

Abstract
Escherichia coli sequence types (STs) 69, 73, 95, and 131 are collectively responsible for a large proportion of E. coli urinary tract and bloodstream infections, and they differ markedly in their antibiotic susceptibilities. Here, we describe a novel PCR method to rapidly detect and distinguish these lineages. Three hundred eighteen published E. coli genomes were compared in order to identify signature sequences unique to each of the four major STs. The specificities of these sequences were assessed in silico by seeking them in an additional 98 genomes. A PCR assay was designed to amplify size-distinguishable fragments unique to the four lineages and was validated using 515 E. coli isolates of known STs. Genome comparisons identified 22 regions ranging in size from 335 bp to 26.5 kb that are unique to one or more of the four predominant E. coli STs, with two to 10 specific regions per ST. These regions predominantly harbor genes encoding hypothetical proteins and are within or adjacent to prophage sequences. Most (13/22) were highly conserved (>96.5% identity) in the genomes of their respective ST. The new assay correctly identified all 142 representatives of the four major STs in the validation set (n = 515), with only two ST12 isolates misidentified as ST95. Compared with MLST, the assay has 100% sensitivity and 99.5% specificity. The rapid identification of major extraintestinal E. coli STs will benefit future epidemiological studies and could be developed to tailor antibiotic therapy to the different susceptibilities of these dominant lineages.
INTRODUCTION
Extraintestinal pathogenic Escherichia coli (ExPEC) strains are frequent pathogens, causing infections spanning a great range of severity (1, 2). They are responsible for 70 to 90% of acute community-acquired uncomplicated urinary infections, 85% of asymptomatic bacteriuria cases, and >60% of recurrent cystitis infections (3). E. coli is also one of the major pathogens of bloodstream infections, with mortality rates of 10 to 30%; in the United Kingdom, it has been the most common cause of bacteremia in most years since 1990, showing year-on year increases and now accounting for almost one-third of all bacteremias (see www.hpa.org.uk) (4). Successful treatment has been complicated by a rise in the prevalence of antibiotic-resistant strains.
DNA profiling, e.g., by multilocus sequence typing (MLST), has advanced our understanding of ExPEC lineages, and several international studies have reported the predominance of sequence types (STs) 69, 73, 95, and 131 among large collections of ExPEC from human infections (5–8). In the United Kingdom, recent regional studies reported the consistent prevalences of these four STs among ExPEC from urinary and bloodstream infections. Collectively, they comprised 45% of the ExPEC strains from community and hospital urine samples recovered in 2007 to 2009 and 2007 to 2008 in Northwest (NW) England and the East Midlands, respectively, as well as 58% of those from bacteremias in northern England in 2010 to 2012 (9–11). The antibiotic resistance profiles of these STs differ markedly: members of STs 69, 73, and 95 remain largely susceptible to antibiotics and rarely have resistance to extended-spectrum cephalosporins in particular, whereas members of ST131 show increasing resistance to multiple antibiotic classes and account for 80 to 90% of multiresistant ExPEC infections (10, 12, 13). In particular, ST131 isolates often carry CTX-M-type extended-spectrum β-lactamases (ESBLs), together with fluoroquinolone and aminoglycoside resistances (13–16).
Rapid diagnostics able to identify and distinguish these STs could therefore be used to tailor therapy before conventional susceptibility test data become available. MLST is time-consuming, labor-intensive, and expensive, but simpler molecular strategies might make testing more accessible to diagnostic laboratories. We report here the design and validation of a PCR assay to distinguish E. coli isolates belonging to these ST lineages.
MATERIALS AND METHODS
Comparative genomics to identify ST-specific signature sequences.
A total of 318 publically available E. coli genomes were retrieved from the GenBank database (ftp.ncbi.nlm.nih.gov/genomes/(DRAFT-)Bacteria), and the multilocus sequence type and clonal complex of each genome were deduced in silico. These genomes were then grouped according to their clonal complexes. Predicted genes and intergenic sequences exceeding 50 bp in size from the genomes of published E. coli strains UMN026 (ST69, GenBank accession no. CU928163), CFT073 (ST73, GenBank accession no. AE014075), UTI89 (ST95, GenBank accession no. CP000243), and NA114 (ST131, GenBank accession no. CP002729) were searched one sequence at a time against the database of all 318 E. coli genomes. Sequences unique to one or more of the four ST lineages were identified, based on BLASTn homologies, with a stringent threshold for inclusion of >90% nucleotide identity over the length of the predicted gene(s); these were then grouped into regions according to their proximity to each other on the chromosome of the four ST reference genomes.
Whole-genome sequencing of E. coli isolates.
