

Abstract
Background and Purpose
The transporter, multidrug resistance protein 1 (MRP1, ABCC1), plays a critical role in the development of multidrug resistance (MDR). Ibrutinib is an inhibitor of Bruton's tyrosine kinase. Here we investigated the reversal effect of ibrutinib on MRP1-mediated MDR.
Experimental Approach
Cytotoxicity was determined by MTT assay. The expression of protein was detected by Western blot. RT-PCR and Q-PCR were performed to detect the expression of MRP1 mRNA. The intracellular accumulation and efflux of substrates for MRP1 were measured by scintillation counter and flow cytometry. HEK293/MRP1 cell xenografts in nude mice were established to study the effects of ibrutinib in vivo.
Key Results
Ibrutinib significantly enhanced the cytotoxicity of MRP1 substrates in HEK293/MRP1 and HL60/Adr cells overexpressing MRP1. Furthermore, ibrutinib increased the accumulation of substrates in these MRP1-overexpressing cells by inhibiting the drug efflux function of MRP1. However, mRNA and protein expression of MRP1 remained unaltered after treatment with ibrutinib in MRP1-overexpressing cells. In vivo, ibrutinib enhanced the efficacy of vincristine to inhibit the growth of HEK293/MRP1 tumour xenografts in nude mice. Importantly, ibrutinib also enhances the cytotoxicity of vincristine in primary cultures of leukaemia blasts, derived from patients.
Conclusions and Implications
Our results indicated that ibrutinib significantly increased the efficacy of the chemotherapeutic agents which were MRP1 substrates, in MRP1-overexpressing cells, in vitro, in vivo and ex vivo. These findings will lead to further studies on the effects of a combination of ibrutinib with chemotherapeutic agents in cancer patients overexpressing MRP1.
Introduction
Multidrug resistance (MDR), a phenomenon characterized by the resistance to structurally and mechanistically unrelated drugs, is a major obstacle to the success of cancer chemotherapy. Although the MDR phenotype is mediated through various mechanisms, the overexpression of membrane ATP-binding cassette (ABC) transporters in cancer is considered to be a primary determinant of such drug resistance (Danks et al., 1988; Giaccone et al., 1992). ABC transporters act as the defence system of cancer cells against anticancer drugs via an active efflux mechanism, leading to a reduced intracellular accumulation of substrate drugs thereby rendering them ineffective (Sodani et al., 2012a). Of these ABC transporters, MDR1/P-glycoprotein (ABCB1), breast cancer resistance protein (ABCG2, MXR) and multidrug resistance protein 1 (MRP1, ABCC1) are key components in the development of the MDR phenotype (Cole and Deeley, 1998; Ambudkar et al., 1999; Gottesman et al., 2002).
MRP1, the first identified member of C subfamily of ABC transporters, was cloned from anthracycline-selected human small-cell lung carcinoma cells, H69AR, in 1992 by Cole et al. (1992). MRP1 is expressed in various normal human tissues including lung, spleen, testis, kidney, placenta, thyroid, bladder and adrenal gland (Flens et al., 1996). As a basolaterally located transporter, MRP1 is involved in the transport of GSH, GSH conjugates and organic anions along with neutral and basic organic compounds (Muller et al., 1994; Ishikawa et al., 1997). Moreover, MRP1 is a major transporter for endogenous leukotriene C4, an important modulator of the inflammatory response (Jedlitschky et al., 1994; Wijnholds et al., 1997; 1998; 2000,,). With its wide expression profile, MRP1 is able to protect tissues and eliminate toxic substrates, thereby maintaining homeostasis (Cole et al., 1992). Overexpression of MRP1 in tumours has been clearly associated with clinical drug resistance in leukaemia and solid tumours such as childhood neuroblastomas, lung and oesophageal cancers (Cole et al., 1992; Burger et al., 1994; Nooter et al., 1995; Norris et al., 1996). MRP1 expression was detected in 48 children (86%) and 48 adults (98%) with acute lymphoblastic leukaemia (ALL) (Plasschaert et al., 2005). Previous reports showed that high expression of MRP1 was associated with the MDR phenotype and poor prognosis in both acute myeloid leukaemia (AML) and ALL patients (Plasschaert et al., 2005; Mahjoubi et al., 2008). MRP1 transports a large range of important chemotherapeutic agents such as vincristine, vinblastine, epipodophyllotoxins, doxorubicin and some heavy metal oxyanions thereby conferring MDR (Haimeur et al., 2004). Thus, constant efforts have been made in order to block the transport of such substrates of MRP1, with the help of modulatory agents, but with limited success. Since the discovery of MRP1, several modulators of MRP1 have been identified, including indomethacin (Duffy et al., 1998), probenecid (Gollapudi et al., 1997), the leukotriene receptor antagonists MK571 (Burns et al., 1995) and ONO-1078 (Nagayama et al., 1998; Nakano et al., 1998), but none has been successful in clinical practice. Consequently, it is necessary to develop and identify drugs that can successfully inhibit the activity of MRP1 without producing serious adverse effects.
Previous studies reported that inhibitors of tyrosine kinase (TK), such as imatinib and AG1393, have been shown to interact with human MRP1 and inhibit the transport activity, indicating that inhibitors of TK may also be promising MRP1 inhibitors (Hegedus et al., 2002). Bruton's tyrosine kinase (BTK) is a cytoplasmic enzyme, which plays a key role in B-lymphocyte development, proliferation, differentiation, apoptosis and cell migration (Akinleye et al., 2013). Some preclinical and clinical studies have demonstrated that ibrutinib (PCI-32765), a novel potent inhibitor of BTK, induced impressive responses in B-cell malignancies. Recently, ibrutinib has been approved for therapy of refractory mantle cell lymphoma and chronic lymphocytic leukaemia by the FDA (Novero et al., 2014).
In this study, we investigated the effect of ibrutinib on enhancing the efficacy of chemotherapeutic agents in MRP1-overexpressing cells in vitro, in vivo and ex vivo. These findings demonstrate the re-sensitization of drug-resistant, MRP1-overexpressing cells and leukaemia cells to the chemotherapeutic effects of cytotoxic drugs that are substrates for MRP1.
Methods
Cell lines and cell culture
The human leukaemia cell line HL60 and doxorubicin-selected MRP1-overexpressing cell line HL60/Adr were obtained from Dr Susan E. Bates (NCI, NIH, Bethesda, MD) (Tang et al., 2004); the HEK cell line HEK293/pcDNA3.1 and MRP1-transfected cell line HEK293/MRP1 were acquired from Dr Suresh V. Ambudkar (NCI, NIH, Bethesda, MD) (Muller et al., 2002). All the cells were cultured in DMEM or RPMI 1640 medium supplemented with 10% FBS at 37°C in a humidified atmosphere of 5% CO2.
