

Abstract
Background
Cis-regulatory modules (CRMs), or the DNA sequences required for regulating gene expression, play the central role in biological researches on transcriptional regulation in metazoan species. Nowadays, the systematic understanding of CRMs still mainly resorts to computational methods due to the time-consuming and small-scale nature of experimental methods. But the accuracy and reliability of different CRM prediction tools are still unclear. Without comparative cross-analysis of the results and combinatorial consideration with extra experimental information, there is no easy way to assess the confidence of the predicted CRMs. This limits the genome-wide understanding of CRMs.
Description
It is known that transcription factor binding and epigenetic profiles tend to determine functions of CRMs in gene transcriptional regulation. Thus integration of the genome-wide epigenetic profiles with systematically predicted CRMs can greatly help researchers evaluate and decipher the prediction confidence and possible transcriptional regulatory functions of these potential CRMs. However, these data are still fragmentary in the literatures. Here we performed the computational genome-wide screening for potential CRMs using different prediction tools and constructed the pioneer database, cisMEP (cis-regulatory module epigenetic profile database), to integrate these computationally identified CRMs with genomic epigenetic profile data. cisMEP collects the literature-curated TFBS location data and nine genres of epigenetic data for assessing the confidence of these potential CRMs and deciphering the possible CRM functionality.
Conclusions
cisMEP aims to provide a user-friendly interface for researchers to assess the confidence of different potential CRMs and to understand the functions of CRMs through experimentally-identified epigenetic profiles. The deposited potential CRMs and experimental epigenetic profiles for confidence assessment provide experimentally testable hypotheses for the molecular mechanisms of metazoan gene regulation. We believe that the information deposited in cisMEP will greatly facilitate the comparative usage of different CRM prediction tools and will help biologists to study the modular regulatory mechanisms between different TFs and their target genes.
Keywords: epigenetic profile, cis-regulatory module, chromatin regulation, histone modification, sequence conservation, regulatory protein
Background
Differential gene expression distinguishes distinct cell types in the differentiation of cells [1]. Correct temporal and spatial control of gene expression is crucial in different developmental stages in metazoans. In metazoan cells, transcriptional regulation of gene expression is controlled in a modular manner by specific DNA sequences located in the intergenic regions or in the introns [2]. Identifying these regulatory DNA sequences, or the cis-regulatory modules (CRMs), and their functions in gene transcriptional regulation can both expand our understanding of differential gene regulation and have potential application in medicine as well.
Three major CRM functions are known. In metazoan cells, CRMs can function as promoters, enhancers/silencers or insulators [3]. Promoter regulatory DNA sequences direct RNA polymerase to initiate gene transcription [4]. Enhancer/silencer CRMs operationally activate/repress the transcriptional expression of target genes with the help of regulatory proteins bound to them [5]. Finally, insulators block the effect of other enhancers with proteins such as CTCF [6]. Two major factors are closely related to CRM functionality: transcription factor (TF) binding and the shared chromatin epigenetic profiles [3,7-9]. Depending on the epigenetic profiles the regulatory sequences bear, different TFs and regulatory proteins are recruited to CRMs, hence determining the functions of them [7,10]. For example, it has been reported that in promoter regions the RNA polymerase II signals can be identified and in enhancer regions H3K4me and nucleosome depletion can be observed [11].
While experimental approaches allow the determination of CRMs, genome-wide systematic identification of instances of CRMs nowadays still mainly resorts to computational methods due to the time-consuming, small-scale and low-throughput nature of experimental approaches [12-14]. The existing CRM searching computational strategies in Drosophila are based on two features of CRMs, namely the transcription factor binding sites (TFBS) and phylogeny. Conserved homologous and non-coding DNA sequences between related species are thought to be closely related to gene regulation [3]. Some CRM prediction algorithms used the window clustering method or the probabilistic modelling to find regulatory sequences with significant clustering of high densities of TFBSs [15,16]. Others utilized the phylogenetic footprinting on comparative homologous sequences between related species to reveal potential CRMs [3,13]. And many methods are hybrids of the two strategies. Various CRM prediction tools have been developed based on these CRM searching strategies. But the accuracy and reliability of different CRM prediction tools are still unclear. Without comparative cross-analysis of different prediction results and combinatorial consideration with extra experimental information, there is no easy way to assess the confidence of the predicted CRMs. And to use the CRM prediction tools, it requires the users to get fully familiar with these tools and data preprocessing. This further prohibits the comprehensive and comparative usage of these CRM computational methods.
To fill this gap, there is an urgent need to perform the genome-wide CRM screening using different prediction tools for comparative referencing and to manually collect experimental information for assessing the confidence of these potential CRMs. Since most of the prediction algorithms did not rely on the epigenetic features of CRMs, the epigenetic profiles provide excellent resources for assessing the confidence of these prediction results. In Drosophila, there are deposited high-throughput data identifying the epigenetic marks [11]. But the information is still fragmentary in different resources and there is no easy way for combinational visualization of the predicted CRMs and the epigenetic profiles thus far. Hence we performed the genome-wide screening for CRMs using different prediction tools and constructed the pioneer database, cisMEP (cis-regulatory module epigenetic profile database), to deposit the CRM prediction results and to integrate these computationally identified CRMs with manually collected genome-wide experimental epigenetic marks as the confidence assessment resources. Researchers can further assess the CRM prediction confidence by the implemented filters based on the potential regulatory functional annotation or combinations of histone modifications. In addition to the deposited potential CRMs, we also retrieved experimentally verified CRMs from the REDfly database [12] and other literature to distinguish putative and known CRMs. cisMEP aims to provide a user-friendly interface for researchers to understand and decipher the confidence and possible functions of potential CRMs through their epigenetic profiles. We believe that this will facilitate biologists to design subsequent experiments for gene regulatory analysis. cisMEP is available online at http://cosbi3.ee.ncku.edu.tw/cisMEP/.
