

Abstract
Background
The majority of genetic biomarkers for human cancers are defined by statistical screening of high-throughput genomics data. While a large number of genetic biomarkers have been proposed for diagnostic and prognostic applications, only a small number have been applied in the clinic. Similarly, the use of proteomics methods for the discovery of cancer biomarkers is increasing. The emerging field of proteogenomics seeks to enrich the value of genomics and proteomics approaches by studying the intersection of genomics and proteomics data. This task is challenging due to the complex nature of transcriptional and translation regulatory mechanisms and the disparities between genomic and proteomic data from the same samples. In this study, we have examined tumor antigens as potential biomarkers for breast cancer using genomics and proteomics data from previously reported laser capture microdissected ER+ tumor samples.
Results
We applied proteogenomic analyses to study the genetic aberrations of 32 tumor antigens determined in the proteomic data. We found that tumor antigens that are aberrantly expressed at the genetic level and expressed at the protein level, are likely involved in perturbing pathways directly linked to the hallmarks of cancer. The results found by proteogenomic analysis of the 32 tumor antigens studied here, capture largely the same pathway irregularities as those elucidated from large-scale screening of genomics analyses, where several thousands of genes are often found to be perturbed.
Conclusion
Tumor antigens are a group of proteins recognized by the cells of the immune system. Specifically, they are recognized in tumor cells where they are present in larger than usual amounts, or are physiochemically altered to a degree at which they no longer resemble native human proteins. This proteogenomic analysis of 32 tumor antigens suggests that tumor antigens have the potential to be highly specific biomarkers for different cancers.
Keywords: Proteogenomics, biomarker, cancer, tumor antigens, protein-protein interaction, protein expression, gene expression
Background
Cancer cells differ from normal cells by genetic and epigenetic aberrations in a number of cellular functions. The genetic hallmarks of cancer include self-sufficiency in growth signals, insensitivity to growth-inhibition signals, unlimited replicative potential, resistance to apoptosis, sustained angiogenesis, and local tissue invasion and metastasis [1]. In addition, cancer cells display deregulated cell energetics, instability and mutation of the genome, avoidance of immune destruction, and tumor-promoting inflammation [2]. Epigenetic changes include DNA methylation, histone modifications, nucleosome positioning, and miRNA expression [3]. Furthermore, carcinogenesis could be explained by interactions between cancer cells and surrounding tissues [4]. These hallmarks define functional profiles and aberrations that distinguish cancer from normal tissue.
Immune evasion employed by tumors involves multiple cellular and molecular mechanisms. Examples include blocking of the STAT-3 signaling pathway [5], toll-like receptor activation [6], production of immunosuppressive cytokines [7], infiltration of myeloid suppressor cells or regulatory T cells [8,9], activation of immunosuppressive networks [10], and down-regulation of human leukocyte antigen or impairment of antigen processing and presentation [11,12]. Inflammation promotes multiple hallmark capabilities through a spectrum of bioactive molecules that are supplied to the tumor microenvironment in support of the hallmarks of cancer [13-18]. These include growth factors that support proliferative signaling, and survival factors that reduce cell death. Inflammation also promotes the transformation of epithelial cells that enables cancer cells to invade, to resist apoptosis, and to spread to other tissues. Inflammation also promotes factors that stimulate angiogenesis, local tissue invasion, and metastasis.
Tumor antigens (TAs) are tumor proteins that, when expressed in tumors, are recognized by the host immune system. They represent markers that are either specific for individual tumors or are generally overexpressed in tumors as compared to normal tissues [19]. TAs can be neoantigens (tumor-specific antigens) that arise from mutation or RNA splicing. Neoantigens are expressed only by cancer cells and not by normal tissue [20]. Tumor-associated antigens show increase in expression in cancer tissue as compared to normal tissue (e.g. IDO1 [21], HER2 [22], or survivin [23]). Tissue-specific antigens that are recruited and expressed by cancers in specific tissues (e.g. cyclin-A1 [24] or Cancer/Testis Antigen 1B [25]). TAs are targets for cancer diagnostic and therapy that have been studied extensively - more than 1400 clinical trials focusing on TAs have been reported in clinicaltrials.gov as of October 2013 http://www.clinicaltrials.gov.
Most common biomarkers are based on patterns in expression of genes, RNA, proteins, or epigenetic patterns [26]. Examples of cancer biomarkers routinely used in the clinic include α-fetoprotein (AFP) for diagnostics and management of testicular cancer [27], MUC16 (cancer antigen 125 or CA-125) for ovarian cancer [28], ERBB2 (HER2) protein for breast cancer [29], and prostate specific antigen for prostate cancer [30].
The progression of a normal cell towards a neoplastic state comprises a cascade of events that are responsible for inducing tumorigenesis [31]. The conditions of the tumor microenvironment may also contribute to the cellular characteristics of tumor cells, as this microenvironment may induce genetic instability in tumor cells [32].
The advent of high-throughput genomics methods has facilitated a dramatic increase in the number of candidate genetic biomarkers [33]. However, very few new genetic biomarkers have been added to the clinical toolbox in recent years [34]. Approximately 95% of human protein coding genes produce splice variant transcripts increasing the number of potential gene expression biomarker candidates [35]. Individual genetic biomarkers are rarely informative as genetic redundancy provides for high genetic flexibility without necessarily affecting the biological phenotype [36]. The expression of many genes correlate with disease progression in individual patients, but the analyses of large data sets consistently show that only a small subset of these is consistently observed in larger cohorts. The expression of genes may also prove inconsistent over time, since cells respond to changes in environment and undergo transcriptomic changes in different stages of development. While transcriptomic differences are apparent between normal and cancerous tissues, the expression of TAs remains relatively constant through different cancer stages, suggesting that most defining genetic alterations conferring a cancerous potential occur at the early stages of tumorigenesis [37]. In contrast, similar studies of the tumor microenvironment reveal extensive gene expression changes in tumor stromal tissue during cancer progression [38].
The number of protein biomarkers has been growing, owing largely to the advances in mass spectrometry techniques [39]. However, protein biomarkers, similar to genetic biomarkers, are rarely uniquely expressed (or overexpressed) in malignant tissues [34]. At the same time, more than 260,000 protein variants resulting from alternative splicing have been annotated to date [40]. Additionally, a variety of post-translational modifications (PTMs) can alter the structure and function of proteins [41]. Most of the proposed protein biomarkers are not commonly used in clinical application as they do not hold up statistically in large populations, or the cost of assays outweighs their prognostic value [42]. Protein expression profiling has proven useful for stratification of cancer versus normal tissues for invasive ductal carcinoma. Differentially expressed proteins or protein sets can be used as candidate biomarkers for carcinogenesis [43].
Relative to the research effort, the number of verified useful biomarkers is vanishingly small [44] and we need new and improved approaches. The emerging field of proteogenomics seeks to synergistically enrich the value of genomics and proteomics by studying the intersection of the two data sets [45]. True integrative analysis of genomics and proteomics data is a non-trivial task, as the expression of mRNA typically does not always correlate well with the expression of corresponding proteins [46]. The mechanisms of transcriptional and translational regulation have been extensively studied, but are not yet fully understood. However, when the expression of individual genes or pathways correlate with protein expression, this may provide insights into transcriptional effects and the mechanistic basis of statistically derived biomarkers [47]. Proteogenomic profiling has been applied to the study of invasive ductal carcinomas (IDCs) to reveal potential biological events not previously associated with this cancer type. The study of biological networks of differentially expressed proteins and differentially expressed genes revealed patterns that correlate with clinical relapse [48].
In addition to their potential diagnostic and prognostic value as biomarkers, tumor associated antigens (TAAs) and tumor specific antigens (TSAs) have therapeutic potential as targets of cancer immunotherapies [49,50]. In this study, we have combined the analysis of protein and mRNA expression of a selection of well-described TAAs [51,52] to evaluate their proteogenomic potential as biomarkers for IDCs.
