

Abstract
Drosophila epithelial research is at the forefront of the field; however, there are no well-characterized epithelial cell lines that could provide a complementary in vitro model for studies conducted in vivo. Here, a protocol is described that produces epithelial cell lines. The method uses genetic manipulation of oncogenes or tumor suppressors to induce embryonic primary culture cells to rapidly progress to permanent cell lines. It is, however, a general method and the type of cells that comprise a given line is not controlled experimentally. Indeed, only a small fraction of the lines produced are epithelial in character. For this reason, additional work needs to be done to develop a more robust epithelial cell-specific protocol. It is expected that Drosophila epithelial cell lines will have great utility for in vitro analysis of epithelial biology, particularly high-throughput analyses such as RNAi screens.
Keywords: Drosophila, Ras, Tumor suppressor, GAL4/UAS system, Epithelial cell line
1. Introduction
The study of Drosophila epithelia including the imaginal discs, embryonic epidermis, larval midgut, and ovary has proved fertile ground for gene discovery and analysis to understand the mechanisms of cell fate, proliferation, polarity, and movement. What has been lacking in the analysis of Drosophila epithelia is the development of in vitro models. Cell lines have been derived from the imaginal discs but none of these has an overt epithelial character (1, 2). Recently we discovered a rapid way to generate Drosophila cell lines, which offers the promise of developing cell type-specific lines including those from epithelia. Using an approach that has been successful in mammalian tissue culture, we manipulated the expression of oncogenes and tumor suppressors to generate cell lines efficiently. Expression of oncogenic Ras, RasV12, has a profound effect on Drosophila primary cells in culture; they are protected from apoptosis and stimulated to proliferate so that they rapidly give rise to continuous cell lines (3). A small fraction of these lines are epithelial. Blocking the Hippo/Warts tumor suppressor pathway has also produced epithelial cell lines ((3, 4); Simcox, unpublished).
In mammalian systems, there are widely used protocols to generate cell type-specific lines and lines corresponding to mutants. The Drosophila method has already been used to derive cells of a particular mutant strain (5). But its potential to generate cell type-specific cell lines has not been developed. In Drosophila the GAL4/UAS system can target gene expression in a cell type-specific manner (6). In theory, therefore, expressing oncogenic Ras, or inhibiting tumor suppressors, in epithelial cells should promote the generation of epithelial cell lines. It is likely, however, that the success of this will depend on two other developments: a system for conditional expression of the genetic agent and improved culture methods. The first could be achieved in a number of ways, mirroring those that work in mammalian cells, by using temperature-sensitive, drug-inducible, or recombination systems to regulate expression of the transgenes. The second will require the adaptation of existing culture strategies for Drosophila cells, such as surface treatments, strata, or matrices, that promote epithelial cell polarity and development of multicellular structures that more closely resemble their in vivo counterparts. There are certainly incentives for tailoring these methods to Drosophila where cell-based assays for gene discovery, such as RNAi screens, are cost effective and efficient because there are fewer gene redundancies than in vertebrates (7).
2. Materials
2.1. Fly Rearing and Egg Collection
-
Drosophila medium:
Flies are raised in vials (e.g., Drosophila polystyrene vials, Genesee Scientific) on standard cornmeal and molasses food (http://flystocks.bio.indiana.edu/Fly_Work/media-recipes/media-recipes.htm). The food is supplemented with live yeast (e.g., Red Star active dry yeast, Lesaffre Yeast Corporation) to promote the health and fertility of the flies prior to the start of egg collections.
-
Egg laying medium (see Note 1):
Solution A: 83 g dried bakers yeast (e.g., Red Star active dry yeast, Lesaffre Yeast Corporation), 75 mL water, 175 mL vinegar (apple cider, malt, or 4% acetic acid).
Solution B: 17 g agar (e.g., molecular biology grade agar, TEKnova), 485 mL water.
Methylene blue (Sigma-Aldrich®).
Food dye (e.g., McCormick®).
Egg collection dishes: 60 mM Petri dishes.
Egg collection cages: Embryo collection cages—small (FlyStuff. com a division of Genesee Scientific).
Egg rinsing solution, TXN: NaCl (0.7%), Triton X (0.02%) in water.
Paintbrushes: Round, size 6.
Bleach: Diluted with water to 50%.
Sieve: Mesh, Nitex Nylon 120 μM (FlyStuff.com a division of Genesee Scientific).
Tubes for collecting embryos from sieve: 15 mL conical centrifuge tube.