The whole genomes of 98 E. coli isolates were sequenced to validate the specificities of the presumptive ST-specific targets. These organisms had been collected from bloodstream infections across the United Kingdom and Ireland in 2001 (n = 36), 2002 (n = 3), 2003 (n = 3), or 2010 (n = 58) under the ambit of the British Society for Antimicrobial Chemotherapy (BSAC) Bacteremia Resistance Surveillance Programme (www.bsacsurv.org). The sequenced isolates belonged to ST69 (n = 14), ST73 (n = 38), ST95 (n = 24), and ST131 (n = 22) and were randomly chosen to represent the 35 to 40% of all isolates that belonged to these four STs from the 2001 and 2010 collections. Susceptibility data for a panel of antibiotics (amoxicillin, cefuroxime, ceftazidime, cefotaxime, imipenem, ciprofloxacin, gentamicin, and piperacillin-tazobactam) had been determined by BSAC agar dilution methodology at the time of collection (Table 1). Isolates belonging to ST131 exhibited serogroup O25 (n = 19) or O16 (n = 3), and those belonging to ST73 exhibited serogroup O6 (n = 21), O22 (n = 3), O25 (n = 2), O2/O50 (n = 4), O18ab/O18ac (n = 5), or were nontypeable (n = 3); those belonging to the two other STs had not been serotyped. The genomes were sequenced to >30× coverage using the Nextera sample preparation method and the standard 2 × 151- or 2 × 251-base sequencing protocols on a MiSeq instrument (Illumina, San Diego, CA, USA). The reads were trimmed using Trimmomatic to remove low-quality nucleotides, specifying a sliding window of 4, an average Phred quality of 30, and 50 as the minimum length to be conserved (17). The trimmed reads were assembled into contigs using VelvetOptimiser (http://bioinformatics.net.au/software.velvetoptimiser.shtml), with k-mer values from 55 to 75, and mapped against all ST-specific sequences using Bowtie 2 (http://bowtie-bio.sourceforge.net/bowtie2) with SAMtools (http://samtools.sourceforge.net) to produce Binary Alignment Map (BAM) files. The presence and percentages of nucleotide identity for the ST-specific sequences in the assembled contigs were determined by BLASTn or by parsing the variant calling format (VCF) file generated by SAMtools mpileup for each BAM file. Known acquired resistance genes and, in particular, those conferring resistance to β-lactams, sulfonamides, tetracyclines, trimethoprim, and aminoglycosides were sought in contigs by BLASTn, with the reference sequences for the resistance genes obtained from the Comprehensive Antibiotic Resistance Database (http://arpcard.mcmaster.ca) and the NCBI nucleotide database (http://www.ncbi.nlm.nih.gov/nuccore) using the accession numbers described in the supplemental data of Zankari et al (18). Multiresistance was defined as nonsusceptibility, by phenotypic and/or genotypic characterization, to one agent in three or more antimicrobial categories, as proposed by the European Centre for Disease Prevention and Control (http://www.ecdc.europa.eu).
TABLE 1.
Susceptibility results and acquired resistance genes identified in 98 sequenced E. coli genomes of the four major STs
| ST | Yr (n) | No. of genomes | Resistance toa: |
Acquired resistance gene(s) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AMX | CIP | CTX | CAZ | CXM | GEN | IPM | TZP | ||||
| 73 | 2001–2003 (20) | 5 | |||||||||
| 2 | sul, aadA | ||||||||||
| 1 | R | ||||||||||
| 4 | R | blaTEM-1 | |||||||||
| 1 | R | blaTEM-1, sul | |||||||||
| 1 | R | blaTEM-1, sul, aph(6)-Id | |||||||||
| 1 | R | blaTEM-1, dfrA, aadA | |||||||||
| 1 | R | I | blaTEM-1, sul, dfrA, tet(A), aph(6)-Id, aadA | ||||||||
| 1 | R | blaTEM-1,sul, tet(A), aph(6)-Id | |||||||||
| 1 | R | blaTEM-1, sul, dfrA, tet(A), aph(6)-Id, aadA | |||||||||
| 1 | R | blaSHV-1, sul, tet(D), aadA | |||||||||
| 1 | R | R | blaSHV-2, tet(D) | ||||||||
| 2010 (18) | 9 | ||||||||||
| 1 | aadA | ||||||||||
| 1 | sul, aadA | ||||||||||
| 1 | dfrA, aadA | ||||||||||
| 1 | R | blaTEM-1 | |||||||||
| 1 | R | I | blaTEM-1 | ||||||||
| 1 | R | blaTEM-1, sul | |||||||||
| 1 | R | blaTEM-1, tet(A) | |||||||||
| 1 | R | blaSHV-1, sul, tet(D), aadA | |||||||||
| 1 | R | R | R | blaOXA-1, tet(A), aadA | |||||||
| 95 | 2001 (10) | 4 | |||||||||
| 1 | dfrA, tet(A) | ||||||||||
| 1 | R | blaTEM-1 | |||||||||
| 2 | R | blaTEM-1, sul, dfrA, aph(6)-Id | |||||||||
| 1 | R | blaTEM-1, sul, aadA | |||||||||
| 1 | R | blaTEM-1, sul, aph(6)-Id | |||||||||
| 2010 (14) | 5 | ||||||||||