Results and discussion
Translation of mRNA in IDC tissue
The central dogma of molecular biology states that DNA is transcribed into RNA, which is in turn translated into protein. From this general rule, it is often assumed that a certain amount of DNA makes an equal, or at least proportional, amount of RNA, which in turn makes an equal or proportional amount of protein. However, it is increasing clear that a plethora of transcriptional and translational regulatory mechanisms affect the dynamics of the central dogma. Examples of transcriptional regulation include: miRNA interference [53]; epigenetic factors, such as methylation [54]; and alternative splicing [55]. Translational regulation affects the rates of degradation for different proteins [56], as well as PTMs [57]. Many of these regulatory mechanisms have been used in efforts to characterize new biomarkers for various cancers [58-61]. Although most of these mechanisms are not yet fully understood, they modulate transcription and translation in all cells.
The correlation of expression between 1066 gene/product pairs from 23 human cancer cell lines of various origins and cancer types was examined [62]. This study reported 169 genes for which mRNA and protein expression correlated (at threshold of ρ > 0.455). The study, involving the analysis of biological function ontologies, found that within the set of 169 genes, ontologies relating to the cytoskeleton and adherent junctions in the cellular compartment, cellular motility, and other maintenance-related categories were significantly enriched [62].
Dysregulation of cellular processes is a feature of cells undergoing tumorigenesis, and transcriptional and translational mechanisms are likely to be dysregulated as well. Given the heterogeneous nature of cancer, it is unlikely that transcription and translation modulation is homogenous in different cancers. This prompted us to examine the correlation between gene expression and protein expression in the Cancer Genome Atlas (TCGA) consortium IDC tissues, to provide an IDC specific context for our analysis of the unpaired sets of mRNA and protein expression data between normal, preinvasive and invasive breast cancer tissue.
Our results are consistent with the notion that it is insufficient to study only the expression of mRNA when searching for TAs as potential immunotherapy targets, but protein expression also needs to be considered. We calculated Spearman's ρ for the mRNA/protein expression pairs of the 86 genes examined by TCGA. Twenty-nine genes were found to have a ρ > 0.455, six of which are TAs (Figure 1, top panel). Although the number of examined proteins is too small to draw global conclusions (global patterns of translation in IDC tissue still remain to be elucidated in comprehensive analyses), we found that of 13 examined TAs, mRNA and protein expressions only correlate in six. This observation is consistent with the results reported in [62,63].
Figure 1.
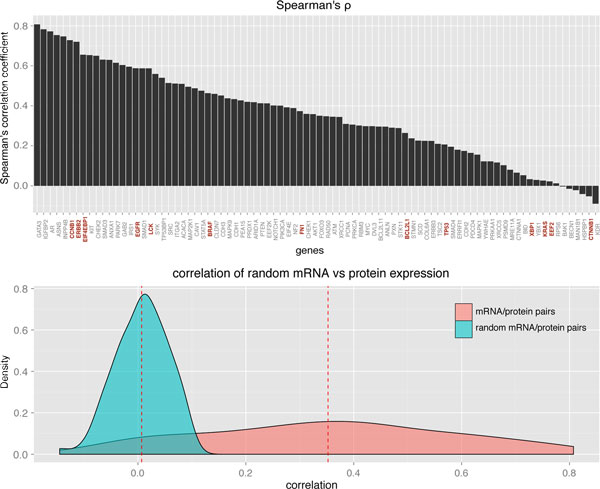
Top panel: Spearman's rank correlation between mRNA expression and protein expression of 86 genes in 404 IDC patients. The TAs are highlighted in red. Bottom panel: density distribution of correlation coefficients of mRNA vs protein expression (pink) and density distribution of correlation coefficients of randomized mRNA vs protein expression (aqua). The dashed red lines mark the mean correlation in each distribution.
The mean correlation of expression of the 86 mRNA/protein IDC pairs was calculated to be 0.35. Randomly paired mRNA and protein expressions yield a mean correlation of approximately zero, indicating no apparent bias in the data. Distributions of correlation coefficients in the normal and randomized data set are shown in Figure 1 (bottom panel).
Expression of tumor antigens on protein level
Although not paired like tissue samples used for the analysis of gene expression, the protein expression data offer valuable information for prescreening and filtering of TA genes for further analysis. With the exception of OAS3, BST2, and SCRN1, all TAs were expressed in at least one of the normal tissue samples [43]. Not all TAs were expressed in the IDC tissue, and only a fraction was consistently expressed in all nine patient samples (Figure 2). These results were not unexpected, since different expression patterns are often observed in different cells of the same tumor [64]. Within the small cohort examined here, 30 of 32 TAs were expressed as proteins in at least two of nine patients. The two TAs, MYO1B and SART3, found to not be expressed in the IDC patient cohort were removed from the list and the remaining 30 TAs were further analyzed.
Figure 2.
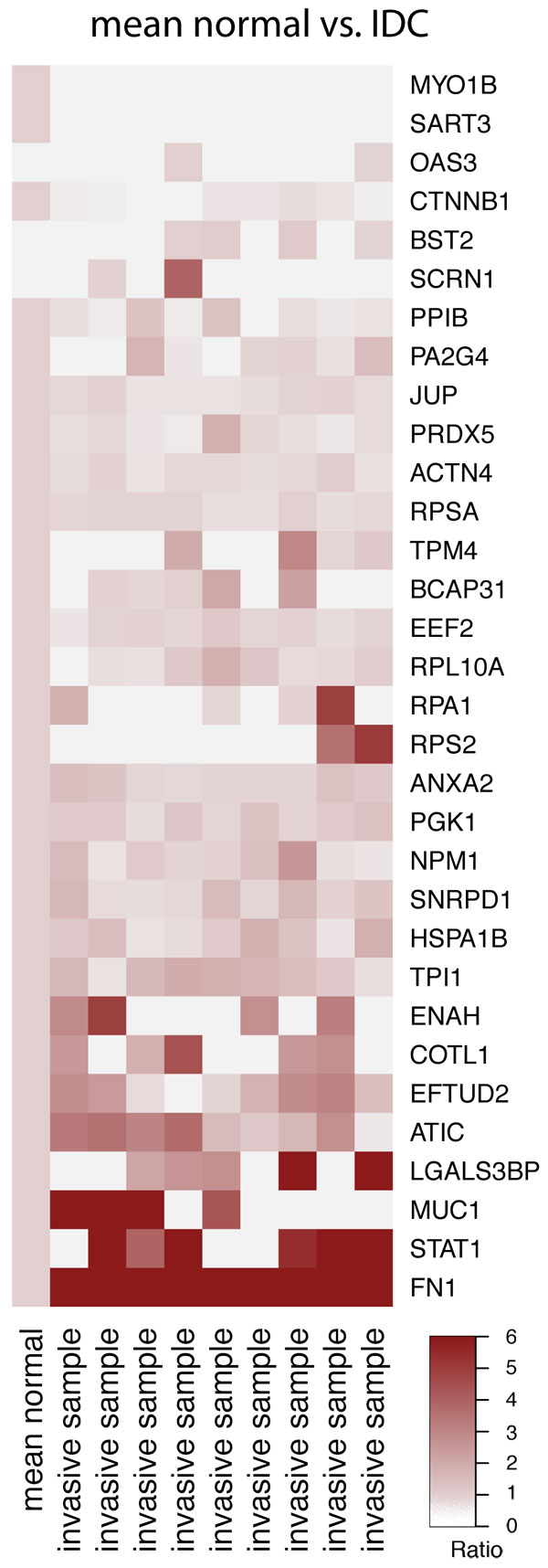
Ratio of protein expression in IDC tissue to mean expression in normal tissue of 32 measured TAs.