Cytotoxicity assay
Cell sensitivity to drugs was analysed using the MTT colorimetric assay as described previously (Shi et al., 2007; Kathawala et al., 2014). Cells were collected and seeded into 96-well plates. To test the effects of ibrutinib, the cells were pre-incubated with or without ibrutinib for 1 h and then with different concentrations of chemotherapeutic agents added into the designated wells. MK571 (50 μM) a known inhibitor of MRP1 was used as a positive control. After 68 h of incubation, 20 μL of MTT solution (4 mg·mL−1) was added to each well and further incubated for 4 h. Then the medium was discarded, 100 μL of DMSO was added into each well to dissolve the formazan crystals. The absorbance was then determined at 570 nm by an OPSYS microplate Reader from DYNEX Technologies, Inc. (Chantilly, VA, USA). Concentrations required to inhibit the growth by 50% (IC50) were calculated from cell survival curves.
[3H]-vinblastine and doxorubicin accumulation assay
The intracellular accumulation of [3H]-vinblastine in MRP1-overexpressing cells was determined as previously described (Cole et al., 1994). Cells were trypsinized and four aliquots (5 × 106 cells) were suspended in the medium and pre-incubated with or without ibrutinib (1 or 5 μM) or MK571 (50 μM) for 1 h at 37°C. Subsequently, these cells were treated with a radioactive substrate [3H]-vinblastine and incubated for another 2 h at 37°C. Later the suspended cells were pelleted at 4°C and washed twice with 10 mL ice-cold PBS. The cells were lysed in 1% SDS, and cell-associated radioactivity was determined using a liquid scintillation counter; the results obtained were expressed as pmol vinblastine per 106 cells. Each sample was placed in scintillation fluid and radioactivity was measured using a Packard TRI-CARB 1900CA liquid scintillation analyser from Packard Instrument Company Inc. (Downers Grove, IL, USA) (Sodani et al., 2012b). Intracellular accumulation of doxorubicin in MRP1-overexpessing HL60/Adr cells and their parental HL60 cells was examined by flow cytometry as previously described (Zheng et al., 2009). Briefly, the cells were collected and 1 × 106 cells of each HL60 and HL60/Adr cell lines were suspended in medium and pre-incubated with or without ibrutinib or MK571 for 2 h at 37°C. Later, 10 μM of doxorubicin was added to the cells and further incubated for another 2 h at 37°C. The cells were then washed with ice-cold PBS twice before reading it using a flow cytometer and a minimum of 20 000 events.
[3H]-vinblastine and doxorubicin efflux assay
To measure drug efflux, cells were pre-incubated with 5 μM ibrutinib or 50 μM MK571 for 1 h as described in the accumulation experiment. [3H]-vinblastine was then added to the samples and further incubated for a period of 2 h, after which the cells were washed with ice-cold PBS and then supplemented with fresh medium with or without ibrutinib (5 μM) or MK571 (50 μM) at 37°C. After 0, 30, 60 and 120 min, equal aliquots of cell suspension were removed and immediately washed twice with ice-cold PBS. The cells were then collected and lysed for radioactivity detection. Each sample was placed in scintillation fluid and radioactivity was measured as described previously (Patel et al., 2013). The efflux of doxorubicin in MRP1-overexpessing HL60/Adr cells and their parental HL60 cells was examined by flow cytometry as previously described (Tang et al., 2014).
Western blot analysis
Western blot analysis was performed to detect the expression levels of MRP1 protein after treatment with 5 μM ibrutinib for 0, 24, 48 and 72 h. Cells were collected and lysed with lysis buffer (pH 7.4, containing 1% Triton X-100 and 0.2% SDS) (Yan et al., 2011). The cell lysates were kept on ice for 20 min followed by centrifugation at 8573× g for 15 min at 4°C. The supernatant was separated and stored at −80°C until required for the experiment. BCA assay kit from Thermo Fischer Scientific Inc. (Rockford, IL, USA) was used to quantify the concentration of protein in each sample. Equal amounts of protein (80 μg) from various treatments were resolved by SDS-PAGE and transferred onto PVDF membranes through electrophoresis. The membranes were then blocked with 5% non-fat milk dissolved in TBST buffer (10 mmol·L−1 Tris-HCL, 150 mmol·L−1 NaCl and 0.1% Tween20 pH 8.0) for 2 h at room temperature. The membranes were incubated overnight with the primary monoclonal antibody against MRP1 (at a 1:200 dilution), or β-actin (at a 1:1000 dilution) at 4°C and then were incubated with HRP-conjugated secondary antibody (1:1000 dilution) for 2 h at room temperature. After washing the membranes three times with TBST, the protein–antibody complex was detected by enhanced chemiluminescence detection system (Amersham, NJ, USA). The expression of β-actin was used as a loading control (Sodani et al., 2012b).
RT-PCR and Q-PCR
Total mRNA was isolated by Trizol Reagent RNA extraction kit purchased from Molecular Research Center (Cincinnati, OH, USA). Q-PCR was performed with a Bio-Rad CFX96 real-time system (Hercules, CA, USA). The PCR primers used in this study are summarized in Supporting Information Table S1. The gene encoding GAPDH was used as a control. The amplification products were analysed on 1% agarose gels. Relative quantification of ABCC1 was performed using the 2-ΔΔCt method (Livak and Schmittgen, 2001). All experiments were repeated three times.
Animals
All animal care and experimental procedures complied with the the Animal Welfare Act and other federal statutes and were approved by the Institutional Animal Care and Use Committee at St. John's University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 28 animals were used in the experiments described here.
Male athymic NCR (nu/nu) nude mice, 5–6 weeks old and weighing 19–25 g, were purchased from Taconic Farms (NCRNU-M, Homozygous, Albino colour) and were used for developing the MRP1-overexpressing tumour xenograft model. All mice were maintained on a 12 h light/12 h dark cycle, with free access to sterilized food and water, and were maintained at the St. John's University Animal Facility.
Nude mouse xenograft model
Tumour xenografts of MRP1-overexpressing HEK293/MRP1 cells in nude mice were developed from earlier studies performed at our facility (Tiwari et al., 2013; Kathawala et al., 2014). HEK293/MRP1 cells (1 × 107) were injected s.c. under the armpits of the mice. The mice were randomized into four groups when the xenografts reached a mean diameter of 5 mm (n = 7) and treated with one of the following regimens: all treatments given every other day for 7 days (i) vehicle (autoclaved water); (ii) ibrutinib (30 mg kg−1, p.o.); (iii) vincristine (0.4 mg·kg−1, i.p.); and (iv) ibrutinib (30 mg·kg−1, p.o., given 1 h before giving vincristine) + vincristine (0.4 mg·kg−1, i.p.).
The concentration of vincristine was chosen according to Huang et al. (2008). Body weight, tumour dimensions, feeding behaviour and locomotor activity of each mouse were recorded every other day. On the last day of the experiment, the mice were killed and xenografts were dissected, weighed and measured as previously described (Mi et al., 2010). The body weights of the animals and the two perpendicular diameters (A and B) were recorded every other day, and tumour volume (V) was estimated according to the following formula:
Patient samples
This study was approved by the Ethics Review Committee at Sun Yat-Sen University. Bone marrow blood (4mL) was obtained from 10 patients with diagnosed AML or ALL, according to the FAB classification (Mihic-Probst et al. 2007) after informed consent. Patient characteristics are summarized in Supporting Information Table S2. Leukaemia blasts were isolated using Ficoll–Hypaque density gradient centrifugation and cultured in RPMI-1640 medium containing 20% FBS. Western blot was performed to detect the MRP1 expression of the patient samples.
