

Abstract
Patients with resected stage II–III cutaneous melanomas remain at high risk for metastasis and death. Biomarker development has been limited by the challenge of isolating high-quality RNA for transcriptome-wide profiling from formalin-fixed and paraffin-embedded (FFPE) primary tumor specimens. Using NanoString technology, RNA from 40 stage II–III FFPE primary melanomas was analyzed and a 53-immune-gene panel predictive of non-progression (area under the curve (AUC)=0.920) was defined. The signature predicted disease-specific survival (DSS P<0.001) and recurrence-free survival (RFS P<0.001). CD2, the most differentially expressed gene in the training set, also predicted non-progression (P<0.001). Using publicly available microarray data from 46 primary human melanomas (GSE15605), a coexpression module enriched for the 53-gene panel was then identified using unbiased methods. A Bayesian network of signaling pathways based on this data identified driver genes. Finally, the proposed 53-gene panel was confirmed in an independent test population of 48 patients (AUC=0.787). The gene signature was an independent predictor of non-progression (P<0.001), RFS (P<0.001), and DSS (P=0.024) in the test population. The identified driver genes are potential therapeutic targets, and the 53-gene panel should be tested for clinical application using a larger data set annotated on the basis of prospectively gathered data.
INTRODUCTION
Metastatic melanoma is a devastating illness, taking the lives of over 48,000 people worldwide per year (Manolio et al., 2013). Patients who have had a stage II or stage III melanoma excised remain at high risk for progression and death because micro-metastases may have spread to other body sites before resection. Stage II and III patients face an approximate 50% risk of death, as compared with <10% risk for stage I disease and >90% risk for stage IV disease (Balch et al., 2009).
Critical prognostic features describing a primary melanoma lesion include depth, ulceration, and mitotic rate, all included in the American Joint Committee on Cancer staging system. Stage II disease is defined as melanomas of at least 1.01mm in depth with ulceration or at least 2.01mm in depth without ulceration. The 2009 American Joint Committee on Cancer guidelines also identified mitotic rate as an independent predictor of survival that is more significant than depth in melanomas under 1mm. Patients are classified as stage III, regardless of tumor depth, if they have any local lymph node metastasis, cutaneous metastasis, and/or “satellite lesions” defined as microscopic foci of tumor separated from the dominant mass indicating subclinical cutaneous metastasis (Balch et al., 2001, 2009).
For patients diagnosed with stage II melanoma, the best test available to further estimate risk is a sentinel lymph node biopsy. Patients with subclinical nodal metastasis may be upstaged to stage III if the node is positive (Morton et al., 2006; Wong et al., 2012). Stage III disease, however, is highly heterogeneous. Five-year survival ranges from 87% for stage III patients with one nodal micro-metastasis and a primary lesion less than 2mm down to 36% for stage III patients with four or more involved nodes (Balch et al., 2010). Patients with stage IIC disease (negative sentinel node but primary lesion 4mm or greater or 2mm with ulceration) have a 5-year survival of only 48% (Balch et al., 2009). Thus, a deep primary melanoma confers a worse prognosis than a microscopic focus of melanoma in the sentinel node, likely due to hematogenous spread (Balch et al., 2009). There is a clear need for prognostic tools for patients with resected stage II–III melanoma, both for surveillance and for stratification for clinical trials of newer adjuvant therapies such as anti-CTLA4 and anti-PD1.
Evidence shows that the phenomenon of immune surveillance has a key role in human solid tumors (Bindea et al., 2011; Fridman et al., 2011; Ott et al., 2013). Thus, the immunoscore, a grading system whereby tumor-infiltrating lymphocytes (TILs) are enumerated and classified through staining for CD3 and CD8, is a proposed biomarker for cancer progression (Ascierto et al., 2013). In primary melanoma, it has historically been known that the presence of TILs confers a more favorable prognosis (Clemente et al., 1996; Azimi et al., 2012). Two factors limit widespread clinical application of TIL quantification. First, TIL quantification is subject to observer variability (Busam et al., 2001). Second, the majority of patients have “non-brisk” TILs, an intermediate category that offers little prognostic information (Azimi et al., 2012). A barrier to the development of molecular markers beyond TILs has been the clinical standards requiring formalin-fixed and paraffin-embedded (FFPE) specimens for diagnosis of primary melanoma, which complicates isolation of RNA for transcriptome-wide profiling (Bogunovic et al., 2009). Whole-genome arrays performed in frozen tissues and, more recently, in FFPE tissues, have characterized melanomas across multiple stages as high or low risk, and differentially expressed genes include immune genes (Winnepenninckx et al., 2006; Harbst et al., 2012; Mann et al., 2013).
