

Patient MSV, a 61-year-old woman born in Penteado, Alagoas, Brazil and residing in São Paulo was hospitalized for decompensated heart failure (HF).
Approximately 1 year before, she experienced dyspnea with moderate effort, and there was marked worsening over the last 3 months associated with orthopnea and lower limb edema.
Laboratory tests from when the patient was first seen at the InCor emergency unit (in March 2013) revealed: 3.3 million red blood cells/mm³; hemoglobin 10.9 g/dL; hemocyte 33%; MCV (mean corpuscular volume), 100 fL; leukocytes, 4,260/ mm³ (3,067 neutrophils, 213 eosinophils, 43 basophils, 809 lymphocytes and 128 monocytes); platelets, 146,000/mm³; CK-MB, 1.37 ng/mL; troponin I, 0.081 ng/mL; urea, 26 mg/ dL; creatinine, 0.92 mg/dL; sodium 140 mEq/L; potassium 4.9 mEq/L; magnesium, 1.5 mEq/L; C-reactive protein, 6.31 mg/L; B-type natriuretic peptide, 282 pg/mL; PT (INR), 1.2; APTT, 0.97 ratio.
Radiography (in March 2013) showed an increase in the pulmonary vasculature and slightly increased cardiac area with middle arc rectification. (Figure 1).
Figure 1.
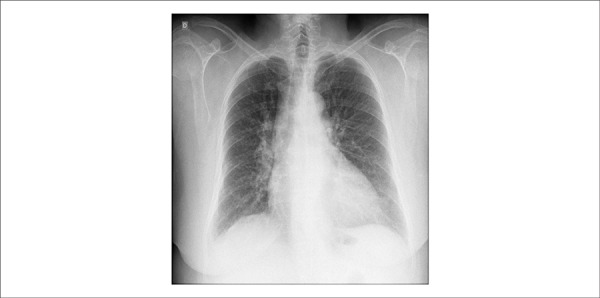
Patient chest X ray: increased pulmonary vasculature and slightly increased cardiac area, with middle arc rectification.
After administration of medicines, HF improved; however, after 1 week, the symptoms worsened and the patient sought emergency medical attention and was hospitalized (on August 6, 2013). She also complained of burning precordial discomfort accompanying dyspnea on exertion. She stated that there were no palpitations, syncope, arterial hypertension, diabetes mellitus, or dyslipidemias. The patient reported use of anti-inflammatory drugs for treating carpal tunnel syndrome.
The physical examination (on August 6, 2013) revealed increased jugular venous pressure, heart rate of 100 bpm, and blood pressure of 117 × 59 mmHg. Pulmonary auscultation revealed decreased vesicular breath sounds at the lung bases and sizzling rales until the middle third; cardiac auscultation revealed no changes. The abdomen had no visceromegaly or ascites, and there was discrete edema in the lower limbs.
Chest radiography (on August 6, 2013) revealed bilateral opacification of the costophrenic sinuses, increased pulmonary vasculature, and increased heart size (Figure 2).
Figure 2.
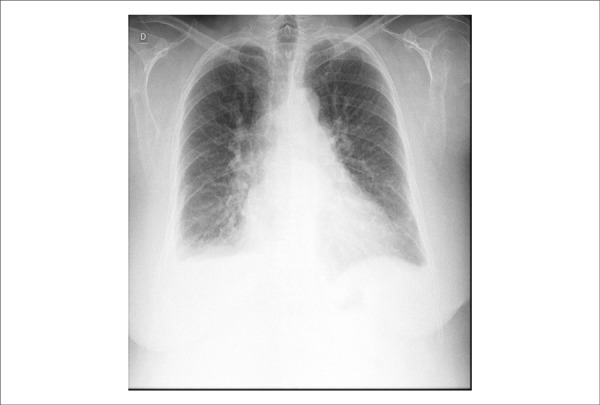
Chest X ray: bilateral opacification of the costophrenic sinuses, increased pulmonary vascular markings, and increased heart area.
The electrocardiogram (ECG, on August 7, 2013) showed sinus rhythm, a heart rate of 78 bpm, low voltage QRS complexes, and diffuse ventricular repolarization (Figure 3).