Construction and contents
Collection of systematic computational CRM screening results and construction of cisMEP
We used currently available computational CRM prediction tools to perform and to collect genome-wide systematic screening for potential CRMs and gathered different experimentally-identified epigenetic profiles as confidence assessment and functional deciphering resources. For CRM identification, we adopted the minimal sequence definition proposed by the REDfly database for the comparative collection of potential CRMs identified by different CRM screening tools. We further retrieved experimentally verified CRMs from the REDfly database and other literatures to distinguish putative and known CRMs. To provide an easy-to-use interface for integrative viewing and retrieving these data, we constructed the cisMEP database to deposit the information. The construction of cisMEP is sketched in Figure 1 and details of the collection and statistics of experimental epigenetic data can be found in the following sections. The collection of potential CRMs are stated as follows.
Figure 1.
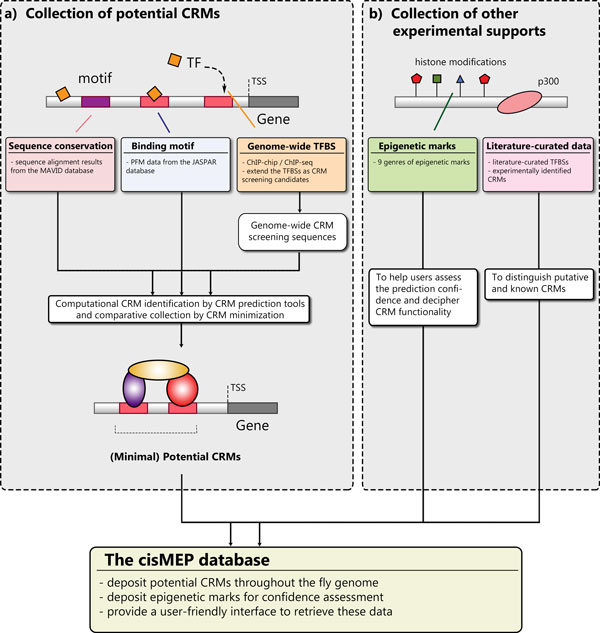
Construction of the cisMEP database. cisMEP gathered the computationally identified CRMs and collected the literature-curated TFBS location data and nine genres of epigenetic data. (a) We have performed different CRM prediction algorithms and gathered these identified potential CRMs for comparative cross-referencing. (b) The genomic epigenetic marks are mapped to the locations of these potential CRMs for prediction confidence assessment and potential CRM function determination. We further retrieved the literature-curated CRMs to distinguish putative and known CRMs.
To perform the systematic genome-wide CRM screening, we selected five CRM prediction tools which have been verified for Drosophila and are still publicly available [13]: cisModule [17], cisPlusFinder [18], ClusterBuster [16], MCAST [15] and MultiModule [19]. These methods were based on TFBS clustering or comparative phylogeny. For MCAST and ClusterBuster, statistic models constructed from the features of TFBS clustering are used to search for significant regulatory sequences. In these two methods, the transcription factor binding motifs are required and should be represented in the PFM format. For cisPlusFinder and MultiModule, CRM prediction is based on sequence conservation across multiple species. And in cisModule, both the binding motifs and the sequence conservation under purifying selection are utilized in a hidden Markov model. Different input data and data preprocessing are required for running all these tools (Figure 1-a).
CRMs carry out their functions by recruiting different regulatory proteins to regulatory motifs within the CRM sequences. Hence to generate candidate sequences for genome-wide CRM screening, we collected the genome-wide TFBS data from the modENCODE project [20]. 39,339 TFBSs identified by the ChIP-chip or ChIP-seq techniques were used to generate the candidate sequences for screening genome-wide potential CRMs. A candidate sequence for CRM searching is defined by the TFBS sequence and extra 3 kb sequences extended from upstream and downstream of the TFBS respectively. The choice of extra 6 kbp is due to the fact that the sequence lengths of experimentally identified CRMs range from a few hundred base-pairs to a few thousand base-pairs long and the default sliding window in most prediction tools are generally 200-1000 base-pairs [13]. To generate the prediction results, we retrieved the genome sequence information of Drosophila melanogaster from Flybase (Version 5.51) [21], the sequence alignment results of Drosophila melanogaster, Drosophila yakuba, Drosophila ananassae, Drosophila virilis and Drosophila pseudoobscura from the MAVID multiple alignment database [22] and the PFM data of TFBS motifs from the JASPAR database [23]. Default parameter settings were used in these prediction tools and the time-consuming genome-wide CRM screening using different available CRM prediction tools was made possible with the assistance of parallel computing techniques.
Different computational CRM searching algorithms may provide different potential CRMs. A predicted CRM is of higher confidence if more CRM screening algorithms find it to be a potential CRM. Hence for comparative referencing, we adopted the minimal sequence definition proposed by the REDfly database. For a group of overlapping potential CRMs, we classified these CRMs into nested sets and took the minimal sequences in the nested sets as the representing CRMs (See Figure 2). In total, there are 410,364 (minimal) potential CRMs deposited in cisMEP. For these potential CRMs, we further distinguish putative and known CRMs from them based on the collected experimentally verified CRMs (Figure 1-b). Finally, we related a potential CRM to those genes regulated by the literature-curated TFBSs or the literature-curated CRMs residing within the range of this CRM, if available. We further linked the CRM annotation to the gene annotation by finding those genes within 100kbp upstream or downstream the potential CRMs. The linkage information of the CRM annotation and the gene annotation can be visualized in the feature of "Transcriptional Regulation Inference Map" in the detailed CRM information page.
Figure 2.