| 1 | sul, dfrA, aph(6)-Id, aadA | ||||||||||
| 1 | sul, dfrA, tet(A), aadA | ||||||||||
| 2 | R | blaTEM-1 | |||||||||
| 1 | R | R | blaTEM-1 | ||||||||
| 1 | R | blaTEM-1, sul, dfrA, aph(6)-Id | |||||||||
| 1 | R | I | blaTEM-1 | ||||||||
| 1 | R | blaTEM-30, sul, dfrA, tet(A), aph(6)-Id, aadA | |||||||||
| 1 | R | blaTEM-1,sul, dfrA, tet(A), aadA | |||||||||
| 69 | 2001–2003 (5) | 1 | sul, dfrA, tet(A), aadA | ||||||||
| 1 | R | blaTEM-1, sul, dfrA, tet(A), aph(6)-Id, aadA | |||||||||
| 2 | R | blaTEM-1, sul, dfrA, tet(A), aph(6)-Id | |||||||||
| 1 | R | blaTEM-1, sul, tet(A), aph(6)-Id | |||||||||
| 2010 (9) | 2 | ||||||||||
| 1 | R | blaTEM-1 | |||||||||
| 1 | R | blaTEM-1, sul, dfrA | |||||||||
| 2 | R | blaTEM-1, sul, dfrA, aph(6)-Id, aadA | |||||||||
| 2 | R | I | blaTEM-1, sul, dfrA, tet(A), aph(6)-Id, aadA | ||||||||
| 1 | R | blaTEM-1, sul, dfrA, tet(A), aph(6)-Id, aadA | |||||||||
| 131 | 2001–2003 (5) | 1 | |||||||||
| 1 | R | blaTEM-1 | |||||||||
| 1 | R | blaTEM-1, sul, aph(6)-Id | |||||||||
| 1 | R | blaTEM-1, tet(A) | |||||||||
| 1 | R | R | blaTEM-1, sul, dfrA, aadA | ||||||||
| 2010 (17) | 1 | R | |||||||||
| 1 | R | sul, dfrA, aadA | |||||||||
| 1 | R | R | sul, dfrA, aadA, ant(2″)-Ia | ||||||||
| 2 | R | blaTEM-1, tet(A) | |||||||||
| 1 | R | R | blaTEM-1, aac(3)-IId | ||||||||
| 1 | R | R | blaTEM-1, tet(A) | ||||||||
| 1 | R | R | R | blaTEM-1, sul, dfrA, tet(A), aph(6)-Id, aadA | |||||||
| 1 | R | R | I | blaOXA-1, tet(A), aac(6′)-Ib-cr | |||||||
| 1 | R | R | R | R | blaCTX-M-15, sul, dfrA, aadA | ||||||
| 1 | R | R | R | R | blaCTX-M-15, blaOXA-1, sul, dfrA, aac(6′)-Ib-cr, aadA | ||||||
| 1 | R | R | R | R | I | blaCTX-M-15, blaOXA-1, sul, dfrA, aac(6′)-Ib-cr, aadA | |||||
| 2 | R | R | R | I | R | I | blaCTX-M-15, blaOXA-1, sul, dfrA, tet(A), aac(6′)-Ib-cr, aadA | ||||
| 1 | R | R | R | R | R | R | I | blaCTX-M-15, blaOXA-1, sul, dfrA, tet(A), aac(6′)-Ib-cr, aadA, aac(3)-IIa | |||
| 1 | R | R | R | R | R | I | blaCTX-M-15, blaOXA-1, blaTEM-1, tet(A), aac(6′)-Ib-cr | ||||
| 1 | R | R | R | I | R | I | blaCTX-M-15, blaOXA-1, blaTEM-1, sul, dfrA, tet(A), aac(6′)-Ib-cr, aadA | ||||
AMX, amoxicillin; CIP, ciprofloxacin; CTX, cefotaxime; CAZ, ceftazidime; CXM, cefuroxime; GEN, gentamicin; IMP, imipenem; TZP, piperacillin plus 4 mg/liter tazobactam; R, resistant; I, intermediate. The empty cells indicate susceptibility. Susceptibility was defined according to BSAC/EUCAST breakpoints.
Multiplex PCR for dominant ExPEC lineages.
Primers were designed to match highly conserved regions and to produce PCR products of different sizes from four ST-specific sequences (regions 1, 9, 19, and 21) that were selected based on their specificities and genetic environments (see Results). Amplification mixtures contained each of the eight primers (Table 2) at final concentrations of 0.2 μM, used purified genomic DNA as a template, and were performed with the following cycling conditions: an initial denaturation at 94°C for 3 min, 30 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s, and one final cycle of 72°C for 5 min.
TABLE 2.
Primer sequences used in the PCR assay for the detection of major E. coli STs
| Primer | Sequence | Size (bp) | Positions | Region, ST, E. coli strain (GenBank accession no.) |
|---|---|---|---|---|
| ST73_for | TGGTTTTACCATTTTGTCGGA | 490 | 2001936–2001916 | 1, ST73, CFT073 (AE014075) |
| ST73_rev | GGAAATCGTTGATGTTGGCT | 2001447–2001466 | ||
| ST131_for | GACTGCATTTCGTCGCCATA | 310 | 4344866–4344847 | 19, ST131, NA114 (CP002797) |
| ST131_rev | CCGGCGGCATCATAATGAAA | 4344565–4344584 | ||
| ST95_for | ACTAATCAGGATGGCGAGAC | 200 | 3124468–3124487 | 9, ST95, UTI89 (CP000243) |
| ST95_rev | ATCACGCCCATTAATCCAGT | 3124668–3124649 | ||
| ST69_for | ATCTGGAGGCAACAAGCATA | 104 | 3053203–3053222 | 21, ST69, UMN026 (CU928163) |
| ST69_rev | AGAGAAAGGGCGTTCAGAAT | 3053306–3053287 |
Validation of the PCR assay was conducted using 515 E. coli isolates of diverse STs that were previously identified to the ST level using the MLST scheme available from the University of Warwick E. coli MLST website (http://mlst.warwick.ac.uk/mlst/dbs/Ecoli) (19). They included isolates from human and animal (cattle or poultry) origins from three different countries, as summarized in Table 3. The sensitivities and specificities of the new PCR compared to those of MLST are presented as percentages.
TABLE 3.