Figure 3.
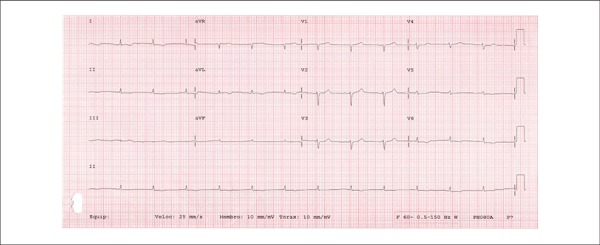
ECG showing sinus rhythm, a heart rate of 78 bpm, low voltage QRS complexes, and diffuse ventricular repolarization.
The following was revealed by the echocardiogram (on August 8, 2013): aortic sinus, 29 mm; left atrium, 42 mm; septal thickness, 16 mm and posterior wall thickness, 12 mm; left ventricle (diastole/systole), 42/29 mm; ejection fraction, 57% (Figure 4). The left ventricle showed marked hypertrophy without segmental changes. The filling pattern was restrictive and not reversible with the Valsalva maneuver (Figure 5). The right ventricle had preserved systolic function without valvular dysfunction. There were signs of pulmonary hypertension, and pulmonary artery pressure was estimated to be at 50 mmHg. The E (mitral protodiastolic speed)/E’ (protodiastolic velocity of the mitral annulus) ratio was 15 (normal value < 8), indicative of the left atrial pressure above 20 mmHg.
Figure 4.
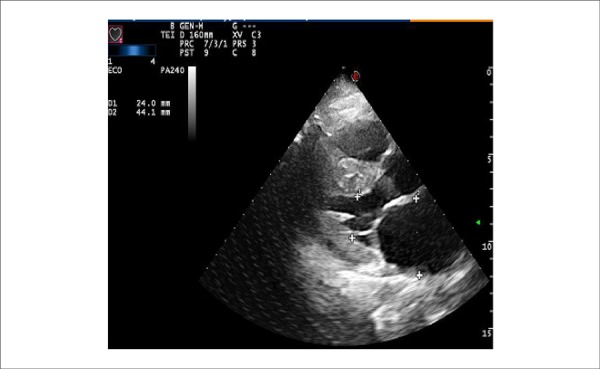
Transthoracic ECHO: marked increase in the left atrium diameter.
Figure 5.
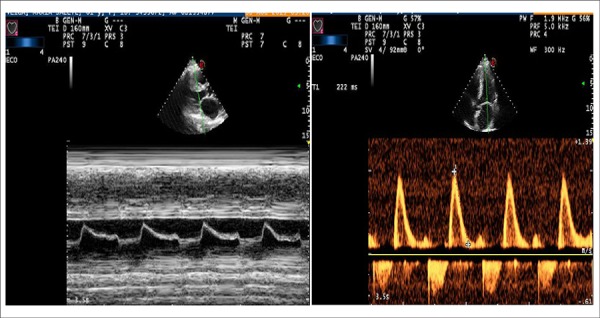
Left panel: Transthoracic ECHO M mode, mitral valve with the Christmas tree pattern, early closure of the mitral valve, and increased end diastolic pressure in the LV. Right panel: restrictive-type pattern ventricular filling.
Laboratory tests revealed anemia, renal failure, and hypothyroidism (Table 1).
Table 1.