Data analysis
All experiments were repeated at least three times. Microsoft Office Excel 2010 (Microsoft Corp., Redmond, WA, USA) and the statistical software SPSS16.0 (IBM, Armonk, NY, USA) were used in data processing. ImageJ (NIH, Bethesda, MD, USA) was used to analyse the greyscale ratio of Western blot. Statistical significance was determined using the Student's t-test. Significance was determined at P < 0.05 or P < 0.01.
Materials
[3H]-vinblastine sulphate (25 Ci·mmol−1) was purchased from American Radiolabeled Chemicals (St. Louis, MO, USA). Ibrutinib was obtained from ChemieTek (Indianapolis, IN, USA). PCI 29732 was purchased from Medchem Express (Shanghai, China). Vincristine was purchased from LC laboratories (Woburn, MA, USA). Monoclonal antibodies anti-MRP1 (sc-18835) and anti-β-actin (sc-8432) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). DMEM and RPMI-1640 were products of Gibco BRL (Grand Island, NY, USA), vinblastine, doxorubicin, paclitaxel, 5-FU, cisplatin, MK571, penicillin/streptomycin, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), DMSO and other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, USA).
Results
Cytotoxic effect of ibrutinib on MRP1-overexpressing cells and their parental-sensitive cells
Prior to investigating cytotoxicity of ibrutinib, we performed Western blots to determine the expression of MRP1 protein in the cells used in our experiments. High levels of MRP1 were expressed in HEK293/MRP1 and HL60/Adr cells (Figure 1B). The cytotoxicity of ibrutinib was evaluated in MRP1-overexpressing cells and parental-sensitive cells by MTT assay. The IC50 values of ibrutinib in these two sets of cells were >10 μM, and more than 85% of the cells survived at the concentration of 5 μM ibrutinib (Figure 1C and D). Based on these results, ibrutinib at a concentration of 5 μM was chosen as the maximum concentration for combination treatment with anticancer drugs, known to be MRP1 substrates.
Figure 1.

Cytotoxicity of ibrutinib in drug-resistant and their parental, drug-sensitive cells. (A) The chemical structure of ibrutinib; (B) Western blot showing the expression of MRP1 in HEK293/MRP1 and HL60/Adr cells; (C) concentration–response curves of HL60 and HL60/Adr cells treated with ibrutinib alone; (D) concentration–response curves of HEK293/pcDNA3.1 and HEK293/MRP1 cells treated with ibrutinib.
The effect of ibrutinib on reversing MRP1-mediated MDR in MRP1-ovexpressing cells
The cytotoxicity of MRP1 substrates such as vincristine, vinblastine, doxorubicin or non-MRP1 substrates, such as cisplatin, paclitaxel and 5-FU, was evaluated in the presence or absence of ibrutinib in HEK293/pcDNA3.1 and HEK293/MRP1 cells. As shown in Table 1, the MRP1-overexpressing HEK293/MRP1 cells exhibited resistance to MRP1 substrates such as vincristine, vinblastine and doxorubicin, compared with HEK293/pcDNA3.1 cells. Ibrutinib at 1 or 5 μM significantly sensitized HEK293/MRP1 cells to the MRP1 substrates but not to cisplatin, paclitaxel or 5-FU. The sensitizing effect of ibrutinib was more potent than that of MK571, a known inhibitor of MRP1 (Table 1). Similarly, ibrutinib significantly potentiated the cytotoxicity of MRP1 substrate drugs in another MRP1-overexpressing cell line, HL60/Adr leukaemia cells. However, ibrutinib did not alter the cytotoxicity of these drugs in HL60 cells (Table 2). Therefore, our results suggested that ibrutinib sensitized MRP1-overexpressing cells to antineoplastic drugs that are substrates of MRP1.
Tables of Links
| TARGETS |
|---|
| Transportersa |
| MRP1 (ABCC1) |
| Enzymesb |
| BTK, Bruton's tyrosine kinase |
| LIGANDS | |
|---|---|
| 5-FU | MK571 (verlukast) |
| Cisplatin | Paclitaxel |
| Doxorubicin | Vincristine |
| Ibrutinib | |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b,).
Table 1.
The effect of ibrutinib on reversal of MRP1-mediated MDR in HEK293/pcDNA3.1 and HEK293/MRP1 cells
| IC50 ± SD (nM) (fold resistance) | ||
|---|---|---|
| Compounds | HEK293/pcDNA3.1 | HEK293/MRP1 |
| Vincristine | 2.37 ± 0.67 (1.00) | 21.7 ± 3.22 (9.16) |
| +Ibrutinib 1 μM | 2.19 ± 0.40 (0.92) | 8.39 ± 1.01 (3.54)** |
| +Ibrutinib 5 μM | 1.84 ± 0.35 (0.78) | 2.51 ± 0.55 (1.06)** |
| +MK571 50 μM | 2.07 ± 0.32 (0.87) | 4.21 ± 0.43 (1.78)** |
| Vinblastine | 1.05 ± 0.20 (1.00) | 6.92 ± 1.02 (6.59) |
| +Ibrutinib 1 μM | 0.90 ± 0.11 (0.86) | 3.45 ± 0.66 (3.29)** |
| +Ibrutinib 5 μM | 0.80 ± 0.23 (0.76) | 1.55 ± 0.21 (1.47)** |
| +MK571 50 μM | 0.83 ± 0.39 (0.79) | 2.39 ± 0.49 (2.28)** |
| Doxorubicin | 21.3 ± 2.26 (1.00) | 115 ± 7.93 (5.39) |
| +Ibrutinib 1 μM | 20.7 ± 3.11 (0.97) | 67.1 ± 8.40 (3.14)** |
| +Ibrutinib 5 μM | 22.9 ± 1.76 (1.07) | 27.8 ± 2.01 (1.30)** |
| +MK571 50 μM | 21.9 ± 2.05 (1.03) | 58.8 ± 5.29 (2.76)** |
| Cisplatin | 1502 ± 120 (1.00) | 1932 ± 200 (1.29) |
| +Ibrutinib 1 μM | 1405 ± 200 (0.94) | 1912 ± 125 (1.27) |
| +Ibrutinib 5 μM | 1258 ± 179.50 (0.84) | 1761 ± 220 (1.17) |
| +MK571 50 μM | 1402 ± 210.38 (0.93) | 1848 ± 176 (1.23) |
| Paclitaxel | 7.03 ± 2.01 (1.00) | 6.33 ± 1.41 (0.90) |
| +Ibrutinib 5 μM | 6.11 ± 1.02 (0.87) | 5.50 ± 1.34 (0.78) |
| +MK571 50 μM | 5.84 ± 1.15 (0.83) | 4.99 ± 1.44 (0.71) |
| 5-FU | 14 418. ± 21265 (1.00) | 20 185 ± 3463 (1.40) |
| +Ibrutinib 5 μM | 15 270 ± 3016 (1.06) | 22 658 ± 4334 (1.57) |
| +MK571 50 μM | 16 369 ± 2264 (1.14) | 23 049 ± 5746 (1.60) |
Cell survival was determined by MTT assays. Data shown are means ± SDs of at least three independent experiments performed in triplicate. The fold resistance of MDR (values given in parentheses) was calculated by dividing the IC50 values of substrate in HEK293/MRP1 cells in the presence or absence of inhibitor, or HEK293/pcDNA3.1 cells with inhibitors, by the IC50 of HEK293/pcDNA3.1 cells without inhibitor.