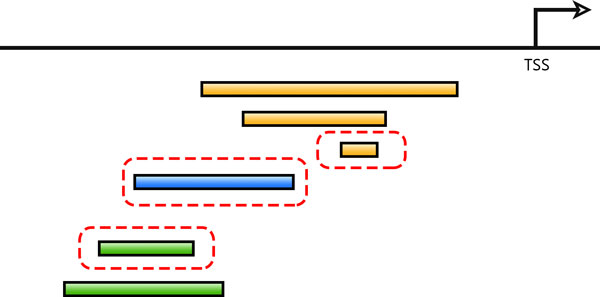
The sketch plot for minimizing overlapping CRMs. The potential CRMs predicted by different CRM screening tools were grouped into nested sets and the minimal sequences in the nested sets were selected as the representing CRMs. In the figure, CRMs forming a nested set are in the same colour and the minimal representing CRM is circumscribed with a dash frame.
Collection and statistics of experimentally identified TFBSs and epigenetic data
To provide confidence assessment for the potential CRMs in cisMEP (Figure 1-b), we collected the literature-curated transcription factor binding site (TFBS) data and nine genres of epigenetic data, including two deduced chromatin state models, chromatin accessibility, cross-species sequence conservation, experimentally identified promoter regions, polycomb group target regions, annotated regulatory elements, histone variant nucleosomes, RNA polymerase II binding and pausing sites, and 40 kinds of histone modifications. The data statistics are stated as follows.
Literature-curated CRMs and TFBSs
Experimentally identified CRMs were retrieved from the REDfly v3.0 database [12] and CAD [24] (CRM Activity Database). REDfly collected experimentally verified CRMs in the literature that were unambiguously demonstrated to regulate target gene expression through reporter gene assays. And the CAD database collected the spatio-temporal information of gene expression driven by CRMs from the REDfly 2.0, literature surveys and their own experiments. In total, we retrieved 1,877 CRMs for 470 genes in Drosophila melanogaster from these two databases. We excluded those CRMs with unspecified target genes. Literature-curated TFBSs were mainly gathered from the REDfly database. In REDfly, curated TFBSs were discovered by DNaseI footprinting assays and electrophoresis mobility shift assays (EMSA).
Data of chromatin states
Chromatin is composed of DNA molecules and associated proteins. Binding of certain chromatin binding proteins and the post-translational modifications on histones are the major determinant of functionality of regulatory sequences [2,25,26]. By integrating the chromatin protein binding data and some detected histone modifications with computational machine learning models, the global potential principal chromatin types can be revealed [25,26]. Filion et al. [25] used 53 selected chromatin components and the hidden Markov model to generate five principal chromatin states. And Kharchenko et al. [26] produced a chromatin landscape of nine chromatin states based on 18 histone modifications and the hidden Markov model. We retrieved 8,428 chromatin regions annotated by the chromatin five state model and 20,734 chromatin regions annotated by the nine chromatin combinatorial patterns. By mapping of these global chromatin states to the CRM regions, we can have the potential picture of the transcriptional regulatory functions of the potential CRMs.
Data of chromatin accessibility
It is known that gene transcriptional expression is occurring at multiple different time periods throughout development. Hence understanding the temporal manner of CRMs are vital for deciphering the CRM regulatory functionality [27]. Chromatin accessibility have been shown to be related to the spatio-temporal behaviour of gene expression patterns [28]. The genome-scale mapping of the DNaseI hypersensitive sites provides the global chromatin accessibility and the regulatory DNA landscape [28,29]. We adopted the data of chromatin accessible regions which were experimentally identified by the increased DNaseI hypersensitive sites from the work of Thomas et al. [28] and Li et al. [29]. In their work, temporal behaviours of chromatin accessibility were measured for five different developmental stages (stage 5, 9, 10, 11 and 14) in the Kc167 cell line under 5% FDR control. In total, 105,485 regulator accessible chromatin regions were deposited in cisMEP for the five developmental stages in Drosophila.
Sequence conservation
The primary reason for cross-species sequence conservation is thought to be purifying selection. This results in the fact that sequences that are significantly more similar than would be expected are prone to have critical regulatory functions [30]. CRMs are often built up of highly conserved sequences that are hard-wired into the genomic sequences. Hence analysis of the cross-species sequence conservation can reveal that many of the cis-regulatory sequences are actually complex regions representing several enhancers, or else are only enhancer fragments. We adopted the cross-species conservation scores from the work of Siepel et al. [30,31]. They have developed a cross-species multiple sequence alignment tool called phastCons, which is based on a two-phase phylogenetic hidden Markov model, to identify conserved elements in multiple-aligned sequences. From their trained and calibrated phylo-HMM model, phastCons generates base-by-base conservation scores for cross-species sequences. The conservation score represents the probability that the sequence is in a conserved element under the trained phylo-HMM model. For Drosophila, the conservation scores generated by phastCons were the sequence conservation measurement for the genomes of Drosophila melanogaster and 12 other fly species.
Experimentally identified promoters
Core promoters are critical DNA regions for gene regulation and transcription initiation. Some CRMs function as gene promoters [3]. Hoskin et al. [4] have identified a high resolution map of sequences of promoter regions by three high-resolution experiments: cap analysis of gene expression (CAGE) tags, RNA ligase mediated rapid amplification of cDNA ends (RLM-RACE) reads and cap-trapped expressed sequence tags (ESTs). The promoters are grouped into three categories: validated (V), supported (S) and RACE-only (R). The V group contains promoters defined by two or more experimental data. The S group includes promoters with a CAGE peak or at least three RACE reads. We retrieved 12,454 promoters for 8,008 genes from their work and mapped them to the potential CRM regions as the promoter-type regulatory sequences.
Polycomb group binding data
Polycomb group (PcG) proteins are those regulatory proteins repressing the transcription of particular target genes. PcG repression of genes requires specific cis-regulatory sequences, forming the silencer CRMs or polycomb response elements (PREs) [32]. Many PcG target genes tend to bear both repression-associated protein binding regions and H3K4me3 [33]. We retrieved the PcG target regions from the work of Schwartz et al. [9]. Two types of PcG target regions were classified: Class I regions contain the regions with binding signals of PC protein, E(Z) protein and H3K27me3; Class II regions consist of regions with weaker PC binding and H3K27me3 signals, but without detectable E(Z) signals. 338 class I PcG target regions were collected and 116 class II PcG target regions were gathered for use in this database.