2.2. Cell Culture
Tissue culture medium: 100 mL Schneider’s insect medium (Sigma-Aldrich®) supplemented with 11.25 mL heat-inactivated fetal bovine serum (FBS) (56°C for 30 min) (e.g., Atlanta Biologicals®, see Note 2) and 1.25 mL antibiotic mix (Penicillin–Streptomycin liquid, Life Technologies™).
Trypsin–EDTA solution (1×) (Sigma-Aldrich®).
Freezing medium: 50% Schneider’s medium:30% serum:20% dimethyl sulfoxide (DMSO).
Homogenizer: Glass 5 mL tissue grinder with Teflon pestle.
Tissue culture flasks, 25 cm2.
Cryovials.
2.3. Antibody Staining
Primary antibody: Anti-DE-cadherin (dCad2, Developmental Studies Hybridoma Bank, University of Iowa).
Secondary antibody: Rodamine conjugated anti-rat (Jackson ImmunoResearch).
Tissue-culture coverslips: Thermenox™.
Chamber slide: Lab-Tek™ 4-well chamber slide.
4% Paraformaldehyde: Dilute in PBS from 16% solution (Electron Microscopy Sciences).
PBS + 0.2% Triton™ X-100 (PBTX).
Block solution: 5% Normal Goat Serum (NGS) in PBS.
VectaShield® Mounting Medium (Vector Laboratories).
2.4. Transfection and RNAi
Transfection was performed using Effectene® transfection reagent (Qiagen).
A 500 bp dsRNA against the argos gene was generated using in vitro transcription (Megascript, Ambion®).
3. Methods
3.1. Modulation of Oncogenes and Tumor Suppressors to Establish Cell Lines
3.1.1. Expression of Oncogenic RasV12 with the GAL4–UAS System
The GAL4/UAS system directs expression of UAS-transgenes under the control of the transcription factor GAL4 (6). There is a library of the so-called GAL4 driver lines in which GAL4 is expressed in a large number of different spatial and temporal patterns. In each strain the GAL4 gene is under the control of different genomic enhancers. We have used the Act5C-GAL4 strain in our experiments. Act5C is a broadly expressed cytoplasmic actin gene and therefore directs expression of GAL4 in many cell types. Flies from a genetic strain carrying a UAS-RasV12 transgene are crossed to flies of an Act5C-GAL4 strain and in response, RasV12 is expressed in many cell types. The embryos are organism lethal but when homogenized and grown in vitro, the Act5C-GAL4; UAS-RasV12 cells are viable and indeed outcompete cells of other genotypes and dominate the culture at confluence. For this reason there is no requirement to sort the embryos to select the desired genotype prior to setting up the primary cultures (see Note 3). The method can easily be adapted to generating mutant cell lines by using genetics to make strains in which only the homozygous mutant cells are also Act5C-GAL4; UAS-RasV12 (5). While the method primarily gives rise to lines of cells with a spindle shape, two RasV12-expressing lines with epithelial character have been generated; one of these expresses both RasV12 and wtsdsRNA (3).
3.1.2. Effect of Reduction of Tumor Suppressor Function
We assayed the effects of reducing the function of some tumor suppressors using mutants or RNAi. Blocking two pathways proved effective: PI3K/Pten or Warts (Wts) ((3); Justiniano and Simcox, unpublished). Three Pten− cell lines have been established; none of these has epithelial character. We tested the effect of knocking down wts with RNAi, wtsdsRNA, and mutation and found that both promoted the establishment of cell lines ((3); Simcox, unpublished). Two epithelial cell lines have been generated, one is mutant for wts (Simcox, unpublished) and, as mentioned above, one expresses both RasV12 and wtsdsRNA.