P < 0.05
P < 0.01 significantly different from values obtained in the absence of inhibitor.
Table 2.
The effect of ibrutinib on reversal of MRP1-mediated MDR in HL60 and HL60/Adr cells
| IC50 ± SD (nM) (fold resistance) | ||
|---|---|---|
| Compounds | HL60 | HL60/Adr |
| Vincristine | 12.6 ± 2.33 (1.00) | 2021 ± 191(161) |
| +Ibrutinib 1 μM | 13.2 ± 1.51 (1.05) | 900 ± 120 (72)** |
| +Ibrutinib 5 μM | 9.12 ± 2.82 (0.72) | 138 ± 11.3 (11.0)** |
| +MK571 50 μM | 10.5 ± 1.90 (0.83) | 190 ± 20.4 (15.1)** |
| Doxorubicin | 59.2 ± 14.90 (1.00) | 3890 ± 340 (65.8) |
| +Ibrutinib 1 μM | 52.3 ± 8.11 (0.88) | 1671 ± 210 (28.2)** |
| +Ibrutinib 5 μM | 48.1 ± 5.27 (0.81) | 624 ± 100 (10.6)** |
| +MK571 50 μM | 49.8 ± 4.56 (0.84) | 705 ± 70.4 (11.9)** |
| Cisplatin | 1804 ± 201 (1.00) | 2101 ± 321 (1.16) |
| +Ibrutinib 5 μM | 1726 ± 229 (0.96) | 2255 ± 219 (1.25) |
| +MK571 50 μM | 1623 ± 219 (0.90) | 2076 ± 302 (1.15) |
| Paclitaxel | 7.71 ± 1.02 (1.00) | 6.65 ± 1.20 (0.86) |
| +Ibrutinib 5 μM | 8.16 ± 1.45 (1.05) | 6.74 ± 0.89 (0.87) |
| +MK571 50 μM | 7.28 ± 2.10 (0.94) | 7.90 ± 1.48 (1.02) |
| 5-FU | 35 999 ± 4005 (1.00) | 38 037 ± 4185 (1.06) |
| +Ibrutinib 5 μM | 31 653 ± 5392 (0.88) | 39 627 ± 5205 (1.10) |
| +MK571 50 μM | 39 9757 ± 6198 (1.11) | 41 696 ± 5531 (1.16) |
Cell survival was determined by MTT assays. Data shown are means ± SDs of three independent experiments performed in triplicate. The fold resistance of MDR (values given in parentheses) was calculated by dividing the IC50 values of substrate in HL60/Adr cells in the absence or presence of inhibitor, or the IC50 values of substrate in HL60 cells with inhibitor, by IC50 of HL60 cells without inhibitor.
P < 0.05
P < 0.01 significantly different from values obtained in absence of inhibitor.
The effect of ibrutinib on intracellular accumulation of [3H]-vinblastine and doxorubicin in MRP1-overexpressing cells
To elucidate the mechanism underlying reversal of drug resistance, we measured the effect of ibrutinib on the accumulation of [3H]-vinblastine in the cells. The intracellular levels of [3H]-vinblastine were measured in the presence or absence of ibrutinib in HEK293/pcDNA3.1 and HEK293/MRP1 cells. The intracellular accumulation levels of [3H]-vinblastine in HEK293/MRP1 cells were significantly lower than those in HEK293/pcDNA3.1 cells, after incubation with [3H]-vinblastine for 2 h (Figure 2A). However, the intracellular accumulation levels of [3H]-vinblastine in HEK293/MRP1 cells were increased significantly after treatment with ibrutinib. Furthermore, the increased effect produced by ibrutinib at 5 μM was comparable to that of MK571 at 50 μM. However, neither ibrutinib nor MK571 increased the intracellular accumulation of [3H]-vinblastine in HEK293/pcDNA3.1 cells (Figure 2A). These results suggested that ibrutinib is likely to affect the efflux function of the MRP1 transporter. Similarly, flow cytometry analysis showed that ibrutinib increased the intracellular accumulation of doxorubicin in HL60/Adr cells but not in the parental HL60 cells (Supporting Information Fig. S1A).
Figure 2.
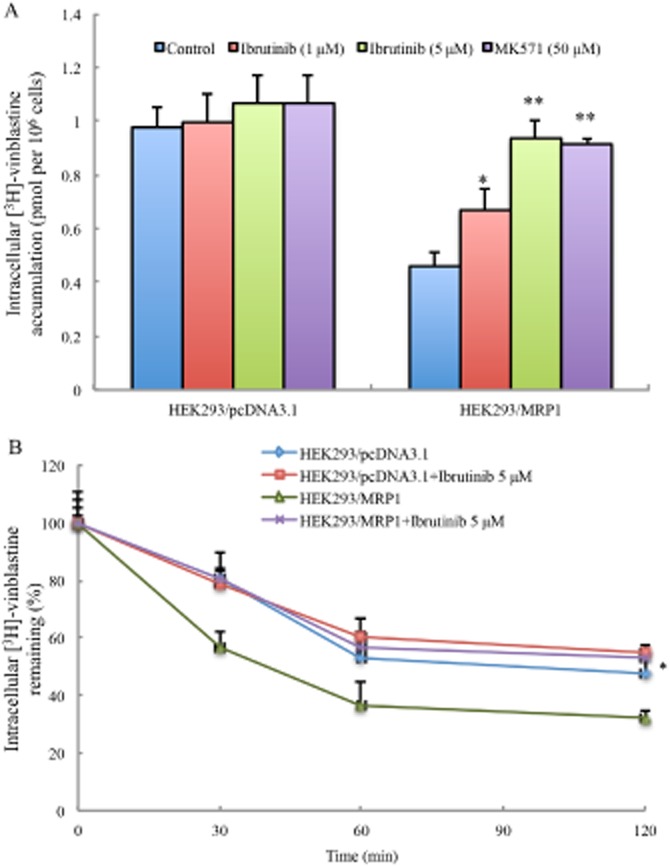
Ibrutinib increased the intracellular accumulation of [3H]-vinblastine by inhibiting efflux function of MRP1. (A) The accumulation of [3H]-vinblastine was measured in HEK293/pcDNA3.1 cells and HEK293/MRP1 cells with or without ibrutinib; MK571 was used as a known inhibitor of MRP1. Data shown are means (with SD) of triplicate determinations. * P < 0.05, ** P < 0.01, significantly different from control. (B) The effects of ibrutinib on the efflux of [3H]-vinblastine from HEK293/pcDNA3.1 and HEK293/MRP1 cells was measured. A time-dependent decrease of intracellular [3H]-vinblastine was found (0, 30, 60 and 120 min). Data shown are means ± SDs from three independent determinations, in triplicate. * P < 0.05, significantly different from control.