Data of annotated regulatory elements
The comprehensive annotation of the regulatory elements in chromatin provides possible functionality of the DNA regulatory sequences in CRMs. In the work of Nègre et al. [5], they produced a regulatory map of the Drosophila melanogaster genome on the basis of chromatin features, histone deacetylases and site-specific transcription factor binding signals produced by ChIP-chip, ChIP-sequencing or RNA-sequencing techniques across different stages of embryonic, larval, pupal and adult development. The data retrieved and used in cisMEP are as the following: 9,058 promoters predicted based on the co-occurrence of H3K4me3 and RNA polymerase II in embryos; 81 enhancers predicted by the binding locations of CREB binding protein (CBP) and RNA polymerase II; 4,774 Class I insulators identified by the binding of CTCF/CP190/BEAF-32 proteins and 2,911 Class II insulators identified by the binding of SU(HW) protein; 537 PREs associated with histone deacetylases (HDACs). These predicted cis-regulatory maps provide insight into CRM functions.
Histone variant nucleosome data
Eukaryotic DNA is packaged to form nucleosome particles [34]. The location of nucleosomes and the replacement of histones in nucleosomes play vital roles in gene transcriptional regulation and epigenetic inheritance [35]. We gathered two genome-wide detected nucleosome replacement data, the H3.3 replacement and the H2A.Z (also known as H2Av) map, to depict the nucleosome organization in the Drosophila genome. H3.3 are known to be related to abundant RNA polymerase II and methylated H3K4 [36]. H2A.Z were reported to be linked to an open and uniform chromatin architecture at promoter regions [37]. In cisMEP, we retrieved 617,304 regions with H2A.Z replacement reported by the ChIP-chip technique and 415,119 regions reported to contain the bulk nucleosome (DNA sequences contain any combinations of H2A and H2A.Z) from the work of Mavrich et al. [37]. For H3.3 replacement patterns, the original biotin pull-down ratios (including H3.3/wild type and H3.3/H3) were adopted from the work of Mito et al. [36] and were mapped to the chromosomal locations of the potential CRMs.
Data related to the RNA polymerase II activity
RNA polymerase II is the main molecule responsible for gene transcription. The activity of this polymerase suggests active gene transcription [8]. Hence we collected two types of data representing the RNA polymerase II activity: the regions with RNA polymerase II binding signals and the pausing sites of RNA polymerase II. 9,126 RNA polymerase II binding regions reported by ChIP-sequencing were adopted from the modENCODE project [8] and 3,729 RNA polymerase II pausing regions were collected from the work of Mavrich et al. [37].
Data identifying histone modifications and chromatin binding proteins
Post-translational modifications of nucleosomal core histones play critical roles in altering chromatin structure and creating target sites for proteins acting on chromatin, thereby regulating gene transcriptional expression [11]. Chromatin binding proteins may function as histone-modifying enzymes related to certain activator or repressor recruitment modifications. Or they may act as the chromatin structure remodelling regulators [7]. In cisMEP, we collected 40 different datasets providing the histone modification regions or chromatin protein binding target regions from the work of Kellner et al. [38] and the modENCODE project [8]. The .bed files of histone modification data or chromatin protein binding regions identified by the ChIP-chip or the ChIP-sequencing technologies were mapped to the CRM locations. We further categorized the histone modifications and chromatin binding proteins by their potential functionality according to the literature. Details of the functional classification can be seen in Additional File 1.
Confidence measures for the deposited potential CRMs
In the near future, it will become easier to conduct larger-scale transgenic experiments in Drosophila. Hence it would be desirable to rank the CRMs so that experimentalists can prioritize constructs for transgenic experiments. We have provided two confidence measures to rank the deposited potential CRMs in cisMEP. The first confidence measure is the histone mark confidence score. The histone mark confidence score is calculated by taking the arithmetic mean of the normalized binding scores (the M-values calculated in the ChIP-chip data analysis [8]) of the histone marks overlapping with the CRM. The higher the histone mark confidence score is, the stronger signals of the histone marks are observed. The second confidence measure is the conservation confidence score. This is provided by the phastCons score [30,31] that specifies the sequence conservation among different Drosophila species. Highly conserved CRMs are considered to be more confident potential regulatory elements.
Implementation of the web service of cisMEP
The database is constructed using the PHP language with the CodeIgniter MVC framework. Epigenetic profiling data was deposited through MySQL. Charts displaying the epigenetic profiles for CRMs were produced by the JQuery tool Jqplot.
Utility and discussion
Database interface
The cisMEP database provides three basic functions for users to select potential CRMs of interest and to view the epigenetic profiles for deciphering potential functionality of the chosen one: (I) search mode, searching for potential CRMs of specific genes or querying potential CRMs with specified annotated functions; (II) browse mode, browsing the database for the computationally identified CRMs deposited in this database; (III) download feature, downloading the plain text file of epigenetic profiles for CRMs. The cisMEP web interface provides all entries to these functions.
In the search mode, users can key in the gene of interest to find all potential CRMs related to this gene. Alternatively users can also select the annotated functions defined by the region classification of Nègre et al. [5] and Müller et al. [32] or specify the intended histone modification combination to find the deposited potential CRMs with the specified epigenetic features. Users can further set the confidence filters of the histone mark confidence and conservation confidence to filter out only those potential CRMs above certain confident probability scores. In the search result page, the computationally identified CRMs satisfying the specified constrains will be listed in a table. In the table, the number of algorithms that specify this sequence as a potential CRM, the number of literature-curated TFBSs residing in the potential CRM region, the number of CRM literature supports for the regulatory region, the histone mark confidence and the conservation confidence for the potential CRM are listed. cisMEP also provides the browse mode for users to browse all the deposited CRM prediction results in this database. In the browse mode, users can browse the CRM predictions on different chromosomes or genes. Whether through the search mode or the browse mode, the user can now click the link of "detail" to select the CRM of interest to view the details of the functionally categorized epigenetic profiles for this regulatory sequence.