However, no prognostic immune biomarkers are currently available and despite the critical role of the immune system in melanoma progression, we do not have a practical way to translate this concept into clinical application. To address this need, we used NanoString technology to profile a targeted set of 446 immune-related genes in primary melanoma tumors (Fortina and Surrey, 2008). We report that expression levels of a 53-gene panel (subset of the 446 genes screened) predict non-progression and prolonged recurrence-free survival (RFS) and disease-specific survival (DSS) in two independent patient populations with resectable stage II–III melanoma. Our results suggest that larger-scale prospective studies should be conducted to define genomic immune biomarkers in primary melanoma tumors.
RESULTS
Characterization of 446-gene panel enriched for immune function in a training set of 40 stage II–III primary FFPE melanomas
NanoString is the most reliable technology capable of robustly quantitating RNA levels for hundreds of genes in FFPE tissues, and we used it to assess transcription levels across a broad range of immune- and/or melanoma-related genes. We identified 446 genes of interest for profiling in the melanoma samples on the basis of a PubMed search of the literature (schema Figure 1a; Supplementary Tables S1 and S2 online). Clinical characteristics of the training set are shown in Figure 1b. Pathology databases at Geisinger Medical Center (GMC, Danville, PA) and the Icahn School of Medicine at Mount Sinai (MSSM, New York, NY) were screened for primary stage II and III melanoma tumors. Patients were scored as “progressors” if they presented with unresectable and/or systemic (stage IV) disease at any time during follow-up. Patients were scored as “non-progressors” if they remained free of melanoma during the entire follow-up period of at least 24 months (median time to censor 61 months). Progression was selected as an end point rather than survival to avoid bias introduced by subsequent treatments known to prolong survival in the metastatic setting, whereas recurrence per se was not selected as an end point because patients who have an isolated resectable loco-regional recurrence remain at relatively low risk of further progression (approximately 50%), making the recurrence less clinically significant in these patients (Francken et al., 2008).
Figure 1. In all, 446 immune-related genes were profiled using NanoString technology, and a subset of 53 genes predicts clinical non-progression in two independent sets of patients.

In (a), a diagram describes the schema for selection of the 446 genes. (b) The clinical characteristics of patients in the training set. In (c), relative levels of mRNA expression for each sample are depicted according to the color scale shown, with each column representing a different patient sample and each row representing one of the 446 genes. Unsupervised hierarchical clustering was performed on both genes and samples. Patients who progressed are labeled in blue and those who did not are labeled in yellow. Blue indicates higher expression and yellow indicates lower expression of each gene in the color scale.
On the basis of these criteria, an initial training set of 47 patients was identified. RNA of sufficient quality for NanoString analysis was obtained in 40 of these cases (85%, see Materials and Methods). Heat map depicts mRNA copy number using unsupervised clustering in the 40 patients (Figure 1c). As shown, the distribution of samples is nonrandom with higher expression of immune genes in patients who did not progress.
Identification of a 53-gene immune panel predictive of non-progression, RFS, and DSS in the training set
Each one of the 446 candidate genes was assessed for its ability to distinguish between progressors and non-progressors using two standard classification methods: random forest and elastic net. A subset of 53 genes was selected as the final gene panel (Figure 2a). Receiver operating characteristic curves were generated using fivefold cross-validation on the training set with a mean area under the curve of 0.920 (Figure 2b). Ten thousand training data sets were then generated by randomly removing eight bootstrapped samples of the data. The removed samples served as the testing data sets. Receiver operating characteristic curves were generated and the distribution of AUCs is shown in Figure 2c. Interestingly, all 53 genes were upregulated in the non-progressors, a bias that significantly deviates from what we would expect by chance (P<0.001).