The effect of ibrutinib on efflux of [3H]-vinblastine in MRP1-overexpressing cells
In order to determine whether the increase in intracellular accumulation of [3H]-vinblastine was due to inhibition of MRP1 efflux, we measured the efflux of [3H]-vinblastine in the presence or absence of ibrutinib at different time points (0, 30, 60 and 120 min) in HEK293/pcDNA3.1 and HEK293/MRP1 cells. In the absence of ibrutinib, there was a rapid efflux of [3H]-vinblastine from HEK293/MRP1 cells, resulting in decreased accumulation of [3H]-vinblastine, compared with the HEK293/pcDNA3.1 cells. However, in the presence of ibrutinib, the MRP1 transporter was blocked, leading to decreased efflux of [3H]-vinblastine at different time points (Figure 2B). The intracellular levels of [3H]-vinblastine at 0 min of the efflux time point was set as 100% and, over 120 min, there was a time-related reduction in intracellular [3H]-vinblastine in HEK293/MRP1 cells in the absence of ibrutinib. When HEK293/MRP1 cells were incubated with ibrutinib, the levels of intracellular [3H]-vinblastine were increased at all time points. Our results also showed that ibrutinib significantly inhibited the efflux of doxorubicin in HL60/Adr cells (Supporting Information Fig. S1B). These data indicated that ibrutinib inhibits the efflux function of the MRP1 transporter and significantly increased accumulation of substrates.
The effect of ibrutinib on the expression levels of mRNA and protein of MRP1
The sensitization effect of ibrutinib could be achieved either by antagonizing the function of MRP1 or decreasing the expression levels of MRP1. We performed RT-PCR, Q-PCR and Western blot to analyse the effect of ibrutinib on the expression levels of mRNA and protein of MRP1. As shown in Figure 3, the expression levels of mRNA or protein of MRP1 were not significantly altered in HEK293/MRP1 cells after treatment with ibrutinib for 24, 48 and 72 h. The ratios of MRP1/β-actin were proportional to the MRP1 protein levels (Figure 3). The result suggested that the sensitization effect of ibrutinib was not caused by a decrease in expression of MRP1.
Figure 3.
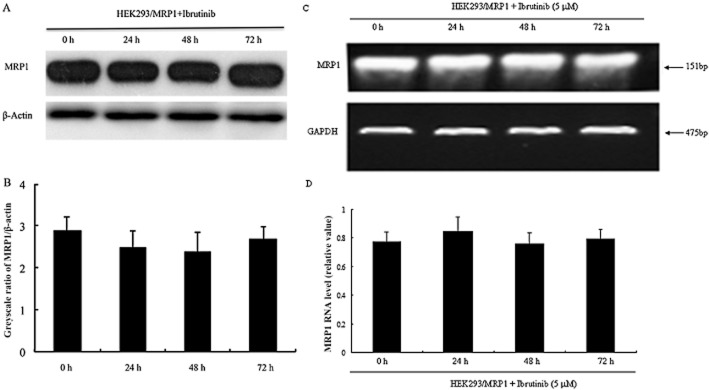
Ibrutinib did not alter the expression levels of MRP1 in MDR cells. (A) The effect of ibrutinib on the protein levels of MRP1 was tested after HEK293/MRP1 cells were treated with 5 μM ibrutinib for 0, 24, 48 and 72 h and Western blots were performed. (B) ImageJ was used to analyse the greyscale ratios of MRP1/β-actin. The greyscale ratios were proportional to the MRP1 protein levels. (C) RT-PCR was performed to confirm mRNA levels in HEK293/MRP1 cells. (D) Q-PCR was further conducted to test mRNA levels in HEK293/MRP1 cells. The differences were not statistically significant (P > 0.05).
The effect of ibrutinib on reversing MRP1-mediated MDR in the nude mouse xenograft model
A model of MDR in vivo, xenografts of HEK293/MRP1 cells in athymic mice, was used to estimate the efficacy of ibrutinib to reverse MRP1-mediated vincristine resistance. There was no significant difference in tumour size between groups treated with saline and ibrutinib. The group treated with vincristine (0.4 mg kg−1, i.p.) showed mild reductions of the growth rate of tumours, compared that in the control group, treated with saline (Figure 4A and B). However, the group treated with ibrutinib (30 mg kg−1, p.o.) in combination with vincristine (0.4 mg kg−1, i.p.) showed a greater inhibition of tumour growth than in the animals treated with saline, vincristine or ibrutinib alone (Figure 4A and B). The mean weights of tumours removed from mice at the end of the experiment (Figure 4C) showed inhibition by vincristine alone and a significantly greater inhibition after the combination treatment (ibrutinib + vincristine). In addition, there was no significant difference in body weight or in phenotypic changes between the treatment groups after the course of therapy (Figure 4D). Our results suggested that ibrutinib in combination with vincristine did not increase the in vivo toxicity, but improved the anti-tumour efficacy of vincristine in this HEK293/MRP1 cell xenograft model.
Figure 4.

Ibrutinib enhanced the effect of vincristine on the growth of an MRP1-overexpressing HEK293/MRP1 cell xenograft model in athymic nude mice. (A) Images of excised HEK293/MRP1 tumour tissues from different mice are shown on the 20th day after implantation (n = 7). (B) Changes in tumour volume with time in MRP1 xenograft model are shown. (C) Mean (with SD) weights of the excised HEK293/MRP1 tumours from the mice treated with vehicle, ibrutinib, vincristine or combination of ibrutinib and vincristine (n = 7 each group). *P < 0.05 versus vehicle group; #P < 0.05 versus the paclitaxel group. (D) Average percentage of body weight change after treatments.
The effect of ibrutinib on the cytotoxicity of vincristine on patient-derived leukaemic blast cells that overexpress MRP1
MRP1 is expressed in both AML and ALL patients (Mahjoubi et al., 2008). Therefore, we tested the expression of MRP1 and analysed the cytotoxicity of vincristine, with or without treatment of ibrutinib, in leukaemic blast cells derived from patients. Our results demonstrated that 3 of 9 patient samples displayed detectable expression of MRP1 (Figure 5A). Treatment of cultures of these leukaemia blast cells with 5 μM ibrutinib sensitized them to the effects of vincristine, in these three samples (Figure 5B–D). Our findings suggested that combination with ibrutinib and vincristine could have significant clinical value, although further clinical studies are necessary.
Figure 5.

Ibrutinib increased the cytotoxicity of vincristine in patient-derived primary cultures of leukaemic blast cells with MRP1 overexpression. Bone marrow samples were collected from nine newly diagnosed patients'. (A) The expression of MRP1 in primary cultures of leukaemia blasts was determined by Western blot. Samples from three patients showed higher levels of MRP1. (B, C, D) Enhancement by ibrutinib of vincristine cytotoxicity in primary cultures of leukaemia blasts with MRP1 overexpression from the three patients with high MRP1 levels. Data shown are means ± SDs for independent determinations in triplicate.
Discussion and conclusions
The kinase BTK is an essential cytoplasmic component of the B-cell receptor signalling pathway, which has become an attractive target for cancer chemotherapy (Akinleye et al., 2013). Ibrutinib, a novel BTK-targeting inhibitor, has shown significant activities across a variety of B-cell neoplastic disorders in preclinical models and clinical trials (Sharma, 2013). The US FDA has approved ibrutinib for the treatment of patients with mantle cell lymphoma or chronic lymphocytic leukaemia (Dolgin, 2013). In addition, several clinical trials are being conducted to evaluate the synergistic effect of ibrutinib administered in combination with rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone in newly diagnosed patients with non-germinal centre B-cell subtype of diffuse large B-cell lymphoma (http://clinicaltrials.gov).