In the detail page, users can assess the prediction confidence and decipher possible transcriptional regulatory functions for the chosen CRM. cisMEP summarizes the details of the selected potential CRM. The summary elucidates the screening algorithms which this potential CRM satisfied as well as whether it is a putative or known CRM based on literature (Figure 3-a). Next a figure, called the epigenetic profile map, consisting of the genome tracks of the CRM, its functional regions and its epigenetic profiles is provided (Figure 3-c). In the epigenetic profile map, nine types of epigenetic data as described in the Content Section are rendered as horizontal tracks (except for the ratio plots of H3.3 replacements and the genomic distribution of conservation scores) if available. Users can also re-render the axis of the chromosome coordination to zoom in and zoom out the epigenetic tracks of the given CRM (Figure 3-b). Detailed descriptions of specific epigenetic tracks are provided as tool-tips when the users point to them. Below the epigenetic profile plot, we provide the "Transcriptional Regulation Inference Map" that helps visualize the orientations and spacings of TFBSs within the CRMs and helps demonstrate the related gene annotation to this potential CRM and the TFBSs (Figure 3-d). From this map, users can infer the possible transcriptional regulation mechanisms from these TFBS and gene locations. For the raw data of these epigenetic tracks, users can refer to the tables below the chart (Figure 3-e). All tables presented in cisMEP can be downloaded easily as the .csv format or as the .pdf format (Figure 3-f).
Figure 3.
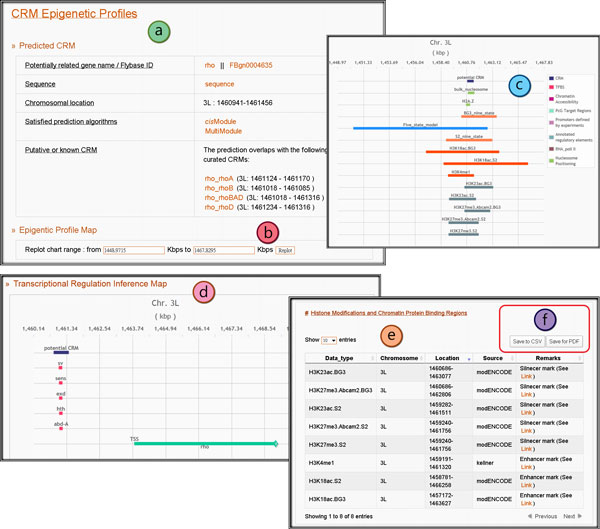
The CRM epigenetic profile page. (a) The summary table for the selected potential CRM. (b) The function to zoom in and zoom out the epigenetic profile chart. (c) The epigenetic profile chart for the selected CRM. Epigenetic profiles are rendered as bar tracks if available. (d) The transcriptional regulation inference map for the selected CRM. In this map, TFBSs and gene annotation related to this predicted CRM are shown for transcriptional regulation mechanism inference. (e) The tables of the detailed epigenetic data for the selected CRM. (f) The download function. Tables provided in cisMEP can be downloaded as the .csv format or the .pdf form by the users. In this figure, only parts of the data are shown due to space limitations.
Issues related to cisMEP
cisMEP collects different types of epigenetic data for assessing the confidence of the potential CRMs and deciphering the possible CRM functionality. Yet this type of data integration may result in a potential systematic bias. In systems biology, a cellular system is perturbed and measured by high-throughput technologies. Thus an understanding of the system is based on the integration of different high-throughput assays [39]. But this type of data integration often suffers from the system bias caused by the fact that different high-throughput data were performed on different cell states and cell types [40,41]. Since this type of systematic bias is still unavoidable [40], some critical points are worthy of notice when using the epigenetic profiles for deciphering CRM functionality.
Correct temporal and spatial control of gene expression is crucial in different developmental stages in metazoans. cisMEP collects the chromatin accessibility data for different developmental stages and hence provides the temporal behaviour of CRMs. While genome-wide epigenetic data are still not available for all possible combinations of cell types and stages, in cisMEP, we have marked out the developmental stages and the cell types for which the high-throughput experiments were performed. Hence in deciphering the CRM functionality, users can also take care of the different cell types for data integration in order not to run the risk of deducing the incorrect inferences.
Besides the potential bias that may result in data integration, due to the nature of high-throughput data, there are still inherent false positives and false negatives in the high-throughput experiment results [39,40], causing the incompleteness in the experimental data support. This insufficiency will be overcome when more and more experiment data are generated. While these data are still not totally complete now, updated version of the experimental validated promoters will be published [4,8] and we will keep cisMEP up-to-date with these newly published datasets when more genome-wide data are released.
It is well known that epigenetic marks such as histone variant nucleosomes, histone modifications, and protein binding events are highly dynamic. While looking at these events in a time-sensitive and condition-specific manner is very important for deciphering CRM functionality, the dynamic behaviour of gene expression and CRM regulatory functionality can be considered only in the scope of gene regulatory networks [27]. Construction of the gene regulatory network using the TF-bound CRMs reveals the properties and dynamics of CRM regulation. But challenges remain to be solved in modelling the gene regulatory networks on CRMs [27]. In the current version of cisMEP, we aimed to deposit computationally identified CRMs and to provide epigenetic profiles for CRM prediction confidence assessment and potential CRM functionality deciphering. Gene regulatory networks on CRMs will be incorporated in the future updating plan when further advances in gene regulatory network modelling on CRMs are available.