3.2. Primary Cultures
3.2.1. Egg Collection and Processing
Two hundred males and 200 virgin females of the desired genotypes are crossed and kept in well-yeasted vials or bottles (at normal density) for 2–3 days (see Note 4). The flies are transferred to laying cages with sterile egg-collection plates (see Note 5). To make the plates, autoclave solutions A and B for 20 min and mix (see Subheading 2.1, step 2). Add 10 mg methylene blue or edible food-coloring drops to provide contrast for the eggs, and pour into 60 mm Petri dishes. Eggs are collected overnight in the dark at room temperature (22°C). The eggs are dislodged from the surface of the plate using a soft paintbrush and TXN (see Subheading 2.1, step 5). The suspended eggs are poured into a small sieve that retains eggs but allows smaller particles to pass through (see Note 6). The eggs are rinsed in the sieve with a stream of TXN from a squirt bottle to wash off any debris. The sieve is reversed over a 15 mL conical tube and the eggs are transferred into the tube by a stream of TXN. At this point further manipulations are done in a sterile hood. The TXN is removed and the eggs are dechorionated (the eggshell is removed) in 7 mL 50% bleach. The tube is inverted about three times to suspend the eggs in the bleach solution and then the eggs are allowed to settle. After 3 min, the bleach is removed and the eggs are washed with TXN (the success of the dechorionation step can be checked by examining the eggs under a dissecting microscope—they should lack the anterior appendages that are part of the eggshell and appear shiny due to the exposed vitelline membrane). The packed volume of the eggs is estimated at this point, as this will determine how many primary cultures to establish; 100 μL of packed eggs is sufficient for 2–3 primary cultures.
3.2.2. Homogenization and Plating
The eggs are transferred to a new sterile 15 mL conical tube by aspiration from the bottom of the first tube. This reduces the amount of bleach carried over. The eggs are rinsed in TXN three more times and transferred to a 5 mL glass homogenizer. The eggs are rinsed once in sterile water and once in cell culture medium (see Subheading 2.2, step 1). Care should be taken at these steps to avoid losing eggs, as without the detergent present in the TXN, they tend to stick together, float on the surface, and stick to the pipette. About 3 mL of culture medium is added and the pestle is inserted carefully to minimize trapped air at the interface with the medium. The embryos are homogenized with three gentle strokes keeping the pestle submerged even at the top of the upstroke, so as not to introduce air bubbles. The pestle is set aside (but kept sterile) and large clumps and unbroken embryos are allowed to settle. The supernatant is removed into a 15 mL conical tube. Fresh medium (3 mL) is added to the homogenizer, which is flicked to resuspend any clumps that are compressed at the bottom. The homogenization is repeated with slightly more vigorous strokes and a twist at the bottom of the tube to break up the pellet if necessary. The homogenate is pooled with the first fraction and the tube is spun in a benchtop clinical centrifuge for 2 min (room temperature at 1,380×g). The supernatant is removed and the pellet is washed with two more changes of fresh medium. The washing removes yolk and other subcellular particles. The final pellet is resuspended in a small quantity of medium and divided into one or more T-flasks (25 cm2), each with 3 mL of medium. The number of flasks depends on the starting number of embryos (see Subheading 3.2.1) (see Note 6). Flasks are placed in an incubator at 22°C (or 25°C for faster growth). No CO2 is required for growing fly cells.
3.2.3. Care of Primary Cultures
Primary cultures are checked periodically and the medium is changed after 10 days for the first time and thereafter about every 10–14 days. Checking the cultures every few days initially is important as small fraction may be infected with yeast, mold, or bacteria and should be discarded. In our experience infections are not a major factor but additional surface sterilization of the eggs can be performed by rinsing with 70% EtOH after the bleaching step.
3.2.4. Epithelial and Other Cell Types Found in Primary Cultures
The development of Drosophila primary cultures has been described in detail (8-11). Briefly, many differentiated cell types appear including muscle, nerve, fat, epithelial, and spindle-shaped cells. The notable difference between cultures with cells expressing RasV12 and wild-type cultures is that more of these latter two cell types, epithelial and spindle-shaped, are present. Figure 1a, b shows typical RasV12-expressing cultures with patches of these cell types.
Fig. 1.
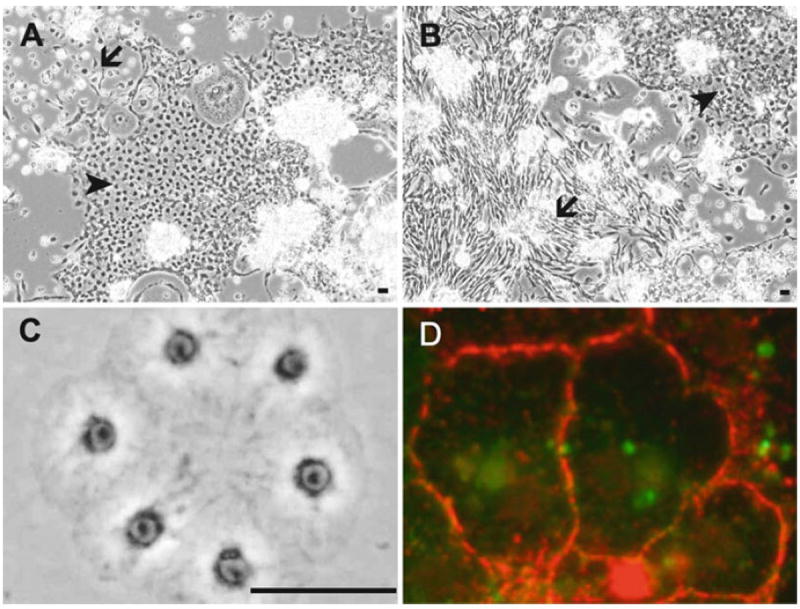
Drosophila epithelial cells in primary cultures and established cell lines. (a and b) Primary cultures showing patches of epithelial (arrow) and spindle-shaped (arrowhead) cells. The cultures are about 70% confluent and ready for the first passage. (c) Cells of the RasV12; wtsdsRNA line are forming a small sheet in a new subculture soon after plating. (d) Cells of the wts line stained for E-cadherin. E-cadherin accumulates at the cell boundary. Bar in a–c, 10 μm.