Tumor antigen gene expression patterns of normal, preinvasive and invasive IDC tissues
We examined differential expression of TA mRNAs between normal and preinvasive IDC tissue for the TAs expressed as proteins in the IDC tissue. Of the 30 TAs expressed in IDC tissue (excluding MYO1B and SART3), mRNA expression data was measured for 28 TAs (not measured for RPSA and HSPA1B). Five TAs displayed consistent down regulation between the normal and preinvasive tissues, nine TAs displayed up regulation and 16 showed no significant difference between the two types of tissue (p < 0.05). Fold change to median expression of significantly differentially expressed TAs is shown in Figure 3 (left panel). A comparison of TA expression between normal and invasive tumor tissues revealed ten up regulated TAs and two down regulated TAs (Figure 3, right panel).
Figure 3.
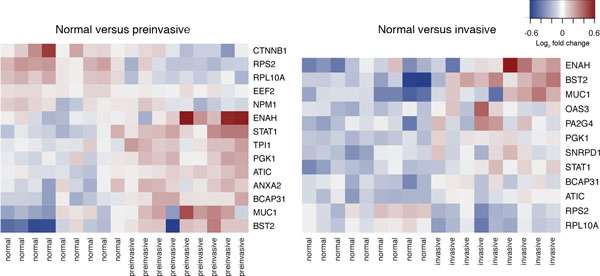
The heat map based on log2 transformed fold change from median expression of TA mRNA. Shown in these heat maps are TAs that are significantly differentially expressed between normal tissue and preinvasive tissue (left) and between normal tissue and invasive tissue (right) (p < 0.05). Red corresponds to up regulated and blue corresponds to down regulated transcripts.
The cytoplasmic ribosomal subunits RPS2 and RPL10A were down regulated in both preinvasive and invasive tissues (Figure 3). These patterns, although not consistent with the increased protein synthesis by tumor cells, have been observed previously in breast cancer and colorectal cancer studies [38,65]. The gene expression of these TAs is typically up regulated in cancer, but it was down regulated in the IDC tissue. Sgroi and colleagues noted that the mechanisms by which ribosomal proteins contribute to tumorigenesis are relatively poorly understood [38] but it is known that changes in components of translational mechanism increase cancer risk [66]. The down regulation of these particular ribosomal subunits may be indicative of qualitative rearrangement of the ribosomal proteins as a whole, meaning that one should be careful in interpreting these observation points as potentially causal.
Seven TA genes were up regulated in both preinvasive and invasive tissues as compared to the normal tissue. The genes of the STAT protein family encode a series of signal transducers and transcription activators. STAT1 is up regulated in both preinvasive and invasive IDC tissues, as compared with the normal tissue. This pattern has been associated with the faster progression from ductal carcinoma in situ to invasive carcinoma, most likely by inducing immunosuppression in the tumor microenvironment [67]. STAT1, which is typically dormant in normal tissue, is also associated with aggressive growth and chemotherapy resistance [68]. Interestingly, there is very little difference between fold changes of STAT1 expression between preinvasive and invasive tissues compared with the normal samples, indicating that the effects of STAT1 are already present in the preinvasive tissue.
Likewise, the bone marrow stromal cell antigen 2 (BST2) is up regulated in the IDC. The up regulated BST2 gene has been proposed as a biomarker for bone metastasis of breast cancer [69]. Similar patterns have been observed in tamoxifen resistant breast cancer cells, where up regulation of BST2 gene expression correlated to increased invasiveness and metastasis, regulated and activated by STAT3 [70].
Increased expression of phosphoglycerate kinase 1 (PGK1) was also observed in both preinvasive and invasive tissues. PGK1 is a major enzyme in glycolysis and facilitates ATP production under hypoxic conditions [71]. The elevated levels of PGK1 have previously been associated with an increased invasiveness of gastric cancer [72]. PGK1 was reported to facilitate the release of the anti-angiogenic enzyme, angiostatin [73].
Mucin 1 (MUC1) is an oncoprotein that is overexpressed in 90% of breast cancer patients and its gene is amplified in 40% of the patients. Overexpression of this protein has been linked to tamoxifen resistance [74]. The activities of MUC1 leading to tamoxifen resistance are many: it contributes to the activation of the PI3K-AKT pathway known to be involved in apoptosis [75]. It activates the MEK/ERK pathway, a regulator of cellular growth and apoptosis in breast cancer [76]. It also activates the Wnt/β-catenin pathway involved in cell proliferation and migration as well as formation and maintenance of cancer stem cells [77], and it activates STAT pathways associated with cellular growth and inflammation [78].
We also found the cytoskeleton regulatory protein, ENAH, to be overexpressed in both preinvasive and invasive tissues. EHAH is non-detectable in normal tissues, but was found to be weakly expressed in the low risk benign lesions, and was overexpressed in the high-risk benign breast lesions [79].
ATIC, the product of the purH gene, is involved in the final steps of de novo synthesis of purine [80]. Imbalances in the biosynthesis and metabolism of purine is linked with progression of a number of cancer types [81]. ATIC inhibition has been explored as a therapeutic strategy in breast cancer patients [82], as has the potential of ATIC as a TA [83].
The function of B-cell receptor-associated protein 31 (BCAP31) is relatively unknown. It has been proposed to be involved in CASP8-mediated apoptosis, where its cleavage product is a strong inducer of apoptosis. However, caspase resistant types of BCAP31 have been observed and the lack of cleavage during apoptosis leads to reduced apoptotic potential [84]. BCAP31 has previously been reported to be up regulated in the breast cancer tissue as compared to normal tissue [85].
Individual expression profiles
Although there are similarities between TA genes differentially expressed in preinvasive and invasive tissues, there were also discrepancies between patients, even in this small cohort. The analysis of four patients for whom expression was measured in normal, preinvasive, and invasive tissues revealed that there are only a few clear patterns of expression (Figure 4). Only four TA genes, ANXA2, ENAH, ATIC, and STAT1, were consistently up regulated in both IDC tissue types in all four patients. Two TA genes, CTNBB1 and RPL10, were consistently down regulated in both IDC tissues in all four patients. No complex patterns, i.e. genes up regulated in one IDC tissue type and down regulated in the other, or vice versa, were observed. However, the expression of some TA genes were consistent across tissue types, but not across patients; for example, LGALS3BP is strongly expressed in patient 1, but down regulated in patient 4.
Figure 4.

Heat map based on log2 transformed fold change of gene expression in preinvasive and invasive IDC tissue compared with expression in normal tissue in four individual patients.
Pathway-based assessment of expression patterns
To evaluate the potential biological impact of aberrantly expressed TAs, we examined their involvement in canonical pathways [86]. From the analyses of differential mRNA expression in the four individual expression profiles, it is clear that heterogeneity of mRNA expression exists even when examining a very limited number of proteins. Although only approximately half of the TA genes were significantly differentially expressed in the full cohort, all 30 TAs were aberrantly expressed in at least one of the four individual profiles, and all 30 were expressed at protein level. Of these 28 have entries in the STRING database.
The functional relationship between the 28 TAs was examined using the STRING database. An interaction confidence threshold of 0.5 yielded four functionally related groups as well as nine non-related TAs (Figure 5).
Figure 5.
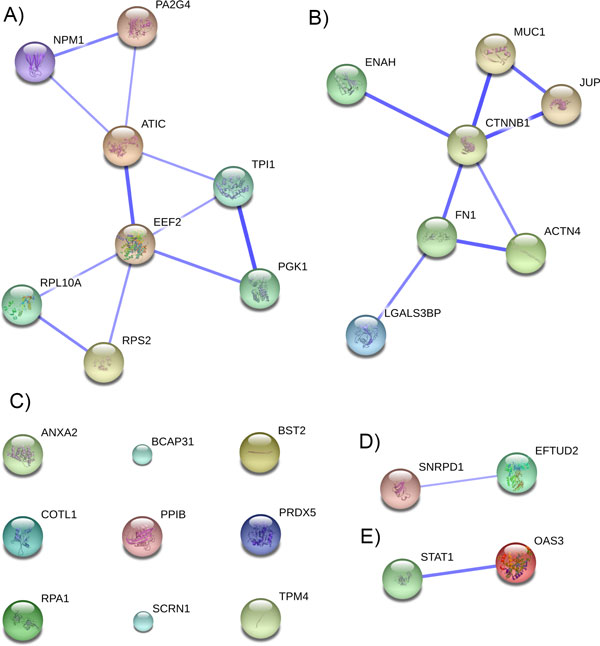
Confidence view of protein-protein interactions within the 28 examined TAs, generated using STRING database. Nodes correspond to TAs and edges correspond to functional interactions. Thicker edges signify higher confidence in the interaction. Only interactions with a confidence score higher than 0.5 were included.