Laboratory tests
| August 7 | August 9 | August 11 | |
|---|---|---|---|
| Erythrocytes/mm3 | 2,700,000 | 2,800,000 | 2,700,000 |
| Hemoglobin (g/dL) | 8.8 | 9.7 | 8.6 |
| Hematocrit (%) | 27 | 29 | 28 |
| MCV (fL) | 100 | 104 | 104 |
| Leukocytes/mm3 | 5,330 | 8,080 | 7,820 |
| Neutrophils (%) | 70 | 79 | 74 |
| Eosinophils (%) | 2 | 1 | 3 |
| Basophils (%) | 0 | 1 | 1 |
| Lymphocytes (%) | 24 | 15 | 18 |
| Monocytes (%) | 4 | 4 | 4 |
| Platelets/mm3 | 147,000 | 202,000 | 179,000 |
| Urea (mg/dL) | 102 | 157 | 184 |
| Creatinine (mg/dL) | 3.62 | 6.02 | 7.93 |
| Glomeruiar filtration rate (mL/min/1.73 m2) | 14 | 8 | 5 |
| TSH (μUI/ml) | 17.8 | ||
| Free T4 (ng/dL) | 1.1 | ||
| Potassium (mEq/L) | 3.9 | 5.7 | 5 |
| Sodium (mEq/L) | 137 | 136 | 130 |
| Venous blood gas | |||
| pH | 7.3 | 7.3 | 7.18 |
| pCO2 (mmHg) | 51 | 42.6 | 51.7 |
| pO2 (mmHg) | 33.5 | 42.9 | 59.5 |
| Bicarbonate (mMol/L) | 24.4 | 20.2 | 18.7 |
| Base excess (mMol/L) | (-) 16 | (-) 5.3 | (-) 9 |
| Ionized calcium (mMol/L) | 1.15 | 1.17 | 1.15 |
| Magnesium (mEq/L) | 1.5 | 2 | 2 |
| D dimer (ng/mL) | 1,162 | 712 | |
| PT (INR) | 0.96 | 1.3 | |
| APTT (ratio time) | 0.96 | 1 | |
| C-reactive protein (mg/L) | 10.81 | 21.98 | 25.68 |
| B-type natriuretic peptide (pg/mL) | 524 | 662 | |
| CK-MB (ng/mL) | 8.56 | 12.26 | |
| Troponin I (ng/mL) | 0.607 | 2.54 | 12 |
| Venous lactate (mg/dL) | |||
| Urine (L) | |||
| Proteins (g/L) | 6 | ||
| Leukocytes/mL | 280,000 |
Diuretics and intravenous dobutamine were administered with initial improvement in pulmonary congestion; however, congestion and oliguria worsened, with a marked increase in urea and creatinine levels (Table 1 and Figure 6). Dialysis was indicated, during which the patient showed signs of bacteremia, and the procedure was stopped. This was followed by worsening dyspnea and cardiac arrest by hypoxia. Resuscitation and orotracheal intubation for respiratory support were started, but there was no response, and the patient died (at 3:45 am on August 12, 2013).
Figure 6.
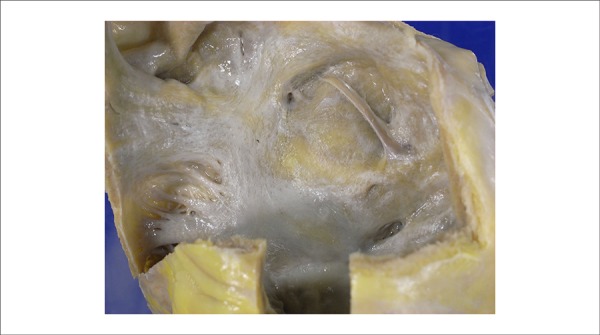
Left aspect of the atrial septum. Irregular yellowish areas correspond to the amyloid deposit.
Clinical aspects
The patient was a 61-year-old woman who developed HF in March 2013, with initial improvement after introduction of a specific treatment. However, 5 months later, she was admitted to the emergency room with decompensated underlying disease, requiring hospitalization.
HF is a complex syndrome resulting from any structural or functional damage to ventricular filling or ejection volume.1 The cardinal manifestations of HF are dyspnea and fatigue, which in turn may cause exercise intolerance, fluid retention, and pulmonary and systemic congestion1. There is no specific diagnostic test for HF because it is primarily a syndrome of clinical diagnosis based on the history and physical exam1. The importance of an active search for risk factors or previous cardiovascular disease should be emphasized in determining the etiology for this syndrome.
The patient returned to the emergency room with worsening dyspnea on exertion and burning chest discomfort. The patient presented no risk factors relevant to HF, except for the recent use of anti-inflammatory drugs, which could be a cause for decompensation of the disease. Physical examination called attention to signs of pulmonary congestion, as well as elevated right atrial pressure, increased jugular venous pressure, and signs of systemic congestion, observed in the edema of the lower limbs. The reduction in lower lung lobe auscultation corresponds to bilateral pleural effusion, which can be associated with HF or possibly with the patient’s underlying disease.