3.3. Establishment of Lines
3.3.1. Subculturing
Once primary cultures reach about 70% confluence they can be subcultured. The lawn of cells covering the surface is rarely even and a judgment needs to be used about the best time to first passage a given culture. Once cultures reach confluence they typically deteriorate very rapidly; on the other hand, sparse subcultures do not do well. Examples of primary cultures ready for subculturing are shown in Fig. 1a, b. In cultures with cells expressing RasV12, the first subcultures can be made starting at about 3 weeks (3). With wild-type cultures this is much longer and more variable (3).
To subculture, remove the medium (save in a labeled 15 mL conical tube).
Rinse the surface with 3 mL trypsin/EDTA, add a new aliquot of trypsin/EDTA, and incubate for 3 min at room temperature.
Squirt the trypsin/EDTA solution over the cells to help release them from the surface and dilute into the saved medium.
Spin the cells for 2 min in a centrifuge (room temperature at 1,380×g).
Plate half the cells in a new flask (i.e., a dilution of 1 in 2). At passage 10–15, cells can be diluted 1 in 4. On a case-by-case basis, cells can be split at higher dilutions.
3.3.2. Designation of a Line as a Continuous Cell Line
In our experience most cell cultures that have been passaged ten or more times can be propagated indefinitely with few exceptions. Two epithelial lines derived from RasV12; wtsdsRNA-expressing or wts mutant cells have been passaged more than 150 times, representing conservatively about 500 population doublings.
3.3.3. Frequency of Occurrence of Epithelial Like Cell Lines
In the primary cultures there are always patches of epithelial cells, but as the cultures are passaged these tend to be outcompeted by the spindle-shaped cells. Since the development of the method we have generated more than 50 cell lines and only three of these are epithelial. To increase the success rate, we are modifying the protocol to favor epithelial cells by using the GAL4/UAS system to induce gene expression only in epithelial cells and changing culture conditions.
3.4. Epithelial Cell Lines
3.4.1. Histochemical Analysis with E-Cadherin
Some cells in primary cultures (Fig. 1a, b) and continuous cell lines (Fig. 1c) have overt epithelial characteristics and form sheets of cells. These cells are positive for E-cadherin, the classical marker of epithelial cells. In Drosophila the gene encoding E-cadherin is called shotgun and there is a monoclonal antibody available that works well to detect the protein in cultured cells (Fig. 1d).
For antibody staining, cells are grown on tissue-culture coverslips or in tissue-culture chamber slides (see Subheading 2.3, step 4).
Cells are washed once in PBS and fixed for 20 min in 4% paraformaldehyde (see Subheading 2.3, step 5).
Cells are rinsed briefly in PBS and washed three times in PBS for 5 min.
PBTX (see Subheading 2.3, step 6) is used to permeabilize the cells. Cells are washed three times in PBS and blocked NGS (see Subheading 2.3, step 7) for 1 h and incubated with primary antibody (1:5) (see Subheading 2.3, step 1) and 5% NGS, overnight at 4°C.
Cells are washed three times in PBS and rhodamine-conjugated secondary antibodies (1:200) (see Subheading 2.3, step 2) are added and incubated for 30 min at room temperature.
Cells are washed three times in PBS and mounted using mounting medium (see Subheading 2.3, step 8). The image shown in Fig. 1d was captured with a Zeiss 510 META Laser Scanning Confocal microscope.