Figure 2. RNA was extracted from 40 FFPE stage II–III primary melanoma specimens, and 53 genes predictive of melanoma progression were identified using elastic net and random forest classifiers.

See Supplementary Figure S1 online for gene selection method. In (a), a bar graph depicts the number of times each of the 53 genes was selected using a leave-8-out cross-validation with bootstrapping. In (b), a mean receiver operating characteristic curve with fivefold cross-validation to predict melanoma progression is shown, mean area under the curve (AUC)=0.92, P<0.001. In (c), distribution of AUC values using a leave-8-out cross-validation with bootstrapping test is shown.
Next, the gene signature was evaluated in the context of known clinically relevant predictors. Stage (P=0.027), depth (P=0.033), and age (P=0.014) significantly correlated with progression by logistic regression, whereas ulceration and mitotic rate trended toward significance (P=0.053 and P=0.062, respectively). TILs, gender, and location of the primary tumor did not significantly correlate with progression. Multivariate logistic regression showed that the 53-gene signature score was the best predictor of progression (P<0.001), with the gene signature contributing significantly to the accuracy in the context of known predictors (P<0.001).
The 53-gene signature was then tested as a predictor of both RFS and DSS using Cox analysis and correlated with both end points (P<0.001) in a univariate model. The 53 genes were lastly examined in the context of known clinical predictors. TILs, mitotic rate, depth, age, and location on an extremity correlated with DSS. Ulceration and stage III disease trended toward significant correlation with diminished DSS (P=0.060 and P=0.072, respectively). In multivariable analysis, the gene signature added significantly to the predictive power of clinicopathologic features for both RFS and DSS (P<0.001).
Identification of a closely related immune module using unbiased coexpression analysis of the GEO database
To further assess the applicability of our findings to melanoma patients, and to define key node genes driving the immune signature, a coexpression network, consisting of 16,745 genes, (Figure 3a) was reconstructed using data from 46 primary melanoma patients (GEO accession ID: GSE15605)26. A 758-gene module (highlighted in yellow in Figure 3a) was found to be the most enriched for both the 53-gene panel and the 446-gene panel. In all, 42 genes were found within the 758-gene module, yielding an enrichment fold of 17.50 with a P-value <2.2e-16. An enrichment fold was similarly computed for the same module against the 446-gene panel where 161 genes were found in the 758-gene module, yielding an enrichment fold of 7.98 with P-value <2.2e-16. The enrichment fold increased over twofold in the more refined set of genes, which indicates a higher correlation among the selected 53 genes compared with the original 446 genes. These data show that the 53-gene panel is closely related to a coexpression module discovered by unbiased network analysis of the GEO database as 42 genes from the panel were found within the 758-gene module. In fact, these 42 genes correlated closely with progression in the training set showing that they contain a significant fraction of the predictive power of the original 53-gene panel (Supplementary Figure S2 online).
Figure 3. Immune response Bayesian network surrounding the 53-gene panel and the 446-gene panel networks.
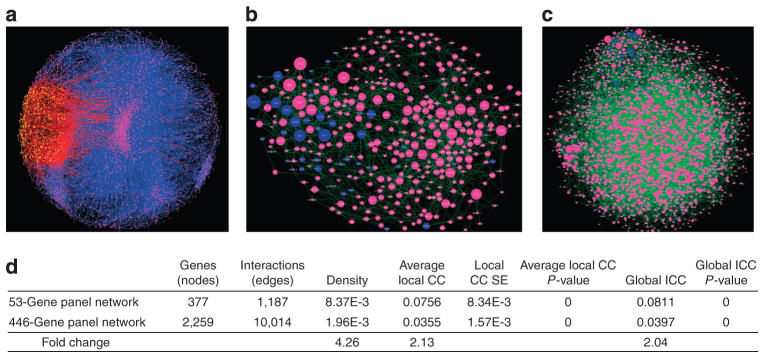
Larger node size indicates larger edge degrees. The 53-gene panel (dark blue) in (a) forms a denser network of gene–protein or protein–protein interactions (green) with neighbor genes (pink) than the network surrounding the 446-gene panel as shown in b. (c) Coexpression network on 46 gene expression profiles in primary melanoma patients. The yellow dots compose a 758-gene module within the entire gene genome (pink). Red lines denote interactions between nodes, involving nodes within the module. (d) Network attributes of the 53-gene panel and 446-gene panel networks. CC, clustering coefficient.