Case study
To show the application of cisMEP, we demonstrate an example describing the epigenetic profiles for one potential CRM residing in the genomic region 3L: 1460941-1461456. This CRM is annotated to be related to the regulation of the gene rhomboid. rhomboid is a protein encoding gene in Drosophila melanogaster that regulates the fly embryonic development in the ventral neurogenic ectoderm. The CRM regulatory clusters for rhomboid gene expression are vital in the formation of patterns of the dorsoventral body axes and the development of the peripheral nervous system [42,43]. We will call this computationally identified CRM as the name of the experimentally verified one, RhoBAD, in the following paragraph for simplicity.
As shown in the summary table, RhoBAD is predicted by the CRM prediction tools of cisModule and MultiModule. And in the epigenetic profiles, we can see that this CRM is enriched in bulk nucleosome (indication of any combination of H2A.Z and H2A) and H2A.Z in embryos, which implies this region is mainly occupied by the histone variant of H2A.Z and suggests the chromatin structure around the promoter region and TSS for actively transcribed genes [37,44]. Enhancer/silencer marks, such as H3K4me1, H3K18ac (marks for enhancers [11,45]), H3K27me3 and H3K23ac (marks for silencers [46,47]) can also be observed in the S2 and BG3 cells. These epigenetic marks imply that this potential regulatory sequence may bear cellular functions and the possible regulatory function of it is gene transcriptional enhancing/silencing. And the conservation score of this predicted CRM reflects the highly conserved tendency of the sequence (conservation probability = 0.56), indicating the potential regulatory functionality of this sequence. From the support of these experimental data, we are quite sure that this CRM prediction is biologically meaningful and may indeed exist and function as the enhancer/silencer CRM for rhomboid in living cells. In fact, by the efforts of molecular biologists, four transcription factors- Abdominal-A (Abd-A), Extradenticle (Exd), Homothorax (Hth) and Senseless (Sens)- have been reported to bind to the DNA sequence regions within the RhoBAD CRM [43]. And experimental evidence has revealed the fact that RhoBAD acts as an enhancer when bound with the Exd/Hth/Abd-A protein complex and acts as a silencer when Sens binds to it [43,48]. The sketch of the epigenetic profiles deposited in cisMEP and the experimental CRM function verification for RhoBAD are shown in Figure 4. This example shows that the epigenetic profiles gathered in cisMEP can help researchers assess the prediction confidence of the deposited potential CRMs, deduce the potential functionality of the computationally identified CRMs and possibly establish the transcriptional regulation mechanism hypotheses of these potential CRMs.
Figure 4.
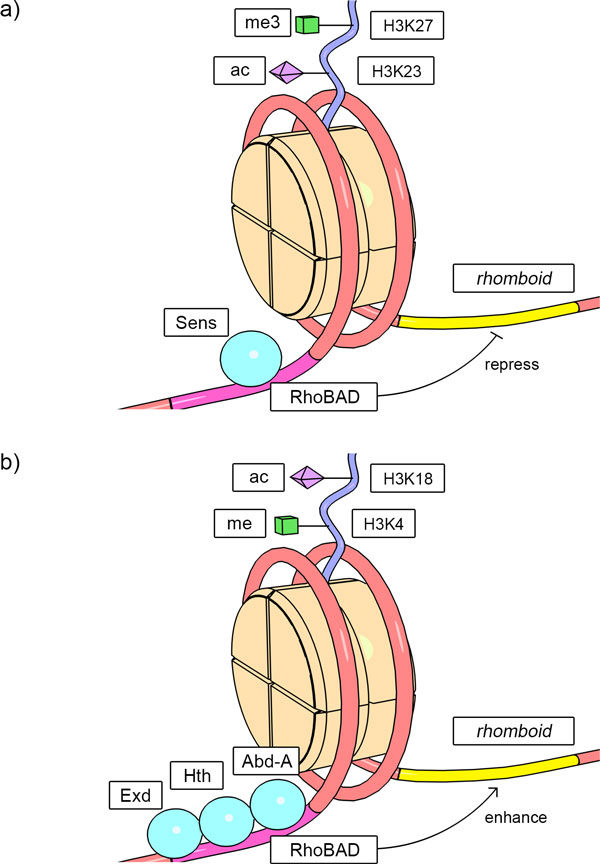
Simplified sketch plot of the epigenetic profiles and experimental results for RhoBAD. The epigenetic profiles suggest that the predicted CRM RhoBAD is of high confidence and acts as an enhancer/silencer for the transcription of rhomboid. From experimental literature evidence, we know that RhoBAD indeed acts as an enhancer when bound with the Exd/Hth/Abd-A protein complex and acts as a silencer when Sens binds to it. (a) Silencer markers (H3K23ac, H3K27me3). (b) Enhancer markers (H3K4me, H3K18ac).
Conclusions
In this study, we constructed a database called cisMEP, which serves to deposit genome-wide systematically screened CRM predictions and to provide the epigenetic profiles as confidence assessment of these potential CRMs. cisMEP has an easy-to-use and user-friendly interface for biologists to search and browse CRMs of interest. With TFBSs, epigenetic profiles and chromatin protein binding information, researchers can thus assess the confidence of the potential CRMs and decipher the possible CRM functions. This will facilitate biologists to unravel the regulatory mechanisms of metazoan gene transcription and to figure out the so-called "histone codon" that may possibly reside in transcriptional regulation. Finally, cisMEP will be updated on a regular basis to keep in accordance with the latest CRM screening tools, literature-curated CRM data and experimental epigenetic data.
Availability and requirements
cisMEP is available at http://cosbi3.ee.ncku.edu.tw/cisMEP/. JavaScript functioning should be enabled in the user-side browsers and the Adobe Flash Player for specific browsers should also be installed. The web interface of cisMEP is fully tested on popular browsers: Microsoft IE9, Google Chrome, Apple Safari and Mozilla Firefox 21. Users are recommended to use these popular browsers for full functionality of cisMEP.