3.4.2. Transfection
The epithelial cells can be transfected using standard methods. We use Effectene® and the recommended procedure for adherent cells. A transfection efficiency of up to about 70% can be achieved for the wts cells.
3.4.3. RNAi
The epithelial cells derived from wts mutants have been subjected to RNAi. Microarray analysis and western analysis show that wts cells express components of the Egfr pathway (not shown), including the receptor and the three zygotic ligands. We tested for the effect of introducing dsRNAs against two secreted factors, the ligand Spitz and its inhibitor Argos. The dsRNA (15 μg) was delivered using transfection, as above. The expression of Argos was greatly reduced (see Fig. 2), whereas Spitz levels were not appreciably reduced (not shown). Variability in gene response is not unexpected, and the success with argos shows that in principle the cells are susceptible to RNAi.
Fig. 2.
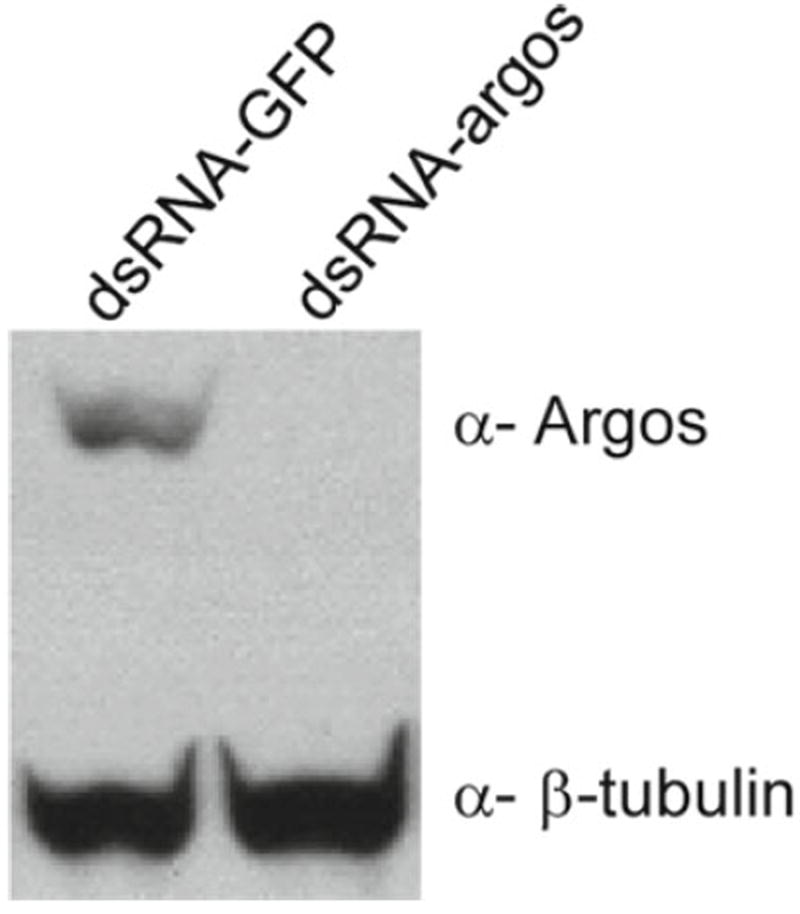
Cells in the wts epithelial cell line are susceptible to RNAi. Treatment with a double-stranded RNA targeting the argos gene (dsRNA-argos), but not a control dsRNA targeting the gfp gene (dsRNA-GFP), reduces expression of Argos (β-tubulin is a loading control).
3.4.4. Freezing Cells for Long-Term Storage
We freeze aliquots of cells from the lines as they evolve, for example, at passages 5, 10, 20, and 30.
Cells are harvested from confluent T-flasks.
The cell pellet is suspended in a small volume of medium (50–100 μL remaining after the medium is removed) and then diluted into 1 mL of freezing medium (see Subheading 2.2, step 3).
This is divided into two aliquots in freezing tubes.
The tubes are placed at −80°C overnight for a “slow freeze” and then transferred to liquid nitrogen for long-term storage.
Multiple aliquots are frozen and one is tested after a few days for cell viability. The tube is defrosted in a beaker of room-temperature water for a “quick defrost.”
To remove the freezing medium, the cells are diluted into 3 mL of cell-culture medium and spun and the pellet is plated in a single T-flask with 3 mL of fresh medium.
Cell viability is assessed by cell growth; typically some cells fail to adhere because they are inviable but many settle and start to proliferate.