Bayesian network shows high connectivity among the 53 genes in the immune panel
To further illustrate the causal regulatory mechanism of immune response, interactions within and around the 53-gene panel were investigated. A neighborhood of genes related to the 53-gene panel was first selected using the knowledge base network tool VisAnt (see Supplementary Methods page 9 online). A Bayesian network was constructed for the VisAnt gene list (which includes the original 53-gene panel) using the melanoma gene expression data set (GSE15605) shown in Figure 3b. A reference Bayesian network was similarly constructed for the 446-gene panel and its neighborhood set (Figure 3c).
Descriptive statistics across each Bayesian network (number of interactions, clustering coefficients, density, and so on) are listed below (Figure 3d). The 53-gene panel Bayesian network is more densely connected with a 4.815-fold change relative to the 446-gene panel Bayesian network. Similarly, the clustering coefficients (both global and local) are over twofold change relative to the 446-gene panel Bayesian network. Therefore, a significant improvement in the connectivity was observed in the 53-gene panel of predictive genes, possibly indicating a more significant biological mechanism.
Bayesian network identifies driver genes with immunesurveillance function
To identify potentially significant driver genes, genes were ranked by their out-degree. The top 25 hub genes in the 53-gene Bayesian network are listed with functional annotation in Supplementary Table S3 online. Hub genes are expressed by infiltrating immune cells implicated in immune surveillance, including CCR5 (Th1 response), CD8a, CD8b, CD3, and IKZF1. Interestingly, the 42-gene subset of our 53 marker genes shows a significantly higher number of regulatory interactions in the immune nodule from the GEO network relative to the other genes (marker genes are indicated by blue nodes in Figure 3b).
Interaction network and coexpression network pathway enrichment analyses
Next, we tested which functional pathways were enriched in our 53-gene panel. The gene list generated by VisAnt, which was later used to construct the Bayesian network, was annotated with Pathway and GO molecular function (full table of results for the 446-gene panel network and 53-gene panel network are in Supplementary Data set S1 online). The top 10 most significant enriched pathways or GO terms are shown in Supplementary Table S4 online for the 446-gene panel network genes and the 53-gene panel network genes, respectively. Interestingly, we see that the smaller network surrounding the 53 genes shows a higher enrichment of biological processes that characterize lymphocyte function and immune surveillance. Moreover, the enrichment fold change (Supplementary Table S4 online) in the top enriched terms for the 53-gene panel network ranges from 5- to 11-fold, whereas the enrichment fold change of the top 10 terms corresponding to the 446-gene panel network (Supplementary Table S4 online) ranges from just 2- to 4-fold. Therefore, a higher functional enrichment was observed in the network induced by the 53-gene panel.
Finally, we sought to determine whether the module identified in GEO correlated functionally with our proposed 53-gene signature. The functional pathways enriched by the yellow module derived from the GEO model (Figure 3c) are listed in Supplementary Data set S1 online. The top 10 enrichment terms for the GEO module are listed in Supplementary Table S4 online and enriched for immune response. These findings show that a module enriched for immune processes known to be implicated in immune surveillance is identified both in two independent melanoma patient populations of matched stage and also in primary melanoma data from GEO.
Confirmation of the 53-gene immune panel in a second test set of 48 stage II–III primary FFPE melanomas for progression, RFS, and DSS
Given the number of genes in the above analysis and the moderate number of samples comprising the test set, there is a danger of over-fitting the classifiers (an overdetermined system) even with statistical procedures such as cross-validation. Therefore, we assembled the independent test set to replicate our classification results from the test set. In all, 57 patients were identified using identical criteria to the training set, with an additional participating institution (NYU), and RNA was successfully extracted from 48 melanomas (84%).
The clinical characteristics of the test population are shown in table, Figure 4a and were generally similar to the training set with the exception of mitotic rate (Supplementary Table S5 online). Logistic regression showed that ulceration (P=0.013) and depth (P=0.044) associated significantly with progression in the test set. Death rates were 43% and 36% in the training and test populations, respectively, generally consistent with the expected death rates based on American Joint Committee on Cancer staging over the follow-up time (median 61 months in the training set and 53 months in the test set).