The four functional groups were further examined for their involvement in the canonical pathways related to the hallmarks of cancer. One cycle of expansion to include related proteins was applied to each of the four groups to yield four functional modules (Figure 6). Further examination of each of the four functional modules and their direct interactants, using MSigDB [87], reveals dysregulation of canonical pathways related to the hallmarks of cancer.
Figure 6.
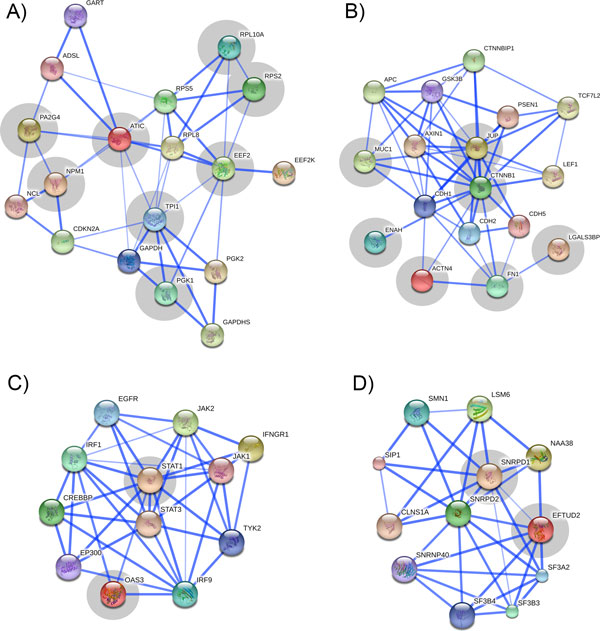
Confidence view of expanded protein-protein interactions within the four functional groups of TAs (highlighted in gray) generated using STRING database. Interacting proteins were added to the TAs using one cycle of expansion. Nodes correspond to the proteins and edges correspond to their functional interactions. The thicker edges signify higher confidence in the interaction. Interactions with a confidence score higher than 0.5 are shown.
Module 1
Expansion of group 1 (Figure 5A) yielded 10 direct functional neighbors at a confidence score of 0.5 (Figure 6A). The resulting PPI is heavily interconnected indicating tight functional homology within the module.
The proteins of functional module 1 overlap in two canonical pathways: PGK1, TPI1, GAPDH, and GAPDHS overlap in the glycolysis pathway, and expression of twelve of the eighteen proteins are known to be positively correlated with BRCA1 expression in BRCA1mut tumors [88]. Both pathways have been extensively studied as potential therapeutic targets in cancer.
Aerobic glycolysis has profound effects on proliferating cells [89]. Abnormal cellular metabolism is a defining feature of cancer [1], and proteins involved in metabolic pathways have both diagnostic and therapeutic potential [90,91]. Numerous enzymes are involved in maintaining elevated rates of glycolysis, making them potential targets of therapeutic agents [92].
Mutations in the BRCA1 gene are associated with faster progression of breast cancer and other cancers [93]. BRCA1 plays a role in DNA repair and genomic stability maintenance. It is a known tumor suppressor, which, when mutated, is linked with the early onset of breast cancer. Network modeling strategies revolving around BRCA1mut revealed a number of genes associated with centrosome dysfunction and thereby increased cancer aggressiveness [88]. Twelve genes in module 1 (six of these are TAs) were involved in the network functionally associated with BRCA1 mutations and centrosome dysfunctions.
Module 2
Module 2 consists of seven TAs (Figure 5B), which when expanded by one cycle in the STRING database has ten interaction partners in a highly connected PPI (Figure 6B). Seven proteins (CTNNB1, APC, GSK3B, AXIN1, LEF1, TCF7L2, PSEN1, CTNNBIP1) overlap in the Wnt pathway, and a subset of these additionally overlaps in the β-catenin pathway. A smaller subgroup consisting of TAs ACTN4, ENAH, and FN1 overlaps in an actin cytoskeleton regulation pathway. Both these pathways are generally regarded to play a role in cancer, and are known to be connected through shared regulators [94].
The Wnt pathway is involved in a number of cancer-relevant cellular processes such as cell division and migration, and cell fate decisions [95]. Elevated levels of β-catenin in the nucleus or cytoplasm of cells, indicates activation of the Wnt pathway, which correlates with poor prognosis in breast cancer patients [96], and detection of β-catenin levels using immunohistochemical staining is an important diagnostic tool [97]. Molecules of the Wnt pathway have been extensively targeted in anticancer treatment modalities, involving small interfering molecules, blocking antibodies, and peptide-based therapies [98].
Module 3
Module 3 (Figure 6C, generated by expansion of the TAs in Figure 5E) is centered on proteins relating to the immune response, as ten of the proteins in this module overlap in immune system related pathways. The predominant immune system pathways in this module are cytokine signaling (specifically IL-6), interferon signaling, and JAK/STAT signaling.
All three pathways have been associated with cancer development. The JAK/STAT pathway is recognized as a modulator of cytokine signaling, and has therefore been associated with a large number of malignancies [99]. In cancer, the JAKs and STATs (particularly STAT3) are often observed to be constitutively activated, which is thought to induce cell proliferation and prevent apoptosis [100]. Additionally, the STATs are known to induce pro-oncogenic inflammation in the tumor microenvironment by promoting pathways such as NF-κB and IL-6-GP130-JAK pathways [101]. A number of NF-κB encoded inflammatory factors, such as IL-6, are activators of STAT3, in turn creating a positive feedback loop, leads to dysregulated action of immune modulating pathways [102].
Another commonly observed immune deficiency observed in cancer cells, is impaired interferon-signaling. Interferons are important modulators of immune response, and defects in interferon signaling are highly detrimental to immune control of cancer cells [103]. Cytokine secretion is critically modulated by JAK/STAT activity [104].
Module 4
Finally, module 4 (Figure 6D) consists of the TAs EFTUD2 and SNRPD1 (Figure 5D), and their ten closest interaction partners. Seven of these proteins overlap in the spliceosome pathway, which plays a central role in pre-mRNA processing and splicing. Splicing in tightly regulated in different tissues and different stages of development, and dysregulation of the spliceosome function may lead to incorrect assembly of exons and nonfunctional translation. As such, alternative splicing plays a significant role in cancer and other malignancies [105]. The spliceosome has therefore been examined for its potential as a therapy target [106]. Spliceosome-related therapy can be directed towards the products of alternative splicing [107] or the spliceosome modulators [108].
Non-interacting TAs
In addition to the four modules, we also observed 9 TAs that do not connect with any other TAs (Figure 5C). These are not necessarily less valuable as targets, but the analyses performed here are much less comprehensive for these TAs. Their interactants and involvement in molecular pathways relating to cancer is summarized in Additional file 1.
Conclusions
Statistical testing for patterns in high throughput mRNA expression data has long been the primary method for defining biomarkers in human cancers. The analyses are constantly refined with inclusion of data from epigenetic experimentation, measurements of ncRNAs, and protein expression, and are expanded with ontology enrichment analyses, pathway analyses, and co-analyses of different data types. Additionally, as experimental methods increase in efficiency and resolution, the bodies of data examined keep growing. A large number of diagnostic and prognostic biomarkers have been reported, and a small number are utilized in clinics, but it is believed by some that the majority of reported biomarker candidates are the result of stochastic noise within data sets [109].