Strains used for the validation of the new PCR assay
| MLST | Country | Origin | No. of strains | PCR product size (bp) | CC by PCR |
|---|---|---|---|---|---|
| ST73 | United Kingdom | Human | 48 | 490 | CC73 |
| SLV73a | United Kingdom | Human | 7 | 490 | CC73 |
| SLV73 | Germany | Human | 1 | 490 | CC73 |
| SLV73 | Germany | Animal | 1 | 490 | CC73 |
| ST69 | United Kingdom | Human | 10 | 104 | CC69 |
| SLV69 | United Kingdom | Human | 4 | 104 | CC69 |
| ST69 | Germany | Human | 1 | 104 | CC69 |
| ST69 | Netherlands | Human | 1 | 104 | CC69 |
| ST69 | United Kingdom | Animal | 2 | 104 | CC69 |
| ST69 | Germany | Animal | 1 | 104 | CC69 |
| ST95 | United Kingdom | Human | 20 | 200 | CC95 |
| SLV95 | United Kingdom | Human | 2 | 200 | CC95 |
| ST95 | Netherlands | Human | 1 | 200 | CC95 |
| ST131 | United Kingdom | Human | 24 | 310 | CC131 |
| ST131 | Germany | Human | 1 | 310 | CC131 |
| ST131 | Netherlands | Human | 17 | 310 | CC131 |
| ST131 | Germany | Animal | 1 | 310 | CC131 |
| Nonmajor | United Kingdom | Human | 146 | ||
| Nonmajor | Germany | Human | 7 | ||
| Nonmajor | Netherlands | Human | 47 | ||
| Nonmajor | United Kingdom | Animal | 50 | ||
| Nonmajor | Germany | Animal | 45 | ||
| Nonmajor | Netherlands | Animal | 76 | ||
| ST12b | United Kingdom | Human | 2 | 200 | CC95 |
SLV, single-locus variant.
These two ST12 isolates were misidentified as ST95.
Nucleotide sequence accession number.
The Illumina sequences generated in this study are deposited and available in the European Nucleotide Archive (ENA) under the study accession no. PRJEB7002 (http://www.ebi.ac.uk/ena/data/view/PRJEB7002).
RESULTS
Antibiotic resistance profiles of major ExPEC sequence types.
The susceptibility data obtained by BSAC agar dilution with EUCAST breakpoints for the 98 ExPEC isolates belonging to STs 69 (n = 14), 73 (n = 38), 95 (n = 24), and 131 (n = 22) from the BSAC Bacteremia Surveillance Collections are shown in Table 1. Most ST69, ST73, and ST95 isolates were either susceptible to all tested antibiotics (45% [34/76]) or were resistant to amoxicillin only (43% [33/76]) (Table 1). Only nine ST69, ST73, and ST95 isolates (12%) showed any greater nonsusceptibilities to the tested antibiotics, and this was confined to intermediate- or low-level resistance to ciprofloxacin (MIC, 1 to 4 mg/liter), piperacillin-tazobactam (MIC, 16 to 32 mg/liter), or cefuroxime (MIC, 16 mg/liter); none were resistant to cefotaxime, ceftazidime, gentamicin, or imipenem. Genome sequence analyses identified blaTEM-1, blaTEM-30, blaSHV-1, blaSHV-2, and blaOXA-1 as the sources of amoxicillin resistance and also detected the presence of sul, dfr, tet, aph(6)-Id, and/or aadA alone or in combination in 38/76 of these genomes, therefore predicting resistance also to sulfamethoxazole (41% [31/76]), trimethoprim (29% [22/76]), tetracycline (26% [20/76]), and/or streptomycin (42% [32/76]) (Table 1). Overall, multiresistance was most prevalent in E. coli ST69, with 79% (11/14) of the isolates predicted to be resistant to at least three classes of antibiotics, compared with 37.5% (9/24) and 24% (9/38) in ST95 and ST73, respectively. Nevertheless, this resistance largely encompassed older antibiotics only.
Isolates belonging to ST131 from 2001 to 2003 were mostly (4/5) resistant to amoxicillin only, with only one isolate also resistant to ciprofloxacin; those from 2010 (n = 17) were resistant mostly to ciprofloxacin (76% [13/17]), with 8/17 also resistant to cephalosporins associated with the presence of blaCTX-M-15, which was often (6/8 cases) accompanied by blaOXA-1, aac(6′)-Ib-cr, sul, and dfrA and, in two isolates, also by blaTEM-1 (Table 1). Resistance to gentamicin, associated with the presence of the aac(3)-IIa, aac(3)-IId, or ant(2′)-Ia genes, was correctly predicted in three ST131 isolates, including the only one with blaCTX-M-15 (Table 1).
Identification of ST-specific DNA target sequences.
Analyses of 318 E. coli genomic sequences deposited in the GenBank database identified 130 different STs among the 4,386 STs recognized so far in the E. coli MLST database (http://mlst.warwick.ac.uk/mlst/dbs/Ecoli). Thirty-two of the 318 sequences collectively belonged to STs 69 (n = 5), 73 (n = 9), 95 (n = 9), and 131 (n = 4) or were single-locus variants of ST69 (n = 2) and ST95 (n = 3). For each of these four STs, a comparative analysis revealed from 2 to 10 regions that were conserved in all representatives of the ST and its single-locus variants (SLVs) and which were found in that ST only (Table 4).
TABLE 4.