Considering the history and the physical examination, we concluded that the patient had a cardiac insufficiency syndrome with the involvement of left and right cardiac chambers.
After analyzing the complementary tests, a significant worsening of renal function was noted in relation to the tests performed in March, as well as a significant drop in hemoglobin values, indicating anemia and acute renal failure. The U/Cr ratio remained < 40, and type I urine results revealed the presence of significant proteinuria, suggesting a parenchymatous process for renal dysfunction. Another relevant fact is the elevation of the myocardial necrosis markers, which may be associated with coronary disease or, more likely, with the kidney disease and decompensated HF, because the patient did not present significant risk factors for ischemic cardiovascular disease.
The intake chest X-ray confirms the findings of the physical examination, demonstrating bilateral pleural effusion and some degree of pulmonary congestion, with cardiac area at the upper limit of normal.
The ECG showed low QRS voltage, most evident in the frontal plane, late R wave progression in the horizontal plane, and diffuse changes in ventricular repolarization. This pattern can be found in some diseases such as decompensated hypothyroidism, pericardial effusion, and chronic obstructive pulmonary disease, in addition to infiltrative cardiomyopathies. Although the thyroid-stimulating hormone (TSH) was elevated (17.1), the value was not high enough to justify a myxedema coma, which would lead to the ECG changes.
At this time, echocardiography would be instrumental in identifying the etiological cause of the decompensation. This examination ruled out the presence of pericardial effusion, showing significant left ventricular hypertrophy with some degree of asymmetry, in addition to indirect signs of restrictive cardiomyopathy2. Myocardial hypertrophy is often associated with hypertension or hypertrophic cardiomyopathy, but both involve normal or increased voltage on the ECG; and therefore, the findings of ventricular hypertrophy associated with reduced voltage on the ECG in the absence of pericardial effusion are unique to infiltrative cardiomyopathies, a group of cardiac diseases within the restrictive cardiomyopathies2.
Among the functional categories of cardiomyopathy (dilated, hypertrophic, and restrictive), restrictive is the least common3. It is defined as a disease of the heart muscle that negatively impacts ventricular filling, with normal or reduced diastolic volumes or diminished in one or both ventricles. Ventricular function remains unchanged, at least early during the condition, and ventricular wall thickness tends to be normal or increased according to the underlying disease2.
The principal pathophysiological finding in restrictive cardiomyopathies (CMP) is diastolic dysfunction. Restricted ventricular filling occurs due to increased filling pressures and loss of ventricular compliance caused by excessive stiffening of the ventricular walls. This change is the result of myocardial fibrosis, inflammation or scarring3.
Clinical manifestations can be either right or left HF, because both ventricles may be affected. In the right ventricular involvement, signs of right-side HF predominated with elevated jugular venous pressure, peripheral edema, and ascites3. When the left ventricle was affected, effort dyspnea was present and chest X ray showed lung congestion. It should be noted that the heart area was preserved in radiography, as there was no ventricular dilatation2.
Atrial dilatation is very common; the ventricular thickness may be normal or increased, diastolic volume may be normal or reduced, and myocardial relaxation is affected3. A diagnosis of restrictive CMP should be expected when there is clinical HF syndrome associated with normal cardiac area and preserved systolic function4. Ventricular filling basically depends on the pressure gradient between the atria and ventricle4. At the beginning of diastole, ventricular filling depends on the atrial pressure, which is increased. In the late phase of diastole, atrial contraction and ventricular compliance are the main determinants4.
Differential diagnosis of restrictive cardiomyopathies is conducted with constrictive pericarditis, which is considered a classic clinical challenge and one with significant implications for therapeutic conduct.3 Both conditions lead to rapid and deep decline in ventricular pressure at the beginning of diastole, with rapid increase in early diastole until it reaches a plateau, leading to a hemodynamic pattern known as the square root sign observed in the atrial pressure curves3. In constrictive pericarditis, there is equalization of the diastolic pressures, whereas in restrictive cardiomyopathy, pressures may vary more than 5 mmHg.
Restrictive cardiomyopathy can result from a wide variety of systemic diseases, some rare in clinical practice and may initially present as HF.