TAs are a group of proteins against which the immune system has been recorded to autologously react. Specifically, they are recognized in cells where they are present in larger than usual amounts, or physiochemically altered to a degree at which they no longer resemble native human proteins. As such, their presence or abundance in cancer cells is often unique and their roles and functions are, in many cases, studied extensively. Proteins that are frequently observed (and autologously recognized by the immune system) in tumor cells can be hypothesized to play a significant role in tumorigenesis. They therefore hold the potential to be highly specific biomarkers for the cancers in which they are observed.
The challenges pertaining to the utility of TA biomarkers are similar to those we face when we statistically filter out potential biomarkers from vast amounts of high throughput genomics data: our understanding of their function and role in cancer must be elevated to a degree where we can account for outliers and exceptions to the general rules known from clinical observations. To achieve this, we analyzed the mRNA and protein expression of 30 TAs in normal tissue versus IDC tissue. We found that all but two TAs were expressed in IDC on the protein level, and a subset of these was aberrantly expressed on mRNA level. We examined their known and proposed roles in cancer by analyzing the TAs and their closest functional counterparts for overlapping participation in canonical pathways. With this approach, we defined four functional modules of TAs and interactants, which overlapped in canonical pathways. The perturbation of these pathways were readily linked to the hallmarks of cancer by querying relevant literature. A previous study of genetic biomarkers in IDC tissue resulted in the identification of approximately 2,000 differentially expressed genes involved in a large number of biological pathways [37]. Among these pathways were those related to the hallmarks of cancer that we have found from analyzing only 30 TA genes.
Currently, 258 TAs are catalogued and annotated in the TANTIGEN database of TAs. Expanding the analysis performed here to the full set of TAs is highly likely to provide additional insights. RNA sequencing is another desirable follow-up study of TAs found to be expressed in a given cancer tissue, as this may reveal known or novel splice isoforms, mutations, or other genetic aberrations. In addition to the diagnostic and prognostic potential of such a study, a catalogue of expressed TAs and variants in a given tumor can be further analyzed for their potential as therapeutic targets and directly applied in personalized treatment modalities.
Methods
Data
Protein expression data
Nine samples of estrogen receptor positive ER+ IDC tissue were analyzed for expression of 1623 proteins using liquid chromatography coupled with mass spectrometry [43]. The tissue samples were obtained using laser capture microdissection from biopsies collected at the Massachusetts General Hospital. In addition to the nine IDC samples, nine (non-paired) normal tissue samples were analyzed for protein expression.
mRNA expression data
The mRNA expression data were extracted from breast cancer biopsies, again collected at the Massachusetts General Hospital [37]. Three different types of samples were collected: normal tissue, preinvasive tissue, and invasive tissue of ER+ IDC. The tissue consisted of cells from the epithelial and stromal compartments of the normal terminal ductal lobular unit. The following samples were paired: nine samples of normal and preinvasive cancer, nine samples of normal and invasive tissue, and four samples for all three tissue types. Tissue samples were obtained using laser capture microdissection and were analyzed for mRNA expression using the Affymetrix whole genome array U133X3P [38].
Tumor antigen data
A list of known TAs was extracted from the TANTIGEN database of TAs http://cvc.dfci.harvard.edu/tadb/. TANTIGEN contains 4245 T cell epitopes found in 258 unique protein TAs (November 2013) collected from the literature. Protein expression was measured for 32 of these TAs in [45]. Of these, gene expression was meassured for 30 TAs [38]. We further analyzed these 30 TAs by proteogenomics.
mRNA and protein expression in IDC tissue
For the analysis of correlation between gene expression and protein expression, we examined 404 IDC tissue samples collected by The Cancer Genome Atlas (TCGA) consortium. Paired mRNA expression and protein expression data was available for 86 gene/protein pairs, of which 13 are known TAs. mRNA expression was extracted using Agilent mRNA expression microarrays, and protein expression was extracted using Reverse Phase Protein Arrays [63].
Analyses
Protein expression analysis
The TAs were extracted from the protein expression data. The nine normal samples were averaged and compared with expression in the nine invasive tissue samples. Log2 ratios of expression of each protein compared with the normal tissue average (normalized to 1 if expressed, or kept at 0 if not expressed) were calculated for each patient.
mRNA expression analysis
Raw probe intensities were background corrected using rma, quantile normalized [110,111], and the probe sets were indexed relative to the median. Fold changes were calculated as individual mRNA expression compared to the median mRNA expression and log2 transformed.
TA genes were extracted from the expression data. Fold changes were calculated between the three types of cancer tissue (normal, preinvasive, and invasive). Genes were examined for consistently differentially expressed genes in the patient cohort using a paired t test. Additionally, gene expression dynamics in the three cancer tissue types were examined in each individual patient.
Annotation
Each TA was characterized for its potential functions in tumorigenesis in each patient to gain an insight into the underlying biology of the observed expression profiles. Information about protein function and role in disease were extracted from OMIM [112], UniProt [113], GeneCards [114], the Human Protein Atlas [115] and UniGene http://www.ncbi.nlm.nih.gov/unigene.
Pathway analysis
Functional neighbors to aberrantly regulated TAs were extracted from the STRING database of protein-protein interactions (version 9.05) [116]. Interactions with > 0.5 confidence score were considered and the resulting protein groups were analyzed for their overlapping involvement in canonical pathways using the molecular signatures database, MSigDB [87]. The confidence score in STRING database is calculated from combined and corrected probabilities using different evidence channels for protein-protein interaction, described in [117].
Correlating mRNA and protein expression
To determine correlation between mRNA expression and protein expression, we calculated Spearman's rank correlation coefficient. The threshold for correlation was set at ρ > 0.455 to compare results with a previous study of correlation between the mRNA and protein expression [62]. The authors used the ρ > 0.455 threshold by determining that the average correlation coefficient of expression in 1000 randomly chosen gene/product pairs from 23 cancer cell lines was 0.001, and that the selected value of ρ of 0.455 provided the 95% confidence interval of the ρ distribution.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
LRO: Conceived of the study, performed the study, and prepared the manuscript.
BC: Contributed to the study of mRNA/protein pairs and critically reviewed manuscript.
OW: Contributed to the study of protein-protein interaction and critically reviewed manuscript.
DCS: Contributed to the study of gene expression and critically reviewed manuscript.
BLK: Contributed to the study of protein expression and critically reviewed manuscript.
VB: Conceived of the study and prepared the manuscript
Supplementary Material
TA, their interactants and involvement in molecular pathways relating to cancer.
Contributor Information
Lars Rønn Olsen, Email: lro@binf.ku.dk.
Benito Campos, Email: benito.campos@med.uni-heidelberg.de.
Ole Winther, Email: winther@binf.ku.dk.
Dennis C Sgroi, Email: dsgroi@mgh.harvard.edu.
Barry L Karger, Email: b.karger@neu.edu.
Vladimir Brusic, Email: vladimir.brusic@nu.edu.kz.
Acknowledgements
This work was supported by the Novo Nordisk Foundation (LRO and OW), the Susan G. Komen Breast Cancer Foundation (BLK and DCS), NIH grant RO1GM15847 (BLK), NIH grant R01CA112021 (DCS), the NCI SPORE in breast cancer at Massachusetts General Hospital (DCS) and the Avon Foundation (DCS). Publication charges for this work was funded by a Novo Nordisk Foundation grant to OW.
This article has been published as part of BMC Medical Genomics Volume 7 Supplement 3, 2014: Thirteenth International Conference on Bioinformatics (InCoB2014): Medical Genomics. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcmedgenomics/supplements/7/S3.