Figure 4. The 53-gene panel was tested using a second independent set of patients.

The clinical characteristics of patients in the test set are shown in (a). In (b), relative levels of mRNA expression for each sample are depicted according to the color scale shown, receiver operating characteristic curve to predict melanoma progression is shown in (c), area under the curve (AUC)=0.787, P<0.001. In (d), distribution of AUC values using a leave-4-out cross-validation test is shown. In (e), Kaplan–Meier curves of survival based on a 21-gene signature and ulceration using a log-rank Mantel–Cox test are shown for test set. Patients with negative gene signature and an ulcerated tumor had significantly diminished survival (P=0.030). TILs, tumor-infiltrating lymphocytes.
We next examined the 53-gene panel in the test set as shown in Figure 4b. A similar pattern of high immune gene expression was observed in non-progressors as in the training set (Figure 1c). Further, the 53-gene signature was able to predict progression in the test set with an area under the curve of 0.787 (P<0.001, Figure 4c). Cross-validation demonstrated that this signature is statistically robust (Figure 4d). When the 53-gene panel was evaluated in the test set in the context of clinicopathologic predictors, multivariate logistic regression demonstrated that the gene signature remained predictive of progression (P<0.001). Ulceration was the only feature that added significantly to the predictive power of the signature (P=0.035).
The 53-gene signature was then examined in terms of RFS and DFS using Cox proportional hazards analysis in the test set and correlated significantly with both (P<0.001 and P=0.024, respectively). No other clinical feature correlated significantly with DSS in the test set of 48 patients with median time to censor of 47 months, although ulceration tended toward significance (P=0.087). Multivariable analysis showed that the best model to predict DSS within the test set included gene signature and ulceration (P=0.019). Ulceration and an unfavorable immune signature identified a population at high risk of death with a median survival of 49 months as compared with 139 months in patients with one or none of these risk factors (Figure 4e, P=0.030). Thus, the immune gene signature enhances the ability of established clinicopathologic features to predict progression and survival in a second independent test population.
Validation of expression data at the protein level and identification of CD2 as an immunohistochemical marker of favorable prognosis
In order to validate mRNA data obtained by NanoString, staining by immunohistochemistry (IHC) was performed. Results were concordant with NanoString results as determined by linear regression for CD2, the most differentially expressed gene between progressors and non-progressors (r=0.799; Figure 5a and c). CD5 and CD4 staining by IHC also correlated with the NanoString data (r=0.666 and r=0.543; Figure 5b and d, respectively). Thus, immunohistochemistry correlated with the mRNA results from NanoString.
Figure 5. Immunohistochemistry (IHC) using anti-CD2 mAb was performed to assess risk of disease progression.

(a) Photographs of a tumor expressing low levels of CD2 from a progressor (left), and a tumor with high CD2 levels from a patient who remained disease free are shown (right). A brisk peritumoral infiltrate is seen on hematoxylin and eosin (H&E) in the tumor that did not progress. Clockwise from top left, scale bar indicates 400 μm, 200 μm, 160 μM (inset: 80 μm), and 200 μm (inset: 80μm). A linear regression model is used to assess correlation in Nanostring with IHC for CD5 (b), CD2 (c), and CD4 (d) in the training set. (e) The average number of CD2-positive cells counted at 40× magnification in eight random HPFs in the training and test sets is shown (P<0.0001).
CD2 was the most differentially expressed gene between progressors and non-progressors within the training set (P= 0.002). Frequency of CD2-positive cells by IHC correlated strongly with melanoma non-progression in the combined populations (P<0.001; Figure 5e). Thus, the NanoString analysis allowed for the identification of a novel immunohistochemical stain that may be predictive of outcomes in patients with completely resected stage II–III melanoma.