List of abbreviations
CRM, cis-regulatory module; TF, transcription factor; ChIP, chromatin immunoprecipitation; TFBS, transcription factor binding site; PcG, polycomb group; PRE, polycomb response element; cisMEP, cis-regulatory module epigenetic profile database
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
THY conceived the research topic. THY and CCW performed the data collection and data processing. THY and PCH constructed the website interface and the database. THY wrote the manuscript and WSW provided essential guidance. All authors have read and approved the final manuscript.
Declarations
The publication of this manuscript was funded by the Aim for the Top University Project awarded to the National Cheng Kung University.
Supplementary Material
Details of the functional classification of the gathered epigenetic marks. Additional file 1 contains the table showing the potential regulatory functions of the epigenetic marks. The 'pubmed' column contains the literature supports for the classified regulatory functions.
Contributor Information
Tzu-Hsien Yang, Email: c102570@gmail.com.
Chung-Ching Wang, Email: stanleyccwang1987@gmail.com.
Po-Cheng Hung, Email: miofantasy@gmail.com.
Wei-Sheng Wu, Email: wessonwu@mail.ncku.edu.tw.
Acknowledgements
This study was supported by National Cheng Kung University and Ministry of Science and Technology of Taiwan (MOST-103-2221-E-006-174-MY2).
This article has been published as part of BMC Systems Biology Volume 8 Supplement 4, 2014: Thirteenth International Conference on Bioinformatics (InCoB2014): Systems Biology. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcsystbiol/supplements/8/S4.
References
- Davidson EH, Erwin DH. Gene regulatory networks and the evolution of animal body plans. Science. 2006;311:796–800. doi: 10.1126/science.1113832. [DOI] [PubMed] [Google Scholar]
- Jeziorska DM, Jordan KW, Vance KW. Semin Cell Dev Biol. Vol. 20. Elsevier; 2009. A systems biology approach to understanding cis-regulatory module function; pp. 856–862. [DOI] [PubMed] [Google Scholar]
- Hardison RC, Taylor J. Genomic approaches towards finding cis-regulatory modules in animals. Nat Rev Genet. 2012;13:469–483. doi: 10.1038/nrg3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins RA, Landolin JM, Brown JB, Sandler JE, Takahashi H, Lassmann T, Yu C, Booth BW, Zhang D, Wan KH. et al. Genome-wide analysis of promoter architecture in Drosophila melanogaster. Genome Res. 2011;21:182–192. doi: 10.1101/gr.112466.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nègre N, Brown CD, Ma L, Bristow CA, Miller SW, Wagner U, Kheradpour P, Eaton ML, Loriaux P, Sealfon R. et al. A cis-regulatory map of the Drosophila genome. Nature. 2011;471:527–531. doi: 10.1038/nature09990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nègre N, Brown CD, Shah PK, Kheradpour P, Morrison CA, Henikoff JG, Feng X, Ahmad K, Russell S, White RA. et al. A comprehensive map of insulator elements for the Drosophila genome. PLoS Genet. 2010;6:e1000814. doi: 10.1371/journal.pgen.1000814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, Lin MF. et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010;330:1787–1797. doi: 10.1126/science.1198374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz YB, Kahn TG, Stenberg P, Ohno K, Bourgon R, Pirrotta V. Alternative epigenetic chromatin states of polycomb target genes. PLoS Genet. 2010;6:e1000805. doi: 10.1371/journal.pgen.1000805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leeuwen F, van Steensel B. et al. Histone modifications: from genome-wide maps to functional insights. Genome Biol. 2005;6:113. doi: 10.1186/gb-2005-6-6-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros IM. Histone modification in Drosophila. Brief Funct Genomics. 2012;11:319–331. doi: 10.1093/bfgp/els029. [DOI] [PubMed] [Google Scholar]
- Gallo SM, Gerrard DT, Miner D, Simich M, Des Soye B, Bergman CM, Halfon MS. REDfly v3. 0: toward a comprehensive database of transcriptional regulatory elements in Drosophila. Nucleic Acids Res. 2011;39(suppl 1):D118–D123. doi: 10.1093/nar/gkq999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J, Teichmann SA, Down TA. Assessing computational methods of cis-regulatory module prediction. PLoS Comput Biol. 2010;6:e1001020. doi: 10.1371/journal.pcbi.1001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, He X. MORPH: probabilistic alignment combined with hidden Markov models of cis-regulatory modules. PLoS Comput Biol. 2007;3:e216. doi: 10.1371/journal.pcbi.0030216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Noble WS. Searching for statistically significant regulatory modules. Bioinformatics. 2003;19(suppl 2):ii16–ii25. doi: 10.1093/bioinformatics/btg1054. [DOI] [PubMed] [Google Scholar]
- Frith MC, Li MC, Weng Z. Cluster-Buster: Finding dense clusters of motifs in DNA sequences. Nucleic Acids Res. 2003;31:3666–3668. doi: 10.1093/nar/gkg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Wong WH. CisModule: de novo discovery of cis-regulatory modules by hierarchical mixture modeling. Proc Natl Acad Sci USA. 2004;101:12114–12119. doi: 10.1073/pnas.0402858101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierstorff N, Bergman CM, Wiehe T. Identifying cis-regulatory modules by combining comparative and compositional analysis of DNA. Bioinformatics. 2006;22:2858–2864. doi: 10.1093/bioinformatics/btl499. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Wong WH. et al. Coupling hidden Markov models for the discovery of cis-regulatory modules in multiple species. Ann Appl Stat. 2007;1:36–65. doi: 10.1214/07-AOAS103. [DOI] [Google Scholar]