The most common specific cause of restrictive cardiomyopathy is amyloidosis; in almost half of cases, restrictive cardiomyopathy may be idiopathic. Amyloidosis is a generally progressive infiltrative systemic disease.3 In its cardiac form, amyloid protein is deposited in the myocardium and can result in restrictive cardiomyopathy, systolic HF, orthostatic hypotension, or conduction system disturbances3. The ECG is greatly valuable in confirming the diagnosis, diffusely showing low QRS voltage complexes that, along with the thickened appearance of the ventricular walls in the echocardiogram, strongly suggest an infiltrative disease3.
In cardiac amyloidosis, in addition to the echocardiographic findings of the restrictive component, some cases may display a peculiar granular and shiny appearance, probably as a result of the amyloid deposit. The diagnosis of amyloidosis must be confirmed by endomyocardial biopsy. In cases of systemic amyloidosis, biopsy can be performed on other tissues such as the rectum, gums, or kidney.
With respect to the treatment, measures to control symptoms related to diastolic HF should be implemented, such as volemic control. Diuretics and vasodilators should be used with caution because these patients' cardiac output is largely dependent on the increased venous pressures4. Digoxin and calcium channel blockers should be avoided because they may bind to the amyloid substance and cause toxicity4. A specific treatment must be determined on the basis of the etiology of amyloidosis. In its primary form (AL), alkylating chemotherapy can be used with or without concomitant use of autologous bone marrow transplant. The average survival of patients with cardiac amyloidosis is estimated to be 5 months4. Heart transplant should be considered, but with a possibility of relapse in a short time and with survival rates of 30% for 5 years3.
During the patent’s hospitalization, the patient’s hemodynamic parameters and dyspnea worsened. Dobutamine and diuretics were started without clinical improvement, and due to progressive worsening of renal function, it was necessary to start dialysis. The patient presented progressive hypoxemia and exhibited cardiopulmonary arrest with pulseless electrical activity. Despite CPR, the patient died. (Dr. Fabio Grunspun Pitta, Dr. Natalia Quintella Sangiorgi Olivetti, Dr. Diego Simões Peniche and Dr. Andrea Maria Dercht)
Diagnostic hypothesis. Underlying disease: amyloidosis. We believe that the terminal event (considering that the PCR was attributed to hypoxemia) was pulmonary embolism associated with cardiogenic shock. (Dr. Fabio Grunspun Pitta, Dr. Natalia Quintella Sangiorgi Olivetti, Dr. Diego Simões Peniche and Dr. Andrea Maria Dercht)
Autopsy
In the autopsy, the weight of the heart was slightly more than the normal weight (390 g; normal = 350 g), with approximately normal-sized cavities, presenting an endocardial surface with irregular, yellowish areas. The atria had increased volume with the endocardial surface being irregularly yellow (Figure 6). The lungs had increased weight and reddened areas.
The microscopic study revealed the presence of amyloid material in the intramyocardial coronary branches; the endocardium and myocardium; particularly, the subendocardial region, the pulmonary artery; and in part of its intraparenchymal branches, the vessels of the renal hilum, and the glomerular capillaries, on the wall of splenic vessels and hepatic portal spaces (Figure 7). Amyloid substance was also found in the skin, tongue, bladder, and most markedly, in the myometrium and the muscular layer of the digestive tract (Figure 8).
Figure 7.

Photomicrographies showing amyloid deposits in the cardiovascular system: myocardial interstitium, pulmonary and renal microcirculation.
Figure 8.
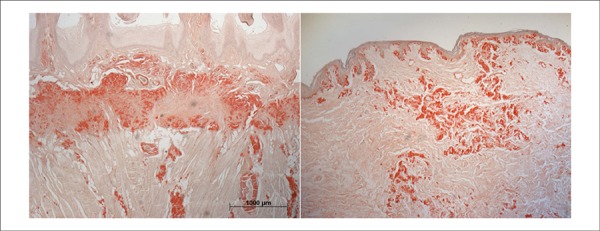
Histological slides showing amyloid deposits outside the cardiovascular system: tongue (left panel) and skin (right panel).