Acknowledgments
I thank Litty Paul for the RNAi experiment. The work was supported by NIH R01GM071856 and an ARRA supplement NIH 3R01GM071856-04S1.
Footnotes
We find that these plates, which incorporate some yeast into the medium, are more appealing to the flies and promote better egg laying than typical egg collection plates that are fruit-juice based. The fruit-juice plates have to be supplemented with killed yeast paste, and even this “sterile” paste can rapidly support the growth of microorganisms, and in our experience leads to more infections in the primary cultures.
We buy serum from a variety of sources based on cost. For the past 10 years we have not checked each new batch for whether it has adverse effects on Drosophila cells. There is certainly no harm in doing so, but in our experience we have never yet had to reject a batch.
Embryos of a desired genotype could be selected prior to setting up the cell cultures by using an embryo sorter (COPAS, Union Biometrica). Transgenes encoding fluorescent markers are used to select for, or against, particular genotypes.
Up to 2,000 flies can be housed in these small-size cages if a fluted sheaf of filter paper is inserted around the inside edge. Also larger cages can be used. Plates should be checked for hatched larvae. These are sometimes present even though the collection period is shorter than the length of embryogenesis because females can retain eggs, some eggs are laid on the cage walls, and the hatched larvae crawl to the plate. It is important to remove these larvae, as their digestive track contents can cause yeast and other infections in the primary cultures. To remove larvae, we flood the plate with TXN and pick them out individually with forceps. Adding the TXN causes the larvae to move and makes them easier to spot. We find that it is well worth the investment of time (about 10 min) to include this step.
Small homemade sieves are constructed from the cutoff cap end of a 15 mL plastic centrifuge tube (about 2 cm of the tube). A small piece of mesh is stretched across the open end and held in place with the cap, which has its center cut out.
There is a limit to the number of embryos that can be processed successfully in a 5 mL glass homogenizer. Too many embryos tend to form a dense clump at the bottom of the homogenizer that cannot be disrupted without causing too much cell damage. Larger volume homogenizers can be used. In our experience, the 5 mL homogenizers work well and are convenient for the scale of work required for establishing cell lines. Three primary cultures can be set up in a typical procedure and as the efficiency of generating cells lines, especially with expression of RasV12, is good, a sufficient number of primary cultures can be set up in four runs of the procedure to guarantee generating 5–10 cell lines.
References
- 1.Ui K, Ueda R, Miyake T. Cell lines from imaginal discs of Drosophila melanogaster. In Vitro Cell Dev Biol. 1987;23:707–711. doi: 10.1007/BF02620984. [DOI] [PubMed] [Google Scholar]
- 2.Peel DJ, Milner MJ. The diversity of cell morphology in cloned cell lines derived from Drosophila imaginal discs. Roux’s Arch Dev Biol. 1990;198:479–482. doi: 10.1007/BF00399059. [DOI] [PubMed] [Google Scholar]
- 3.Simcox A, Mitra S, Truesdell S, Paul L, Chen T, Butchar JP, Justiniano S. Efficient genetic method for establishing Drosophila cell lines unlocks the potential to create lines of specific genotypes. PLoS Genet. 2008;4:e1000142. doi: 10.1371/journal.pgen.1000142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao B, Lei Q, Guan K-L. The Hippo–YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010;24:862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simcox AA, Austin CL, Jacobsen TL, Jafar-Nejad H. Drosophila embryonic ‘fibroblasts’: extending mutant analysis in vitro. Fly (Austin) 2008;2:306–309. doi: 10.4161/fly.7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brand A, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 7.Mohr S, Bakal C, Perrimon N. Genomic screening with RNAi: results and challenges. Annu Rev Biochem. 2010;79:37–64. doi: 10.1146/annurev-biochem-060408-092949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shields G, Dubendorfer A, Sang JH. Differentiation in vitro of larval cell types from early embryonic cells of Drosophila melanogaster. J Embryol Exp Morphol. 1975;33:159–175. [PubMed] [Google Scholar]
- 9.Shields G, Sang JH. Characteristics of five cell types appearing during in vitro culture of embryonic material from Drosophila melanogaster. J Embryol Exp Morphol. 1970;23:53–69. [PubMed] [Google Scholar]
- 10.Cross DP, Sang JH. Cell culture of individual Drosophila embryos. I. Development of wild-type cultures. J Embryol Exp Morphol. 1978;45:161–172. [PubMed] [Google Scholar]
- 11.Eschalier G. Drosophila cells in culture. Academic; New York, NY: 1997. [Google Scholar]