- Washington NL, Stinson E, Perry MD, Ruzanov P, Contrino S, Smith R, Zha Z, Lyne R, Carr A, Lloyd P. et al. The modENCODE Data Coordination Center: lessons in harvesting comprehensive experimental details. Database J Biol Databases Curation. 2011;2011 doi: 10.1093/database/bar023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marygold SJ, Leyland PC, Seal RL, Goodman JL, Thurmond J, Strelets VB, Wilson RJ. et al. FlyBase: improvements to the bibliography. Nucleic Acids Res. 2013;41:D751–D757. doi: 10.1093/nar/gks1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray N, Pachter L. MAVID: constrained ancestral alignment of multiple sequences. Genome Res. 2004;14:693–699. doi: 10.1101/gr.1960404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen C, Chou A, Ienasescu H. et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014;42:D142–D147. doi: 10.1093/nar/gkt997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzen RP, Girardot C, Gagneur J, Braun M, Furlong EE. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature. 2009;462:65–70. doi: 10.1038/nature08531. [DOI] [PubMed] [Google Scholar]
- Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ. et al. Systematic Protein Location Mapping Reveals Five Principal Chromatin Types in Drosophila Cells. Cell. 2010;143:212–224. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T. et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2010;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilczynski B, Furlong EE. Challenges for modeling global gene regulatory networks during development: Insights from Drosophila. Dev Biol. 2010;340:161–169. doi: 10.1016/j.ydbio.2009.10.032. [DOI] [PubMed] [Google Scholar]
- Thomas S, Li XY, Sabo PJ, Sandstrom R, Thurman RE, Canfield TK, Giste E, Fisher W, Hammonds A, Celniker SE. et al. Dynamic reprogramming of chromatin accessibility during Drosophila embryo development. Genome Biol. 2011;12:R43. doi: 10.1186/gb-2011-12-5-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-Y, Thomas S, Sabo PJ, Eisen MB, Stamatoyannopoulos JA, Biggin MD. The role of chromatin accessibility in directing the widespread, overlapping patterns of Drosophila transcription factor binding. Genome Biol. 2011;12:R34. doi: 10.1186/gb-2011-12-4-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S. et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Haussler D. Stat Methods Mol Evol. Springer; 2005. Phylogenetic hidden Markov models; pp. 325–351. [Google Scholar]
- Müller J, Kassis JA. Polycomb response elements and targeting of Polycomb group proteins in Drosophila. Curr Opin Genet Dev. 2006;16:476–484. doi: 10.1016/j.gde.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K. et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Wolffe A. Chromatin: Structure and Function. Access Online via Elsevier; 1998. [Google Scholar]
- Yuan G-C, Liu Y-J, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309:626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- Mito Y, Henikoff JG, Henikoff S. Genome-scale profiling of histone H3. 3 replacement patterns. Nat Genet. 2005;37:1090–1097. doi: 10.1038/ng1637. [DOI] [PubMed] [Google Scholar]
- Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, Tomsho LP, Qi J, Glaser RL, Schuster SC. et al. Nucleosome organization in the Drosophila genome. Nature. 2008;453:358–362. doi: 10.1038/nature06929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellner WA, Ramos E, Van Bortle K, Takenaka N, Corces VG. Genome-wide phosphoacetylation of histone H3 at Drosophila enhancers and promoters. Genome Res. 2012;22:1081–1088. doi: 10.1101/gr.136929.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Galitski T, Hood L. A new approach to decoding life: systems biology. Annu Rev Genomics Hum Genet. 2001;2:343–372. doi: 10.1146/annurev.genom.2.1.343. [DOI] [PubMed] [Google Scholar]
- Hwang D, Rust AG, Ramsey S, Smith JJ, Leslie DM, Weston AD, De Atauri P, Aitchison JD, Hood L, Siegel AF. et al. A data integration methodology for systems biology. Proc Natl Acad Sci USA. 2005;102:17296–17301. doi: 10.1073/pnas.0508647102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang D, Smith JJ, Leslie DM, Weston AD, Rust AG, Ramsey S, de Atauri P, Siegel AF, Bolouri H, Aitchison JD. et al. A data integration methodology for systems biology: experimental verification. Proc Natl Acad Sci USA. 2005;102:17302–17307. doi: 10.1073/pnas.0508649102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves N, Posakony JW. Genetic Programs Activated by Proneural Proteins in the Developing Drosophila PNS. Dev Cell. 2005;8:413–425. doi: 10.1016/j.devcel.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Li-Kroeger D, Witt LM, Grimes HL, Cook TA, Gebelein B. Hox and Senseless Antagonism Functions as a Molecular Switch to Regulate EGF Secretion in the Drosophila PNS. Dev Cell. 2008;15:298–308. doi: 10.1016/j.devcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemette B, Bataille AR, Gévry N, Adam M, Blanchette M, Robert F, Gaudreau L. Variant histone H2A. Z is globally localized to the promoters of inactive yeast genes and regulates nucleosome positioning. PLoS Biol. 2005;3:e384. doi: 10.1371/journal.pbio.0030384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CT, Corces VG. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet. 2011;12:283–293. doi: 10.1038/nrg2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melgar MF, Collins FS, Sethupathy P. et al. Discovery of active enhancers through bidirectional expression of short transcripts. Genome Biol. 2011;12:R113. doi: 10.1186/gb-2011-12-11-r113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle NC, Minoda A, Kharchenko PV, Alekseyenko AA, Schwartz YB, Tolstorukov MY, Gorchakov AA, Jaffe JD, Kennedy C, Linder-Basso D. et al. Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin. Genome Res. 2011;21:147–163. doi: 10.1101/gr.110098.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt LM, Gutzwiller LM, Gresser AL, Li-Kroeger D, Cook TA, Gebelein B. Atonal, Senseless, and Abdominal-A regulate rhomboid enhancer activity in abdominal sensory organ precursors. Dev Biol. 2010;344:1060–1070. doi: 10.1016/j.ydbio.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of the functional classification of the gathered epigenetic marks. Additional file 1 contains the table showing the potential regulatory functions of the epigenetic marks. The 'pubmed' column contains the literature supports for the classified regulatory functions.