E. coli ST-lineage-specific regions identified by genome comparison
| Region by ST, strain (GenBank accession no.) | Positions |
Size (bp) | Presence (% [no./total no.]) in ST: |
PNIa | No. of genes | Function(s) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Start | End | 69 | 73 | 95 | 131 | |||||
| ST73, E. coli CFT073 (AE014075) | ||||||||||
| 1b | 2001254 | 2002243 | 989 | 100 (36/36) | 99.7–100 | 1 | Hypothetical | |||
| 2 | 5116943 | 5127677 | 10,734 | 80.5 (29/36) | 99.9–100 | 9 | Hypothetical | |||
| ST95, E. coli UTI89 (CP000243) | ||||||||||
| 3 | 1397376 | 1399596 | 2,220 | 100 (24/24) | 99.8–99.9 | 2 | Hypothetical | |||
| 4 | 2598930 | 2599265 | 335 | 100 (24/24) | 99.7–100 | 1 | Hypothetical | |||
| 5 | 2900299 | 2902003 | 1,704 | 83 (20/24) | 99.7–100 | 4 | Hypothetical | |||
| 6 | 2925552 | 2928212 | 2,660 | 75 (18/24) | 96.5–96.6 | 6 | Hypothetical and phage related | |||
| 7 | 2941113 | 2942983 | 1,870 | 83 (20/24) | 99.8–100 | 3 | Hypothetical | |||
| 8 | 3114339 | 3117642 | 3,303 | 100 (24/24) | 99.6–100 | 3 | Hypothetical | |||
| 9b | 3124006 | 3126499 | 2,493 | 100 (24/24) | 99.9–100 | 2 | Hypothetical | |||
| 10 | 3861904 | 3866248 | 4,344 | 100 (24/24) | 5 (1/22)c | 99.6–100 | 6 | Hypothetical, putative phosphotransferase, phosphoglycerate, dehydrogenase, and dihydrodipicolinate synthase | ||
| ST131, E. coli NA114 (CP002797) | ||||||||||
| 11 | 290463 | 291668 | 1,205 | 100 (22/22) | 97.8–100 | 1 | Hypothetical | |||
| 12 | 298525 | 299844 | 1,319 | 100 (22/22) | 100 | 2 | Phage related | |||
| 13 | 565237 | 566906 | 1,669 | 100 (22/22) | 99.6–100 | 2 | Putative oxidoreductase | |||
| 14 | 569537 | 576146 | 6,609 | 100 (22/22) | 99.5–100 | 6 | Hypothetical, putative transcriptional regulator, and transketolase | |||
| 15 | 1354460 | 1371763 | 17,303 | 100 (22/22) | 98.8–100 | 21 | Hypothetical, phage related, DNA repair, and transcriptional regulation | |||
| 16 | 1971120 | 1994937 | 23,817 | 86 (19/22) | 97.8–100 | 28 | Hypothetical and phage related | |||
| 17 | 2480863 | 2484064 | 3,201 | 82 (18/22) | 99.1–100 | 5 | Hypothetical and phage related | |||
| 18 | 2928397 | 2930371 | 1,974 | 100 (22/22) | 100 | 1 | Hypothetical | |||
| 19b | 4343837 | 4345117 | 1,280 | 100 (22/22) | 99.3–100 | 1 | Putative manganese transport | |||
| 20 | 4859427 | 4870212 | 10,785 | 86 (19/22) | 99.9–100 | 15 | Phage related and hypothetical | |||
| ST69, E. coli UMN026 (CU928163) | ||||||||||
| 21b | 3051269 | 3077864 | 26,595 | 100 (14/14) | 97.4–100 | 25 | Phage related | |||
| 22 | 1679641 | 1680431 | 790 | 100 (14/14) | 5 (1/22)c | 100 | 0 | Hypothetical | ||
PNI, percent nucleotide identity of ST-specific sequences in corresponding ST genomes determined against sequences from the published ST reference genomes.
Regions used in the PCR assay.
Indicates 91% and 94% nucleotide identities of ST95 region 8 and ST69 region 2 in the ST131 genomes, respectively.
A total of 22 lineage-specific regions were identified and ranged in size from 335 bp to 26.5 kb. Most (14/22) carried genes of unknown function positioned within or adjacent to prophage and phage remnant sequences, which suggests that they were most likely acquired by horizontal transfer early in the evolution of these STs and then retained. Seven ST131-specific regions (regions 11, 12, 14, 15, 16, 17, and 20), five ST95-specific regions (regions 3 to 7), and one (region 21) of two ST69-specific regions encoded diverse phage-related functions or were located within bacteriophage sequences, whereas the ST73 region 2 was flanked by prophage P4 integrase and included nine predicted open reading frames, some of which showed weak homologies (37 to 60% amino acid identities) to unknown proteins described in other Enterobacteriaceae (Table 4). The remaining eight regions, which were not associated with phage sequences, were relatively short (335 to 4,344 bp), with sequences carrying one to six genes embedded in the chromosome, adjacent to genes encoding metabolic functions. Of these, ST95 regions 8 and 9 encode hypothetical proteins, whereas the ST95 region 10 comprises six putative genes with homologies to phosphoglycerate dehydrogenase, dihydrodipicolinate synthase, and phosphotransferase system proteins positioned upstream of an RNA polymerase sigma 32 factor component (Table 4). ST131 regions 13, 18, and 19 and ST73 region 1 each harbored either one or two genes and encode putative oxidoreductase, manganese transport, or hypothetical proteins.
Sequence conservation of ST-specific targets.
The sequences of the 22 ST-specific regions were sought and, where present, extracted from the 98 E. coli genomes sequenced in this study to assess their presence and diversity. These new genome sequences supported the view that 20 of the 22 sequences are lineage specific. However, seven of these ST-specific regions were identified in only 75 to 86% of the genomes of the corresponding ST (Table 4). Notably, ST131 regions 16 and 20 were detected in all ST131 isolates belonging to serogroup O25 but were absent from those of serotype O16, and ST73 region 2 was absent from one-third (7/21) of the ST73 isolates belonging to serogroup O6. ST69 region 22 and ST95 region 10 were detected in the isolates of another lineage (ST131), indicating poor specificity; each was detected once in two different genomes belonging to ST131, with 91 and 94% nucleotide identities, respectively. The 13 other ST-specific regions, which comprised 7/10 ST131-specific, 4/8 ST95-specific, 1/2 ST73-specific, and 1/2 ST69-specific regions, were present in all genomes of the corresponding ST and retained sufficiently conserved sequences (range, 96.6 to 100% identity; Table 4) to justify their investigation as potential diagnostic targets.