References
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg R. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenschein C, Soto AM. The aging of the 2000 and 2011 Hallmarks of Cancer reviews: a critique. J Biosci. 2013;38:651–63. doi: 10.1007/s12038-013-9335-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, Dalton W, Jove R, Pardoll D, Yu H. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- Huang B, Zhao J, Li H, He K-L, Chen Y, Chen S-H, Mayer L, Unkeless JC, Xiong H. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 2005;65:5009–14. doi: 10.1158/0008-5472.CAN-05-0784. [DOI] [PubMed] [Google Scholar]
- Thomas DA, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–80. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, McDermott D, Quiceno D, Youmans A, O'Neill A, Mier J, Ochoa AC. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65:3044–8. doi: 10.1158/0008-5472.CAN-04-4505. [DOI] [PubMed] [Google Scholar]
- Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- Kim R, Emi M, Tanabe K, Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res. 2006;66:5527–36. doi: 10.1158/0008-5472.CAN-05-4128. [DOI] [PubMed] [Google Scholar]
- Cromme F V, Airey J, Heemels MT, Ploegh HL, Keating PJ, Stern PL, Meijer CJ, Walboomers JM. Loss of transporter protein, encoded by the TAP-1 gene, is highly correlated with loss of HLA expression in cervical carcinomas. J Exp Med. 1994;179:335–40. doi: 10.1084/jem.179.1.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicklin DJ, Marincola FM, Ferrone S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol Med Today. 1999;5:178–86. doi: 10.1016/S1357-4310(99)01451-3. [DOI] [PubMed] [Google Scholar]
- Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–61. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- DeNardo DG, Andreu P, Coussens LM. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev. 2010;29:309–16. doi: 10.1007/s10555-010-9223-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klymkowsky MW, Savagner P. Epithelial-mesenchymal transition: a cancer researcher's conceptual friend and foe. Am J Pathol. 2009;174:1588–93. doi: 10.2353/ajpath.2009.080545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–73. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15–33. doi: 10.1007/s10555-008-9169-0. [DOI] [PubMed] [Google Scholar]
- Haen SP, Rammensee H-G. The repertoire of human tumor-associated epitopes--identification and selection of antigens and their application in clinical trials. Curr Opin Immunol. 2013;25:277–83. doi: 10.1016/j.coi.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Van den Eynde BJ, van der Bruggen P. T cell defined tumor antigens. Curr Opin Immunol. 1997;9:684–93. doi: 10.1016/S0952-7915(97)80050-7. [DOI] [PubMed] [Google Scholar]
- Sørensen RB, Berge-Hansen L, Junker N, Hansen CA, Hadrup SR, Schumacher TNM, Svane IM, Becker JC, thor Straten P, Andersen MH. The immune system strikes back: cellular immune responses against indoleamine 2,3-dioxygenase. PLoS One. 2009;4:e6910. doi: 10.1371/journal.pone.0006910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk B, Blevins TL, Wharton JT, Ioannides CG. Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic T lymphocyte lines. J Exp Med. 1995;181:2109–17. doi: 10.1084/jem.181.6.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung JT, Hamilton RL, Okada H, Jakacki RI, Pollack IF. Increased expression of tumor-associated antigens in pediatric and adult ependymomas: implication for vaccine therapy. J Neurooncol. 2013;111:103–11. doi: 10.1007/s11060-012-0998-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsenreither S, Majeti R, Schmitt T, Stirewalt D, Keilholz U, Loeb KR, Wood B, Choi YE, Bleakley M, Warren EH, Hudecek M, Akatsuka Y, Weissman IL, Greenberg PD. Cyclin-A1 represents a new immunogenic targetable antigen expressed in acute myeloid leukemia stem cells with characteristics of a cancer-testis antigen. Blood. 2012;119:5492–501. doi: 10.1182/blood-2011-07-365890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger E, Chen YT, Drijfhout JW, Karbach J, Ringhoffer M, Jäger D, Arand M, Wada H, Noguchi Y, Stockert E, Old LJ, Knuth A. Simultaneous humoral and cellular immune response against cancer-testis antigen NY-ESO-1: definition of human histocompatibility leukocyte antigen (HLA)-A2-binding peptide epitopes. J Exp Med. 1998;187:265–70. doi: 10.1084/jem.187.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusic V, Marina O, Wu CJ, Reinherz EL. Proteome informatics for cancer research: from molecules to clinic. Proteomics. 2007;7:976–91. doi: 10.1002/pmic.200600965. [DOI] [PubMed] [Google Scholar]
- Lange PH, McIntire KR, Waldmann TA, Hakala TR, Fraley EE. Serum alpha fetoprotein and human chorionic gonadotropin in the diagnosis and management of nonseminomatous germ-cell testicular cancer. N Engl J Med. 1976;295:1237–40. doi: 10.1056/NEJM197611252952207. [DOI] [PubMed] [Google Scholar]
- Bast RC, Klug TL, St John E, Jenison E, Niloff JM, Lazarus H, Berkowitz RS, Leavitt T, Griffiths CT, Parker L, Zurawski VR, Knapp RC. A radioimmunoassay using a monoclonal antibody to monitor the course of epithelial ovarian cancer. N Engl J Med. 1983;309:883–7. doi: 10.1056/NEJM198310133091503. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–12. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- Stamey TA, Yang N, Hay AR, McNeal JE, Freiha FS, Redwine E. Prostate-specific antigen as a serum marker for adenocarcinoma of the prostate. N Engl J Med. 1987;317:909–16. doi: 10.1056/NEJM198710083171501. [DOI] [PubMed] [Google Scholar]
- Foulds L. The experimental study of tumor progression: a review. Cancer Res. 1954;14:327–39. [PubMed] [Google Scholar]
- Reynolds TY, Rockwell S, Glazer PM. Genetic instability induced by the tumor microenvironment. Cancer Res. 1996;56:5754–7. [PubMed] [Google Scholar]
- McDermott U, Downing JR, Stratton MR. Genomics and the continuum of cancer care. N Engl J Med. 2011;364:340–50. doi: 10.1056/NEJMra0907178. [DOI] [PubMed] [Google Scholar]
- Brooks JD. Translational genomics: the challenge of developing cancer biomarkers. Genome Res. 2012;22:183–7. doi: 10.1101/gr.124347.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–63. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak MA, Boerlijst MC, Cooke J, Smith JM. Evolution of genetic redundancy. Nature. 1997;388:167–71. doi: 10.1038/40618. [DOI] [PubMed] [Google Scholar]
- Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, Payette T, Pistone M, Stecker K, Zhang BM, Zhou YX, Varnholt H, Smith B, Gadd M, Chatfield E, Kessler J, Baer TM, Erlander MG, Sgroi DC. Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci USA. 2003;100:5974–9. doi: 10.1073/pnas.0931261100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X-J, Dahiya S, Richardson E, Erlander M, Sgroi DC. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res. 2009;11:R7. doi: 10.1186/bcr2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Veenstra TD. Targeted mass spectrometry approaches for protein biomarker verification. J Proteomics. 2011;74:2650–9. doi: 10.1016/j.jprot.2011.04.011. [DOI] [PubMed] [Google Scholar]
- Martelli PL, D'Antonio M, Bonizzoni P, Castrignanò T, D'Erchia AM, D'Onorio De Meo P, Fariselli P, Finelli M, Licciulli F, Mangiulli M, Mignone F, Pavesi G, Picardi E, Rizzi R, Rossi I, Valletti A, Zauli A, Zambelli F, Casadio R, Pesole G. ASPicDB: a database of annotated transcript and protein variants generated by alternative splicing. Nucleic Acids Res. 2011. pp. D80–5. [DOI] [PMC free article] [PubMed]
- Kamath KS, Vasavada MS, Srivastava S. Proteomic databases and tools to decipher post-translational modifications. J Proteomics. 2011;75:127–44. doi: 10.1016/j.jprot.2011.09.014. [DOI] [PubMed] [Google Scholar]