There is good evidence that TK inhibitors have an important interaction with ABC transporters, such as ABCB1, ABCG2 and MRP1. Several TK inhibitors such as lapatinib, gefitinib and erlotinib are also inhibitors of ABCB1 and ABCG2 (Elkind et al., 2005; Shi et al., 2007; Dai et al., 2008). However, little is known to date about the interaction between TK inhibitors and MRP1 and none of the compounds has been approved for clinical use, so far. Because ibrutinib demonstrated a noticeable inhibition of the BTK signal pathway and a promising prospect in combined treatment with traditional anticancer drugs, it was of great interest to investigate the effect of ibrutinib on MRP1-mediated MDR.
As reported previously, the MRP1-overexpressing cell lines, HEK293/MRP1 and HL60/Adr, showed high resistance to vincristine, vinblastine, etoposide and doxorubicin (Wu et al., 2005). In the present research, we showed that ibrutinib, at concentrations showing little or no toxicity, significantly potentiated the cytotoxicity of known MRP1 substrates in MRP1-overexpressing cells. This effect was much more potent than MK571, a well-known inhibitor of MRP1 drug efflux. However, the same concentrations of ibrutinib did not sensitize the parental cells HEK293/pcDNA3.1 or HL60 to the anticancer agents. Furthermore, ibrutinib did not affect the potencies of non-MRP1 substrates such as cisplatin, paclitaxel and 5-FU. Another BTK inhibitor, PCI 29732, also showed reversal of the effects of MRP1, but this effect was much more modest (Supporting Information Fig. S2 and Table S3). Moreover, we have investigated the effects of ibrutinib in primary cultures of patient-derived leukaemic blast cells. Our results indicated that MRP1 was overexpressed in such cells and that ibrutinib sensitized the leukaemia blast cells to the cytotoxicity of vincristine in MRP1-overexpressing samples. These results indicated that ibrutinib significantly sensitized MRP1-overexpressing cells to those chemotherapeutic agents that were also substrates for MRP1.
In order to investigate the potential mechanism(s) by which ibrutinib sensitized the MDR cells to antineoplastic drugs, we examined the effect of ibrutinib on the accumulation of the substrates in MRP1-overexpressing cells. The results showed that ibrutinib increased intracellular accumulation of substrates in these cells. The efflux studies confirmed that this effect was achieved by antagonizing the efflux function of the MRP1 transporter. In further analysis of the mechanisms involved, we performed RT-PCR, Q-PCR and Western blot to determine the mRNA and protein expression levels of MRP1 respectively. The results indicated that ibrutinib did not alter the expression of mRNA or protein of the MRP1 transporter, which eliminated the possibility of down-regulation of protein levels of MRP1 by ibrutinib.
Previous pharmacokinetic studies reported that ibrutinib, at a dose of 14.2 mg·kg−1, could produce a peak plasma concentration of 1.07 μM (Honigberg et al., 2010). Therefore, the dose of 30 mg·kg−1 used in our in vivo experiments may produce a plasma concentration that could reach the concentration range we used for the cellular studies. Thus, we assume that the concentrations of ibrutinib used in reversal experiments in vitro were achievable in tumour tissues after therapeutic treatment. In order to determine whether the in vitro effects of ibrutinib can be translated to the in vivo setting, we examined the effect of ibrutinib on the antitumour activity of vincristine in the model using MRP1-overexpressing HEK293/MRP1 xenografts. Consistent with earlier findings, our present results showed that vincristine alone mildly reduced the tumour volumes. However, the combination of ibrutinib with vincristine resulted in a significantly enhanced antitumour activity of vincristine in this HEK293/MRP1 tumour xenograft model (Figure 4A and B). Additionally, there was no substantial change in body weight in mice treated with the drug combination compared with those with the individual drug treatment alone (Figure 4D). Thus, we were able to demonstrate that ibrutinib significantly potentiated the antitumour effect of vincristine in MRP1-overexpressing HEK293/MRP1 cell xenografts.
In conclusion, we provide the first in vitro, in vivo and ex vivo evidence that ibrutinib could significantly enhance the efficacy of chemotherapeutic agents in MRP1-overexpressing MDR cells, an effect achieved by antagonizing the efflux function of the MRP1 transporter. Although the combination is effective, it should be noted that the cytotoxic effects of doxorubicin reportedly causes alterations in cardiac function and MRP1 localization in the heart does protect the heart from such agents. Thus when combining with MRP1 modulatory agents such as ibrutinib, cardiac function should be monitored carefully. Combination of ibrutinib with substrates of MRP1 could have significant clinical value and further clinical investigation is warranted.
Acknowledgments
We thank Dr Suresh Ambudkar for HEK293/MRP1 cells (NCI, NIH, Bethesda); Drs S.E. Bates and R.W. Robey (NCI, NIH, Bethesda) for HL60 and HL60/Adr cells; we are thankful to ChemieTek (Indianapolis, IN, USA) for supplying free sample of ibrutinib for our study of the project. This work was supported by St. John's University Research Seed Grant (No. 579-1110-7002) for Z.-S. C. and major science and technology project of 863 Program of China (No. 2012AA02A303) for L.-W. F., National Natural Sciences Foundation of China (No. 81072669 and No. 81061160507) for L.-W. F.
Glossary
- ABC
ATP-binding cassette
- BTK
Bruton's tyrosine kinase
- MDR
multidrug resistance
- MRP1
multidrug resistance protein 1
Author contributions
H. Z., A. P., S.-L. M., X. J. L., P.-Q. Y. and Y.-K. Z. performed research and analysed the data. H. Z. and A. P. wrote the manuscript. R. J. K., Y.-J. W. and N. A. assisted in revising the manuscript. Z.-S. C. and L.-W. F. designed the research and revised the manuscript.
Conflict of interest
There are no potential conflicts of interest to be disclosed.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12889
Figure S1 Ibrutinib increased the intracellular accumulation of doxorubicin (DOX) by inhibiting efflux function of MRP1. (A) The accumulation of DOX was measured in HL60 cells and HL60/Adr cells with or without ibrutinib; MK571 was used as a positive inhibitor of MRP1. Columns are the mean of triplicate determinations; bars represent SDs. (B) The effects of ibrutinib on the efflux of DOX from HL60 and HL60/Adr cells was measured. A time-dependent decrease of intracellular DOX was found (0, 30, 60 and 120 min). Data shown were means ± SDs for independent determinations in triplicate. Three independent experiments were performed.
Figure S2 Cytotoxicity of PCI 29732 in drug-resistant and their parental-sensitive cells. Concentration–response curves of HL60 and HL60/Adr cells treated with PCI 29732 alone. Data shown were means ± SDs for independent determinations in triplicate. Three independent experiments were performed.
Table S1 Summery of PCR primers.
Table S2 Patient characteristics.
Table S3 The effect of PCI 29732 on reversal of MRP1-mediated MDR in HL60 and HL60/Adr cells.
