochem Physiol B Biochem Mol Biol. 2012;163:26–36. doi: 10.1016/j.cbpb.2012.05.006. [DOI] [PubMed] [Google Scholar]