- Diamandis EP. The failure of protein cancer biomarkers to reach the clinic: why, and what can be done to address the problem? BMC Med. 2012;10:87. doi: 10.1186/1741-7015-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha S, Imielinski MB, Rejtar T, Richardson EA, Thakur D, Sgroi DC, Karger BL. In situ proteomic analysis of human breast cancer epithelial cells using laser capture microdissection: annotation by protein set enrichment analysis and gene ontology. Mol Cell Proteomics. 2010;9:2529–44. doi: 10.1074/mcp.M110.000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes DF. OMICS-based personalized oncology: if it is worth doing, it is worth doing well! BMC Med. 2013;11:221. doi: 10.1186/1741-7015-11-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renuse S, Chaerkady R, Pandey A. Proteogenomics. Proteomics. 2011;11:620–30. doi: 10.1002/pmic.201000615. [DOI] [PubMed] [Google Scholar]
- Greenbaum D, Colangelo C, Williams K, Gerstein M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003;4:117. doi: 10.1186/gb-2003-4-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigdel TK, Sarwal MM. The proteogenomic path towards biomarker discovery. Pediatr Transplant. 2008;12:737–47. doi: 10.1111/j.1399-3046.2008.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Cha S, Rejtar T, Richardson E a, Karger BL, Sgroi DC. Integrated proteomic, transcriptomic, and biological network analysis of breast carcinoma reveals molecular features of tumorigenesis and clinical relapse. Mol Cell Proteomics. 2012;11:M111.014910. doi: 10.1074/mcp.M111.014910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Old LJ, Chen YT. New paths in human cancer serology. J Exp Med. 1998;187:1163–7. doi: 10.1084/jem.187.8.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, Knuth A, Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science (80-) 1991;254:1643–7. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- Vigneron N, Stroobant V, Van den Eynde BJ, van der Bruggen P. Database of T cell-defined human tumor antigens: the 2013 update. Cancer Immun. 2013;13:15. [PMC free article] [PubMed] [Google Scholar]
- Parmiani G, De Filippo A, Novellino L, Castelli C. Unique human tumor antigens: immunobiology and use in clinical trials. J Immunol. 2007;178:1975–9. doi: 10.4049/jimmunol.178.4.1975. [DOI] [PubMed] [Google Scholar]
- Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–8. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- Skotheim RI, Nees M. Alternative splicing in cancer: noise, functional, or systematic? Int J Biochem Cell Biol. 2007;39:1432–49. doi: 10.1016/j.biocel.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol. 2007;25:117–24. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- Wickner S, Maurizi MR, Gottesman S. Posttranslational quality control: folding, refolding, and degrading proteins. Science. 1999;286:1888–93. doi: 10.1126/science.286.5446.1888. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, Pienta KJ, Rubin MA, Chinnaiyan AM. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–6. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- Buyse M, Loi S, van't Veer L, Viale G, Delorenzi M, Glas AM, D'Assignies MS, Bergh J, Lidereau R, Ellis P, Harris A, Bogaerts J, Therasse P, Floore A, Amakrane M, Piette F, Rutgers E, Sotiriou C, Cardoso F, Piccart MJ. Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. J Natl Cancer Inst. 2006;98:1183–92. doi: 10.1093/jnci/djj329. [DOI] [PubMed] [Google Scholar]
- Spentzos D, Levine DA, Ramoni MF, Joseph M, Gu X, Boyd J, Libermann TA, Cannistra SA. Therapeutic cancer vaccines in combination with conventional therapy. J Clin Oncol. 2004;22:4700–10. doi: 10.1200/JCO.2004.04.070. [DOI] [PubMed] [Google Scholar]
- Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O'Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gry M, Rimini R, Strömberg S, Asplund A, Pontén F, Uhlén M, Nilsson P. Correlations between RNA and protein expression profiles in 23 human cell lines. BMC Genomics. 2009;10:365. doi: 10.1186/1471-2164-10-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas (TCGA) Research Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottoriva A, Spiteri I, Piccirillo SGM, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavaré S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA. 2013;110:4009–14. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Nadano D, Hidaka E, Higuchi K, Kawakubo M, Sato T-A, Nakayama J. Differential expression of ribosomal proteins in human normal and neoplastic colorectum. J Histochem Cytochem. 2003;51:567–74. doi: 10.1177/002215540305100502. [DOI] [PubMed] [Google Scholar]
- Loreni F, Mancino M, Biffo S. Translation factors and ribosomal proteins control tumor onset and progression: how? Oncogene. 2013. [DOI] [PubMed]
- Hix LM, Karavitis J, Khan MW, Shi YH, Khazaie K, Zhang M. Tumor STAT1 transcription factor activity enhances breast tumor growth and immune suppression mediated by myeloid-derived suppressor cells. J Biol Chem. 2013;288:11676–88. doi: 10.1074/jbc.M112.441402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodarev NN, Roizman B, Weichselbaum RR. Molecular pathways: interferon/stat1 pathway: role in the tumor resistance to genotoxic stress and aggressive growth. Clin Cancer Res. 2012;18:3015–21. doi: 10.1158/1078-0432.CCR-11-3225. [DOI] [PubMed] [Google Scholar]
- Cai D, Cao J, Li Z, Zheng X, Yao Y, Li W, Yuan Z. Up-regulation of bone marrow stromal protein 2 (BST2) in breast cancer with bone metastasis. BMC Cancer. 2009;9:102. doi: 10.1186/1471-2407-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi EH, Yoo H, Noh KH, Han S, Lee H, Lee JK, Won C, Kim B-H, Kim M-H, Cho C-H, Ye S. BST-2 is a potential activator of invasion and migration in tamoxifen-resistant breast cancer cells. Biochem Biophys Res Commun. 2013. pp. 685–690. [DOI] [PubMed]
- Daly EB, Wind T, Jiang XM, Sun L, Hogg PJ. Secretion of phosphoglycerate kinase from tumour cells is controlled by oxygen-sensing hydroxylases. Biochim Biophys Acta. 2004;1691:17–22. doi: 10.1016/j.bbamcr.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Zieker D, Königsrainer I, Tritschler I, Löffler M, Beckert S, Traub F, Nieselt K, Bühler S, Weller M, Gaedcke J, Taichman RS, Northoff H, Brücher BLDM, Königsrainer A. Phosphoglycerate kinase 1 a promoting enzyme for peritoneal dissemination in gastric cancer. Int J Cancer. 2010;126:1513–20. doi: 10.1002/ijc.24835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lay AJ, Jiang XM, Kisker O, Flynn E, Underwood A, Condron R, Hogg PJ. Phosphoglycerate kinase acts in tumour angiogenesis as a disulphide reductase. Nature. 2000;408:869–73. doi: 10.1038/35048596. [DOI] [PubMed] [Google Scholar]
- Kharbanda A, Rajabi H, Jin C, Raina D, Kufe D. Oncogenic MUC1-C promotes tamoxifen resistance in human breast cancer. Mol Cancer Res. 2013;11:714–23. doi: 10.1158/1541-7786.MCR-12-0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9:667–76. doi: 10.1023/B:APPT.0000045801.15585.dd. [DOI] [PubMed] [Google Scholar]
- Saini KS, Loi S, de Azambuja E, Metzger-Filho O, Saini ML, Ignatiadis M, Dancey JE, Piccart-Gebhart MJ. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat Rev. 2013;39:935–946. doi: 10.1016/j.ctrv.2013.03.009. [DOI] [PubMed] [Google Scholar]
- Wend P, Holland JD, Ziebold U, Birchmeier W. Wnt signaling in stem and cancer stem cells. Semin Cell Dev Biol. 2010;21:855–863. doi: 10.1016/j.semcdb.2010.09.004. [DOI] [PubMed] [Google Scholar]