References
- Akinleye A, Chen Y, Mukhi N, Song Y, Liu D. Ibrutinib and novel BTK inhibitors in clinical development. J Hematol Oncol. 2013;6:59. doi: 10.1186/1756-8722-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013a;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- Burger H, Nooter K, Zaman GJ, Sonneveld P, van Wingerden KE, Oostrum RG, et al. Expression of the multidrug resistance-associated protein (MRP) in acute and chronic leukemias. Leukemia. 1994;8:990–997. [PubMed] [Google Scholar]
- Burns BM, Taylor JF, Herring KL, Herring AD, Holder MT, Holder DA, et al. Bovine microsatellite dinucleotide repeat polymorphisms at the TEXAN16, TEXAN17, TEXAN18, TEXAN19 and TEXAN20 loci. Anim Genet. 1995;26:208–209. doi: 10.1111/j.1365-2052.1995.tb03174.x. [DOI] [PubMed] [Google Scholar]
- Cole SP, Deeley RG. Multidrug resistance mediated by the ATP-binding cassette transporter protein MRP. Bioessays. 1998;20:931–940. doi: 10.1002/(SICI)1521-1878(199811)20:11<931::AID-BIES8>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Cole SP, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist KC, et al. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992;258:1650–1654. doi: 10.1126/science.1360704. [DOI] [PubMed] [Google Scholar]
- Cole SP, Sparks KE, Fraser K, Loe DW, Grant CE, Wilson GM, et al. Pharmacological characterization of multidrug resistant MRP-transfected human tumor cells. Cancer Res. 1994;54:5902–5910. [PubMed] [Google Scholar]
- Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–7914. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danks MK, Schmidt CA, Cirtain MC, Suttle DP, Beck WT. Altered catalytic activity of and DNA cleavage by DNA topoisomerase II from human leukemic cells selected for resistance to VM-26. Biochemistry. 1988;27:8861–8869. doi: 10.1021/bi00424a026. [DOI] [PubMed] [Google Scholar]
- Dolgin E. Cancer's true breakthroughs. Nat Med. 2013;19:660–663. doi: 10.1038/nm.3245. [DOI] [PubMed] [Google Scholar]
- Duffy CP, Elliott CJ, O'Connor RA, Heenan MM, Coyle S, Cleary IM, et al. Enhancement of chemotherapeutic drug toxicity to human tumour cells in vitro by a subset of non-steroidal anti-inflammatory drugs (NSAIDs) Eur J Cancer. 1998;34:1250–1259. doi: 10.1016/s0959-8049(98)00045-8. [DOI] [PubMed] [Google Scholar]
- Elkind NB, Szentpetery Z, Apati A, Ozvegy-Laczka C, Varady G, Ujhelly O, et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib) Cancer Res. 2005;65:1770–1777. doi: 10.1158/0008-5472.CAN-04-3303. [DOI] [PubMed] [Google Scholar]
- Flens MJ, Zaman GJ, van der Valk P, Izquierdo MA, Schroeijers AB, Scheffer GL, et al. Tissue distribution of the multidrug resistance protein. Am J Pathol. 1996;148:1237–1247. [PMC free article] [PubMed] [Google Scholar]
- Giaccone G, Gazdar AF, Beck H, Zunino F, Capranico G. Multidrug sensitivity phenotype of human lung cancer cells associated with topoisomerase II expression. Cancer Res. 1992;52:1666–1674. [PubMed] [Google Scholar]
- Gollapudi S, Kim CH, Tran BN, Sangha S, Gupta S. Probenecid reverses multidrug resistance in multidrug resistance-associated protein-overexpressing HL60/AR and H69/AR cells but not in P-glycoprotein-overexpressing HL60/Tax and P388/ADR cells. Cancer Chemother Pharmacol. 1997;40:150–158. doi: 10.1007/s002800050640. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- Haimeur A, Conseil G, Deeley RG, Cole SP. The MRP-related and BCRP/ABCG2 multidrug resistance proteins: biology, substrate specificity and regulation. Curr Drug Metab. 2004;5:21–53. doi: 10.2174/1389200043489199. [DOI] [PubMed] [Google Scholar]
- Hegedus T, Orfi L, Seprodi A, Varadi A, Sarkadi B, Keri G. Interaction of tyrosine kinase inhibitors with the human multidrug transporter proteins, MDR1 and MRP1. Biochim Biophys Acta. 2002;1587:318–325. doi: 10.1016/s0925-4439(02)00095-9. [DOI] [PubMed] [Google Scholar]
- Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Jin J, Sun H, Liu GT. Reversal of P-glycoprotein-mediated multidrug resistance of cancer cells by five schizandrins isolated from the Chinese herb Fructus Schizandrae. Cancer Chemother Pharmacol. 2008;62:1015–1026. doi: 10.1007/s00280-008-0691-0. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Li ZS, Lu YP, Rea PA. The GS-X pump in plant, yeast, and animal cells: structure, function, and gene expression. Biosci Rep. 1997;17:189–207. doi: 10.1023/a:1027385513483. [DOI] [PubMed] [Google Scholar]
- Jedlitschky G, Leier I, Buchholz U, Center M, Keppler D. ATP-dependent transport of glutathione S-conjugates by the multidrug resistance-associated protein. Cancer Res. 1994;54:4833–4836. [PubMed] [Google Scholar]
- Kathawala RJ, Sodani K, Chen K, Patel A, Abuznait AH, Anreddy N, et al. Masitinib antagonizes ATP-binding cassette subfamily C member 10-mediated paclitaxel resistance: a preclinical study. Mol Cancer Ther. 2014;13:714–723. doi: 10.1158/1535-7163.MCT-13-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mahjoubi F, Akbari S, Montazeri M, Moshyri F. MRP1 polymorphisms (T2684C, C2007T, C2012T, and C2665T) are not associated with multidrug resistance in leukemic patients. Genet Mol Res. 2008;7:1369–1374. doi: 10.4238/vol7-4gmr482. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi YJ, Liang YJ, Huang HB, Zhao HY, Wu CP, Wang F, et al. Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters. Cancer Res. 2010;70:7981–7991. doi: 10.1158/0008-5472.CAN-10-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihic-Probst D, Kuster A, Kilgus S, Bode-Lesniewska B, Ingold-Heppner B, Leung C, et al. Consistent expression of the stem cell renewal factor BMI-1 in primary and metastatic melanoma. Int J Cancer. 2007;121:1764–1770. doi: 10.1002/ijc.22891. [DOI] [PubMed] [Google Scholar]
- Muller M, Meijer C, Zaman GJ, Borst P, Scheper RJ, Mulder NH, et al. Overexpression of the gene encoding the multidrug resistance-associated protein results in increased ATP-dependent glutathione S-conjugate transport. Proc Natl Acad Sci U S A. 1994;91:13033–13037. doi: 10.1073/pnas.91.26.13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Yong M, Peng XH, Petre B, Arora S, Ambudkar SV. Evidence for the role of glycosylation in accessibility of the extracellular domains of human MRP1 (ABCC1) Biochemistry. 2002;41:10123–10132. doi: 10.1021/bi026075s. [DOI] [PubMed] [Google Scholar]