The bone marrow was hypercellular, with an apparent increase in the amount of plasma cells, although it did not show frank plasmacytosis. Immunohistochemical reactions for light antibody chains (kappa and lambda) in the amyloid deposits yielded negative results.
In addition to amyloidosis, the kidneys exhibited acute tubular necrosis, and the lungs showed areas of hemorrhage, which may have contributed to the patient's death. (Dr. Paulo Sampaio Gutierrez, Dr. Luiz Alberto Benvenuti)
Histopathological diagnosis. Main disease: Amyloidosis, predominantly vascular.
Causa mortis. Heart failure with areas of intra-alveolar bleeding. (Dr. Paulo Sampaio Gutierrez, Dr. Luiz Alberto Benvenuti)
Commentary
When the patient was alive, she was diagnosed with diastolic type HF. Among other possibilities, amyloidosis was suspected, which the autopsy revealed to be the patient's main disease.
Amyloidosis is a rare disease characterized by deposits that correspond to less than 30 proteins, assuming anomalous conformation. Type AA amyloidosis usually does not significantly compromise the heart, accompanying chronic inflammatory diseases that the patient did not have. The coloring of the tissue deposit is sensitive to treatment with potassium permanganate, unlike what occurred in this case. Therefore, deposits associated with transthyretin or light chains of immunoglobulins (kappa or lambda), which make up the most common – or the least rare – proteins deposited in amyloidosis, should be considered. In this case, the protein involved and the type of amyloidosis was not fully clarified. Immunohistochemical reactions for the immunoglobulins yielded negative results. These facts oppose the possibility of this type of protein deposit, but do not rule it out altogether. The bone marrow was hypercellular; it was neither possible to eliminate nor characterize the plasmacytic dyscrasia with certainty, which in any case had no direct connection with amyloidosis. As for transthyretin, we do not have its specific antibody. There are two major subtypes of amyloidosis with this deposit: senile cardiovascular and hereditary. The senile cardiovascular type is far more common in men, rarely appears in patients under age 65, and generally does not cause deposits outside the heart or vessels, as observed in some organs of our patient such as the skin, bladder, uterus, and digestive tract. Still, this cannot be totally ruled out as the patient's underlying disease. Finally, hereditary amyloidosis is rarer than those other types, and has a certain familial nature. Although the patient's family had no history of heart disease, it is not possible to completely rule out this possibility; additionally, there are several other proteins that can cause amyloid deposit. Currently, the most accurate methodology of characterizing the type of deposit is the mass spectra analysis of material obtained in histological slides by laser microdissection5,6.
Section editor: Alfredo José Mansur (ajmansur@incor.usp.br) Associated editors: Desidério Favarato (dclfavarato@incor.usp.br) Vera Demarchi Aiello (anpvera@incor.usp.br)
References
- 1.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Jr, Drazner MH, et al. ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;128(16):1810–1852. doi: 10.1161/CIR.0b013e31829e8807. [DOI] [PubMed] [Google Scholar]
- 2.Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N Engl J Med. 1997;336(4):267–276. doi: 10.1056/NEJM199701233360407. [DOI] [PubMed] [Google Scholar]
- 3.Libby P, Bonow RO, Mann DL, Zipes DP, editors. Braunwald: tratado de doenças cardiovasculares. 9a. ed. Philadelphia: Saunders/Elsevier; 2013. [Google Scholar]
- 4.Salemi V, Fernandes F, Nastari L, Mady C. Mesquita ET, et al. Insuficiência cardíaca com fração de ejeção normal. São Paulo: Atheneu; 2009. Cardiomiopatias restritivas; pp. 197–211. [Google Scholar]
- 5.Picken MM. Modern approaches to the treatment of amyloidosis: the critical importance of early detection in surgical pathology. Adv Anat Pathol. 2013;20(6):424–439. doi: 10.1097/PAP.0b013e3182a92dc3. [DOI] [PubMed] [Google Scholar]
- 6.Esplin BL, Gertz MA. Current trends in diagnosis and management of cardiac amyloidosis. Curr Probl Cardiol. 2013;38(2):53–96. doi: 10.1016/j.cpcardiol.2012.11.002. [DOI] [PubMed] [Google Scholar]