Multiplex PCR design and validation.
Although none of the ST-specific regions identified provided insight into the success of the four STs in causing urinary and bloodstream infections, they constituted a catalog of targets that could be used for rapid molecular detection of the predominant ExPEC STs.
Region 9 for ST95, region 19 for ST131, region 21 for ST69, and region 1 for ST73 were chosen for further development in this role, based on their specificity and high degree of conservation. Sequences predicted to encode a putative manganese transport or hypothetical proteins within the four sets of selected regions were used to design four pairs of primers to amplify fragments of 104 bp, 200 bp, 310 bp, and 490 bp for the detection of ST69, 73, 95, and 131, respectively (Fig. 1). These were then evaluated using DNA from 515 E. coli isolates of known ST (Table 3). All 142 belonging to one of the four major STs or to the same clonal complex (19 of ST69, 57 of ST73, 23 of ST95, and 43 of ST131) yielded a PCR product of the expected size and were correctly assigned to the corresponding ST groups (Table 3). Most (371/373) of the remaining isolates, which belonged to 227 other STs and lineages, yielded no PCR products; the sole exceptions were that two ST12 isolates were misidentified as ST95. Compared with MLST, the newly designed assay thus achieved a sensitivity of 100% and a specificity of 99.5%.
FIG 1.
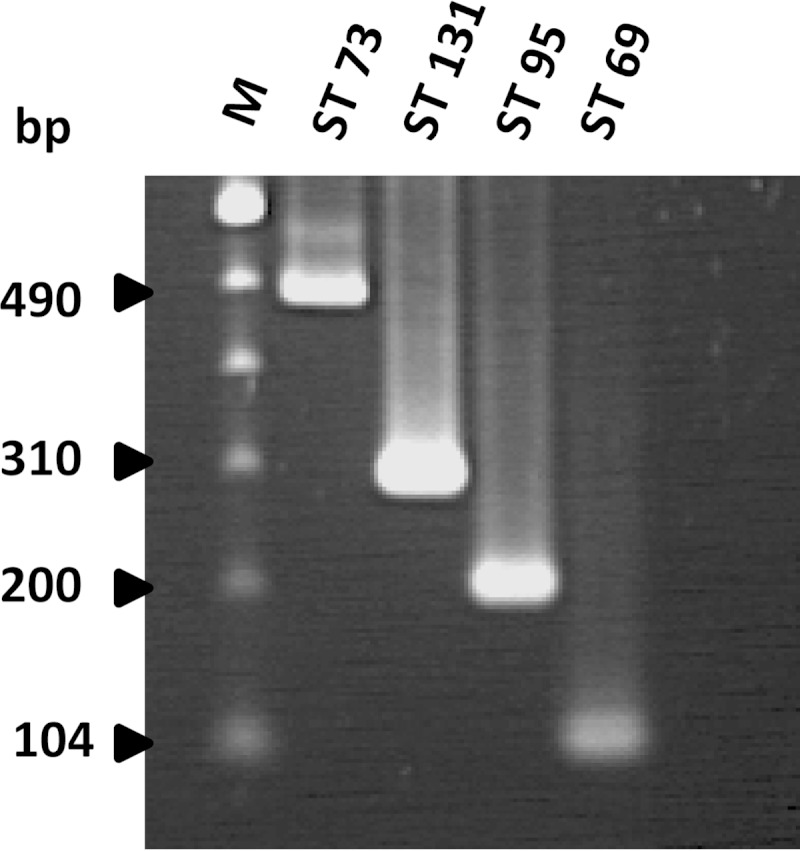
Agarose gel electrophoresis of DNA fragments generated by the new multiplex PCR for the four major E. coli STs. Lane M, 100-bp ladder (Invitrogen, Paisley, United Kingdom).
DISCUSSION
Historically, E. coli has been one of the most antibiotic-susceptible members of the Enterobacteriaceae, but it has now become one of the most resistant. In the United Kingdom, >20% of E. coli isolates from bacteremia are resistant to fluoroquinolones (predominantly through mutations in DNA gyrase) and, despite recent minor declines, about 10% are resistant to third-generation cephalosporins, largely through the production of CTX-M-type ESBLs (20). These proportions compare with 4% fluoroquinolone resistance and 2% cephalosporin resistance at the turn of the century and mirror increases seen across Europe (21). In septic patients, this rising resistance forces clinicians to use carbapenems as empirical therapy, and this, in turn, drives carbapenem resistance.
Previous analyses on large collections of E. coli isolates have shown the predominance of STs 69, 73, 95, and 131 among ExPEC, with these types repeatedly reported to account for 40 to 60% of E. coli urinary tract infections (UTIs) and bloodstream infections in the United Kingdom (9–11, 22). E. coli ST69, ST73, and ST95 have been reported as agents of human UTIs in widely separated geographical areas over many years but are rarely associated with resistance to extended-spectrum cephalosporins. Their continued susceptibilities contrast with those of ST131, which has been associated with a variety of antimicrobial resistances, especially to extended-spectrum cephalosporins, fluoroquinolones, and aminoglycosides (15, 16). Although ST131 was recognized only in 2008, it is clear that it had already been circulating for several years prior, perhaps initially as a susceptible organism, but then it accumulated resistance. Its spread accounts for much of the rise in multidrug-resistant E. coli seen in the last decade (23).