- Nagayama S, Chen ZS, Kitazono M, Takebayashi Y, Niwa K, Yamada K, et al. Increased sensitivity to vincristine of MDR cells by the leukotriene D4 receptor antagonist, ONO-1078. Cancer Lett. 1998;130:175–182. doi: 10.1016/s0304-3835(98)00132-3. [DOI] [PubMed] [Google Scholar]
- Nakano R, Oka M, Nakamura T, Fukuda M, Kawabata S, Terashi K, et al. A leukotriene receptor antagonist, ONO-1078, modulates drug sensitivity and leukotriene C4 efflux in lung cancer cells expressing multidrug resistance protein. Biochem Biophys Res Commun. 1998;251:307–312. doi: 10.1006/bbrc.1998.9472. [DOI] [PubMed] [Google Scholar]
- Nooter K, Westerman AM, Flens MJ, Zaman GJ, Scheper RJ, van Wingerden KE, et al. Expression of the multidrug resistance-associated protein (MRP) gene in human cancers. Clin Cancer Res. 1995;1:1301–1310. [PubMed] [Google Scholar]
- Norris MD, Bordow SB, Marshall GM, Haber PS, Cohn SL, Haber M. Expression of the gene for multidrug-resistance-associated protein and outcome in patients with neuroblastoma. N Engl J Med. 1996;334:231–238. doi: 10.1056/NEJM199601253340405. [DOI] [PubMed] [Google Scholar]
- Novero A, Ravella PM, Chen Y, Dous G, Liu D. Ibrutinib for B cell malignancies. Exp Hematol Oncol. 2014;3:4. doi: 10.1186/2162-3619-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Tiwari AK, Chufan EE, Sodani K, Anreddy N, Singh S, et al. PD173074, a selective FGFR inhibitor, reverses ABCB1-mediated drug resistance in cancer cells. Cancer Chemother Pharmacol. 2013;72:189–199. doi: 10.1007/s00280-013-2184-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasschaert SL, de Bont ES, Boezen M, vander Kolk DM, Daenen SM, Faber KN, et al. Expression of multidrug resistance-associated proteins predicts prognosis in childhood and adult acute lymphoblastic leukemia. Clin Cancer Res. 2005;11:8661–8668. doi: 10.1158/1078-0432.CCR-05-1096. [DOI] [PubMed] [Google Scholar]
- Sharma SP. Targeting the B-cell signalling pathway in CLL and MCL. Lancet Oncol. 2013;14:e343. doi: 10.1016/s1470-2045(13)70313-9. [DOI] [PubMed] [Google Scholar]
- Shi Z, Peng XX, Kim IW, Shukla S, Si QS, Robey RW, et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 2007;67:11012–11020. doi: 10.1158/0008-5472.CAN-07-2686. [DOI] [PubMed] [Google Scholar]
- Sodani K, Patel A, Kathawala RJ, Chen ZS. Multidrug resistance associated proteins in multidrug resistance. Chin J Cancer. 2012a;31:58–72. doi: 10.5732/cjc.011.10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodani K, Tiwari AK, Singh S, Patel A, Xiao ZJ, Chen JJ, et al. GW583340 and GW2974, human EGFR and HER-2 inhibitors, reverse ABCG2- and ABCB1-mediated drug resistance. Biochem Pharmacol. 2012b;83:1613–1622. doi: 10.1016/j.bcp.2012.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang R, Faussat AM, Majdak P, Perrot JY, Chaoui D, Legrand O, et al. Valproic acid inhibits proliferation and induces apoptosis in acute myeloid leukemia cells expressing P-gp and MRP1. Leukemia. 2004;18:1246–1251. doi: 10.1038/sj.leu.2403390. [DOI] [PubMed] [Google Scholar]
- Tang SJ, Chen LK, Wang F, Zhang YK, Huang ZC, To KK, et al. CEP-33779 antagonizes ATP-binding cassette subfamily B member 1 mediated multidrug resistance by inhibiting its transport function. Biochem Pharmacol. 2014;91:144–156. doi: 10.1016/j.bcp.2014.07.008. [DOI] [PubMed] [Google Scholar]
- Tiwari AK, Sodani K, Dai CL, Abuznait AH, Singh S, Xiao ZJ, et al. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett. 2013;328:307–317. doi: 10.1016/j.canlet.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnholds J, Evers R, van Leusden MR, Mol CA, Zaman GJ, Mayer U, et al. Increased sensitivity to anticancer drugs and decreased inflammatory response in mice lacking the multidrug resistance-associated protein. Nat Med. 1997;3:1275–1279. doi: 10.1038/nm1197-1275. [DOI] [PubMed] [Google Scholar]
- Wijnholds J, Scheffer GL, van der Valk M, van der Valk P, Beijnen JH, Scheper RJ, et al. Multidrug resistance protein 1 protects the oropharyngeal mucosal layer and the testicular tubules against drug-induced damage. J Exp Med. 1998;188:797–808. doi: 10.1084/jem.188.5.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnholds J, deLange EC, Scheffer GL, van den Berg DJ, Mol CA, van der Valk M, et al. Multidrug resistance protein 1 protects the choroid plexus epithelium and contributes to the blood-cerebrospinal fluid barrier. J Clin Invest. 2000;105:279–285. doi: 10.1172/JCI8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CP, Calcagno AM, Hladky SB, Ambudkar SV, Barrand MA. Modulatory effects of plant phenols on human multidrug-resistance proteins 1, 4 and 5 (ABCC1, 4 and 5) FEBS J. 2005;272:4725–4740. doi: 10.1111/j.1742-4658.2005.04888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan YY, Zheng LS, Zhang X, Chen LK, Singh S, Wang F, et al. Blockade of Her2/neu binding to Hsp90 by emodin azide methyl anthraquinone derivative induces proteasomal degradation of Her2/neu. Mol Pharm. 2011;8:1687–1697. doi: 10.1021/mp2000499. [DOI] [PubMed] [Google Scholar]
- Zheng LS, Wang F, Li YH, Zhang X, Chen LM, Liang YJ, et al. Vandetanib (Zactima, ZD6474) antagonizes ABCC1- and ABCG2-mediated multidrug resistance by inhibition of their transport function. PLoS ONE. 2009;4:e5172. doi: 10.1371/journal.pone.0005172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Ibrutinib increased the intracellular accumulation of doxorubicin (DOX) by inhibiting efflux function of MRP1. (A) The accumulation of DOX was measured in HL60 cells and HL60/Adr cells with or without ibrutinib; MK571 was used as a positive inhibitor of MRP1. Columns are the mean of triplicate determinations; bars represent SDs. (B) The effects of ibrutinib on the efflux of DOX from HL60 and HL60/Adr cells was measured. A time-dependent decrease of intracellular DOX was found (0, 30, 60 and 120 min). Data shown were means ± SDs for independent determinations in triplicate. Three independent experiments were performed.
Figure S2 Cytotoxicity of PCI 29732 in drug-resistant and their parental-sensitive cells. Concentration–response curves of HL60 and HL60/Adr cells treated with PCI 29732 alone. Data shown were means ± SDs for independent determinations in triplicate. Three independent experiments were performed.
Table S1 Summery of PCR primers.
Table S2 Patient characteristics.
Table S3 The effect of PCI 29732 on reversal of MRP1-mediated MDR in HL60 and HL60/Adr cells.