- Khodarev N, Ahmad R, Rajabi H, Pitroda S, Kufe T, McClary C, Joshi MD, MacDermed D, Weichselbaum R, Kufe D. Cooperativity of the MUC1 oncoprotein and STAT1 pathway in poor prognosis human breast cancer. Oncogene. 2010;29:920–9. doi: 10.1038/onc.2009.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Modugno F, Mottolese M, Di Benedetto A, Conidi A, Novelli F, Perracchio L, Venturo I, Botti C, Jager E, Santoni A, Natali PG, Nisticò P. The cytoskeleton regulatory protein hMena (ENAH) is overexpressed in human benign breast lesions with high risk of transformation and human epidermal growth factor receptor-2-positive/hormonal receptor-negative tumors. Clin Cancer Res. 2006;12:1470–8. doi: 10.1158/1078-0432.CCR-05-2027. [DOI] [PubMed] [Google Scholar]
- Rayl EA, Moroson BA, Beardsley GP. The human purH gene product, 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase. Cloning, sequencing, expression, purification, kinetic analysis, and domain mapping. J Biol Chem. 1996;271:2225–33. doi: 10.1074/jbc.271.4.2225. [DOI] [PubMed] [Google Scholar]
- Weber G. Enzymes of purine metabolism in cancer. Clin Biochem. 1983;16:57–63. doi: 10.1016/S0009-9120(83)94432-6. [DOI] [PubMed] [Google Scholar]
- Martin M, Spielmann M, Namer M, DuBois A, Unger C, Dodwell D, Vodvarka P, Lind M, Calvert H, Casado A, Zelek L, Lluch A, Carrasco E, Kayitalire L, Zielinski C. Phase II study of pemetrexed in breast cancer patients pretreated with anthracyclines. Ann Oncol. 2003;14:1246–52. doi: 10.1093/annonc/mdg339. [DOI] [PubMed] [Google Scholar]
- Shichijo S, Azuma K, Komatsu N, Ito M, Maeda Y, Ishihara Y, Itoh K. Two proliferation-related proteins, TYMS and PGK1, could be new cytotoxic T lymphocyte-directed tumor-associated antigens of HLA-A2+ colon cancer. Clin Cancer Res. 2004;10:5828–36. doi: 10.1158/1078-0432.CCR-04-0350. [DOI] [PubMed] [Google Scholar]
- Nguyen M, Breckenridge DG, Ducret A, Shore GC. Caspase-resistant BAP31 inhibits fas-mediated apoptotic membrane fragmentation and release of cytochrome c from mitochondria. Mol Cell Biol. 2000;20:6731–40. doi: 10.1128/MCB.20.18.6731-6740.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Bestagno M, Burrone O. Molecular Cloning and Characterization of a Transmembrane Surface Antigen in Human Cells. Eur J Biochem. 1996;238:631–638. doi: 10.1111/j.1432-1033.1996.0631w.x. [DOI] [PubMed] [Google Scholar]
- Lawlor K, Nazarian A, Lacomis L, Tempst P, Villanueva J. Pathway-based biomarker search by high-throughput proteomics profiling of secretomes. J Proteome Res. 2009;8:1489–503. doi: 10.1021/pr8008572. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujana MA, Han J-DJ, Starita LM, Stevens KN, Tewari M, Ahn JS, Rennert G, Moreno V, Kirchhoff T, Gold B, Assmann V, Elshamy WM, Rual J-F, Levine D, Rozek LS, Gelman RS, Gunsalus KC, Greenberg RA, Sobhian B, Bertin N, Venkatesan K, Ayivi-Guedehoussou N, Solé X, Hernández P, Lázaro C, Nathanson KL, Weber BL, Cusick ME, Hill DE, Offit K. et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet. 2007;39:1338–49. doi: 10.1038/ng.2007.2. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scatena R, Bottoni P, Pontoglio A, Giardina B. Revisiting the Warburg effect in cancer cells with proteomics. The emergence of new approaches to diagnosis, prognosis and therapy. Proteomics Clin Appl. 2010;4:143–58. doi: 10.1002/prca.200900157. [DOI] [PubMed] [Google Scholar]
- Pauwels EK, Sturm EJ, Bombardieri E, Cleton FJ, Stokkel MP. Positron-emission tomography with [18F]fluorodeoxyglucose. Part I. Biochemical uptake mechanism and its implication for clinical studies. J Cancer Res Clin Oncol. 2000;126:549–59. doi: 10.1007/PL00008465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam SO, Yotsumoto F, Miyata K, Shirasu N, Miyamoto S, Kuroki M. Possible therapeutic targets among the molecules involved in the Warburg effect in tumor cells. Anticancer Res. 2013;33:2855–60. [PubMed] [Google Scholar]
- Futreal PA, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett LM, Haugen-Strano A, Swensen J, Miki Y. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994;266:120–2. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- Akiyama T, Kawasaki Y. Wnt signalling and the actin cytoskeleton. Oncogene. 2006;25:7538–44. doi: 10.1038/sj.onc.1210063. [DOI] [PubMed] [Google Scholar]
- Howe LR, Brown AMC. Wnt signaling and breast cancer. Cancer Biol Ther. 2004;3:36–41. doi: 10.4161/cbt.3.1.561. [DOI] [PubMed] [Google Scholar]
- Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, Pestell RG, Hung MC. Beta-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci USA. 2000;97:4262–6. doi: 10.1073/pnas.060025397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery E, Folpe AL. The diagnostic value of beta-catenin immunohistochemistry. Adv Anat Pathol. 2005;12:350–6. doi: 10.1097/01.pap.0000194628.58501.71. [DOI] [PubMed] [Google Scholar]
- Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- Sansone P, Bromberg J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol. 2012;30:1005–14. doi: 10.1200/JCO.2010.31.8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–88. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- Critchley-Thorne RJ, Simons DL, Yan N, Miyahira AK, Dirbas FM, Johnson DL, Swetter SM, Carlson RW, Fisher GA, Koong A, Holmes S, Lee PP. Impaired interferon signaling is a common immune defect in human cancer. Proc Natl Acad Sci USA. 2009;106:9010–5. doi: 10.1073/pnas.0901329106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JE, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–13. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Alphen RJ, Wiemer EAC, Burger H, Eskens FALM. The spliceosome as target for anticancer treatment. Br J Cancer. 2009;100:228–32. doi: 10.1038/sj.bjc.6604801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laverman P, Roosenburg S, Gotthardt M, Park J, Oyen WJG, de Jong M, Hellmich MR, Rutjes FPJT, van Delft FL, Boerman OC. Targeting of a CCK(2) receptor splice variant with (111)In-labelled cholecystokinin-8 (CCK8) and (111)In-labelled minigastrin. Eur J Nucl Med Mol Imaging. 2008;35:386–92. doi: 10.1007/s00259-007-0604-1. [DOI] [PubMed] [Google Scholar]
- Tazi J, Bakkour N, Soret J, Zekri L, Hazra B, Laine W, Baldeyrou B, Lansiaux A, Bailly C. Selective inhibition of topoisomerase I and various steps of spliceosome assembly by diospyrin derivatives. Mol Pharmacol. 2005;67:1186–94. doi: 10.1124/mol.104.007633. [DOI] [PubMed] [Google Scholar]
- Ratain MJ, Glassman RH. Biomarkers in phase I oncology trials: signal, noise, or expensive distraction? Clin Cancer Res. 2007;13(22 Pt 1):6545–8. doi: 10.1158/1078-0432.CCR-07-2133. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick's Online Mendelian Inheritance in Man (OMIM) Nucleic Acids Res. 2009. pp. D793–6. [DOI] [PMC free article] [PubMed]
- The UniProt Consortium. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013. pp. D43–7. [DOI] [PMC free article] [PubMed]
- Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T, Krug H, Sirota-Madi A, Olender T, Golan Y, Stelzer G, Harel A, Lancet D. GeneCards Version 3: the human gene integrator. Database (Oxford) 2010;2010:baq020. doi: 10.1093/database/baq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, Wernerus H, Björling L, Ponten F. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol. 2010;28:1248–50. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013. pp. D808–15. [DOI] [PMC free article] [PubMed]
- Von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, Jouffre N, Huynen MA, Bork P. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005. pp. D433–7. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TA, their interactants and involvement in molecular pathways relating to cancer.