The antibiotic resistance patterns seen here mirror these more general patterns, with multiresistance, particularly to cephalosporins, ciprofloxacin, and gentamicin, mainly associated with ST131 isolates dating from 2010. In contrast, isolates belonging to ST69 mostly (64% [9/14 cases]) had resistance determinants to amoxicillin, trimethoprim, and sulfamethoxazole, as was also described when this lineage was first recognized in a large apparent outbreak of extraintestinal infections in Berkeley, CA. Similarly, the majority of isolates belonging to ST73 (76% [29/38]) and ST95 (62.5% [15/24]) were fully susceptible or resistant to no more than two classes of antibiotics (24).
The factors behind the long-term circulation of ST73, ST95, and ST69 in the United Kingdom, despite continued susceptibility to most modern antibiotics, need to be investigated further. However, the clear association between ST and the likelihood of resistance reasonably suggests that rapid PCR aiming to detect E. coli ST in the United Kingdom might allow treatment to be optimized before the antibiogram of the causal organism is confirmed by conventional methodology (18 to 48 h). Deploying the assay might reduce the potential misuse of powerful antibiotics by identifying infections caused by ST69, ST73, and ST95 E. coli strains likely to be susceptible to more standard antibiotics. In contrast, the detection of the commonly resistant ST131 would allow treatment to be switched, e.g., to a carbapenem in life-threatening infections or to fosfomycin or nitrofurantoin in an uncomplicated UTI. All remaining STs not identified by this assay would be treated according to local empirical therapy guidelines.
The emergence of resistance in these major STs by the acquisition of new resistance determinants cannot be totally excluded, and therefore, the assay will help to monitor their antibiotic susceptibility profiles, allowing treatment to be adjusted accordingly if needed. The ST-specific targets validated here on a gel-based PCR assay could be easily adapted to more convenient and faster formats, such as real-time PCR or rapid isothermal technologies, which will benefit future epidemiological and surveillance studies. Although several international studies have highlighted the worldwide spread of these four major STs with resistance patterns similar to those found in the United Kingdom, the assay will help in rapidly defining their proportion in E. coli infections at national levels and assessing the significance of its applicability in tailoring therapy according to local antibiotic profiles.
ACKNOWLEDGMENTS
This report was commissioned by the National Institute of Health Research. The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health.
The project was funded by National Institute of Health Strategic Research and Development Fund (project no. 107723).
We thank the BSAC for allowing use of isolates from the BSAC Bacteremia Resistance Surveillance Programme and the SAFEFOODERA Consortium for the use of isolates from EU-SAFEFOODERA project 0817.
D.M.L. is partly self-employed and consults for numerous pharmaceutical and diagnostic companies, including Achaogen, Adenium, Alere, Allecra, AstraZeneca, Basilea, Bayer, BioVersys, Cubist, Curetis, Cycle, Discuva, Forest, GSK, Longitude, Meiji, Pfizer, Roche, Shionogi, Tetraphase, VenatoRx, and Wockhardt. He holds, or has recently held, grants from Achaogen, AstraZeneca, Cubist, GSK, Kalidex, Meiji, Merck, and VenatoRx. He has received lecture honoraria from AOP Orphan, AstraZeneca, Bruker, Curetis, Merck, Pfizer, and Leo and holds shares in Dechra, GSK, Merck, PerkinElmer, and Pfizer amounting to <10% of portfolio value.
REFERENCES
- 1.Johnson JR, Russo TA. 2002. Extraintestinal pathogenic Escherichia coli: “the other bad E coli”. J Lab Clin Med 139:155–162. doi: 10.1067/mlc.2002.121550. [DOI] [PubMed] [Google Scholar]
- 2.Russo TA, Johnson JR. 2000. Proposal for a new inclusive designation for extraintestinal pathogenic isolates of Escherichia coli: ExPEC. J Infect Dis 181:1753–1754. doi: 10.1086/315418. [DOI] [PubMed] [Google Scholar]
- 3.Foxman B, Brown P. 2003. Epidemiology of urinary tract infections: transmission and risk factors, incidence, and costs. Infect Dis Clin North Am 17:227–241. doi: 10.1016/S0891-5520(03)00005-9. [DOI] [PubMed] [Google Scholar]
- 4.Martin GS, Mannino DM, Eaton S, Moss M. 2003. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med 348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 5.Adams-Sapper S, Diep BA, Perdreau-Remington F, Riley LW. 2013. Clonal composition and community clustering of drug-susceptible and -resistant Escherichia coli isolates from bloodstream infections. Antimicrob Agents Chemother 57:490–497. doi: 10.1128/AAC.01025-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banerjee R, Johnston B, Lohse C, Chattopadhyay S, Tchesnokova V, Sokurenko EV, Johnson JR. 2013. The clonal distribution and diversity of extraintestinal Escherichia coli isolates vary according to patient characteristics. Antimicrob Agents Chemother 57:5912–5917. doi: 10.1128/AAC.01065-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaureguy F, Landraud L, Passet V, Diancourt L, Frapy E, Guigon G, Carbonnelle E, Lortholary O, Clermont O, Denamur E, Picard B, Nassif X, Brisse S. 2008. Phylogenetic and genomic diversity of human bacteremic Escherichia coli strains. BMC Genomics 9:560. doi: 10.1186/1471-2164-9-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tartof SY, Solberg OD, Manges AR, Riley LW. 2005. Analysis of a uropathogenic Escherichia coli clonal group by multilocus sequence typing. J Clin Microbiol 43:5860–5864. doi: 10.1128/JCM.43.12.5860-5864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Croxall G, Hale J, Weston V, Manning G, Cheetham P, Achtman M, McNally A. 2011. Molecular epidemiology of extraintestinal pathogenic Escherichia coli isolates from a regional cohort of elderly patients highlights the prevalence of ST131 strains with increased antimicrobial resistance in both community and hospital care settings. J Antimicrob Chemother 66:2501–2508. doi: 10.1093/jac/dkr349. [DOI] [PubMed] [Google Scholar]
- 10.Gibreel TM, Dodgson AR, Cheesbrough J, Fox AJ, Bolton FJ, Upton M. 2012. Population structure, virulence potential and antibiotic susceptibility of uropathogenic Escherichia coli from Northwest England. J Antimicrob Chemother 67:346–356. doi: 10.1093/jac/dkr451. [DOI] [PubMed] [Google Scholar]
- 11.Horner C, Fawley W, Morris K, Parnell P, Denton M, Wilcox M. 2014. Escherichia coli bacteraemia: 2 years of prospective regional surveillance (2010–12). J Antimicrob Chemother 69:91–100. doi: 10.1093/jac/dkt333. [DOI] [PubMed] [Google Scholar]
- 12.Manges AR, Johnson JR. 2012. Food-borne origins of Escherichia coli causing extraintestinal infections. Clin Infect Dis 55:712–719. doi: 10.1093/cid/cis502. [DOI] [PubMed] [Google Scholar]
- 13.Rogers BA, Sidjabat HE, Paterson DL. 2011. Escherichia coli O25b-ST131: a pandemic, multiresistant, community-associated strain. J Antimicrob Chemother 66:1–14. doi: 10.1093/jac/dkq415. [DOI] [PubMed] [Google Scholar]
- 14.Lau SH, Kaufmann ME, Livermore DM, Woodford N, Willshaw GA, Cheasty T, Stamper K, Reddy S, Cheesbrough J, Bolton FJ, Fox AJ, Upton M. 2008. UK epidemic Escherichia coli strains A-E, with CTX-M-15 beta-lactamase, all belong to the international O25:H4-ST131 clone. J Antimicrob Chemother 62:1241–1244. doi: 10.1093/jac/dkn380. [DOI] [PubMed] [Google Scholar]
- 15.Peirano G, Pitout JD. 2010. Molecular epidemiology of Escherichia coli producing CTX-M beta-lactamases: the worldwide emergence of clone ST131 O25:H4. Int. J Antimicrob Agents 35:316–321. doi: 10.1016/j.ijantimicag.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Woodford N, Turton JF, Livermore DM. 2011. Multiresistant Gram-negative bacteria: the role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol Rev 35:736–755. doi: 10.1111/j.1574-6976.2011.00268.x. [DOI] [PubMed] [Google Scholar]
- 17.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. 2012. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu G, Day MJ, Mafura MT, Nunez-Garcia J, Fenner JJ, Sharma M, van Essen-Zandbergen A, Rodríguez I, Dierikx C, Kadlec K, Schink AK, Wain J, Helmuth R, Guerra B, Schwarz S, Threlfall J, Woodward MJ, Woodford N, Coldham N, Mevius D. 2013. Comparative analysis of ESBL-positive Escherichia coli isolates from animals and humans from the UK, The Netherlands and Germany. PLoS One 8:e75392. doi: 10.1371/journal.pone.0075392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livermore DM, Hope R, Reynolds R, Blackburn R, Johnson AP, Woodford N. 2013. Declining cephalosporin and fluoroquinolone non-susceptibility among bloodstream Enterobacteriaceae from the UK: links to prescribing change? J Antimicrob Chemother 68:2667–2674. doi: 10.1093/jac/dkt212. [DOI] [PubMed] [Google Scholar]
- 21.Livermore DM, Hope R, Brick G, Lillie M, Reynolds R, BSAC Working Parties on Resistance Surveillance . 2008. Non-susceptibility trends among Enterobacteriaceae from bacteraemias in the UK and Ireland, 2001–06. J Antimicrob Chemother 62(Suppl 2):ii41–ii54. doi: 10.1093/jac/dkn351. [DOI] [PubMed] [Google Scholar]
- 22.Manges AR, Tabor H, Tellis P, Vincent C, Tellier PP. 2008. Endemic and epidemic lineages of Escherichia coli that cause urinary tract infections. Emerg Infect Dis 14:1575–1583. doi: 10.3201/eid1410.080102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson JR, Tchesnokova V, Johnston B, Clabots C, Roberts PL, Billig M, Riddell K, Rogers P, Qin X, Butler-Wu S, Price LB, Aziz M, Nicolas-Chanoine MH, Debroy C, Robicsek A, Hansen G, Urban C, Platell J, Trott DJ, Zhanel G, Weissman SJ, Cookson BT, Fang FC, Limaye AP, Scholes D, Chattopadhyay S, Hooper DC, Sokurenko EV. 2013. Abrupt emergence of a single dominant multidrug-resistant strain of Escherichia coli. J Infect Dis 207:919–928. doi: 10.1093/infdis/jis933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manges AR, Johnson JR, Foxman B, O'Bryan TT, Fullerton KE, Riley LW. 2001. Widespread distribution of urinary tract infections caused by a multidrug-resistant Escherichia coli clonal group. N Engl J Med 345:1007–1013. doi: 10.1056/NEJMoa011265. [DOI] [PubMed] [Google Scholar]