DISCUSSION
In this work, we tested a candidate panel of 446 genes and defined a proposed biomarker consisting of 53 immune genes associated with non-progression, RFS, and DSS in a training set of 40 patients with completely resected stage II–III melanoma. Next, we identified, using publicly available data and unbiased methods, a module of 758 genes coregulated in melanoma tissues including 42 genes out of the proposed 53-gene panel. The fact that these 758 genes were coregulated, meaning that their expression levels are coordinated across multiple primary melanoma tumors, suggests related biologic function. Bayesian analysis of this coexpression module identified driver genes with key roles in lymphocyte aggregation and activation, including CCR5, CD8, CD3, and IZKF1, showing that this module is related to T-cell activity, specifically the Th1 signaling pathways. Finally, the predictive value of the proposed 53-gene signature was confirmed in a second independent test set of 48 patients, and findings were corroborated by IHC at the protein level with identification of CD2 as a marker of non-progression. These findings should lay the groundwork for the definition of immune biomarkers of clinical utility in primary melanoma tumors on the basis of larger prospective studies.
Notably, our work is consistent with the hypothesis that the immune system is protective against cancer progression and with the concept of the immunoscore proposed by Galon et al. (Galon et al., 2013) whereby careful quantification of immune infiltrates carries prognostic value in multiple tumor types. Although traditional tumor staging focuses only on the characteristics of the tumor, the immunoscore considers the patient’s immune response and thus can provide a more accurate prognosis. Our work is in primary tumors, and our findings certainly do not exclude the possibility that tumors evolve to co-opt the immune system and therefore immune activity may be nefarious in more advanced melanomas. Also, melanoma is not likely caused by a virus or other inflammatory insult, and our findings would suggest that inflammation does not abet the progression of most early-stage melanomas. It is nonetheless striking that the immune genes are generally upregulated in patients who did not progress. There are other contributing features intrinsic to the tumor that affect outcomes, as the clinical course is not likely entirely dictated by the immune system. Thus, the ability of our 53-gene panel to predict survival was improved in the test set by the inclusion of ulceration, generally considered a marker of invasiveness.
Given the current excitement about immunotherapy in the metastatic setting, sorting out which patients have a favorable immune profile is likely to be useful for patient stratification when agents such as anti-PD1 and anti-CTLA4 are tested in the adjuvant setting. A recent report proposed a gene panel predictive of response to a tumor vaccine (Kruit et al., 2013), and it will be interesting to learn which pathways are important to predict response to immunotherapy and how these might relate to the prognostic immune surveillance signature reported here.
Intriguingly, the functions of the genes at the top of the 53-gene panel (Supplementary Table S3 online) focus on T-cell and natural killer functions, as well as leukocyte migration. Meanwhile, driver genes identified on the basis of the GEO module have similar functions. This observation is formalized through the enrichment analysis performed using DAVID (Supplementary Data set S1 online). CD2, a costimulatory molecule and a marker of activation, is expressed on T cells and NK cells. The significance of CD2 is highlighted by the fact that two CD2 ligands, CD53 and CD48, are also found in the 53-gene panel. Other highly differentially expressed genes between progressors and non-progressors include KLRK1 and HLAE, ligands for each other and implicated in NK cell–mediated immunosurveillance, as well as CD4, CD3, LCK, and ITK, genes associated with TCR signaling. CCR5, top hub gene, characterizes the Th1 response and is implicated in leukocyte aggregation to sites of inflammation (Loetscher et al., 1998). Key ligand for CCR5, CCL5, is also included in the 53-gene panel. Other top driver genes, CD8a and CD3, are markers for cytotoxic T-cell infiltration. IKZF1, meanwhile, is a regulator of transcription restricted to lymphocytes and may mediate phenotypic changes important for antimelanoma immunity. There are many reasons why CD2 RNA levels may be more prognostic than CD8 levels, one of them being that CD2 is a marker of activation, whereas CD8 expression is downregulated when T cells are activated. CD2 is also expressed by innate lymphocytes, cells that may have important roles in immune surveillance. (See Supplementary Table S3 online for a referenced list of hub gene functions and corresponding NanoString expression data.)
In summary, we identify a 53-gene panel predictive of melanoma progression in two independent cohorts. We find that 42 of these genes are present in an immune subnetwork found in the GEO database, and that driver genes in this network have key roles in lymphocyte activation and recruitment. This work is based on data gathered retrospectively on the basis of chart reviews from three independent melano-matreatment centers and is therefore preliminary. However, results presented here should be of practical utility in the design of future large-scale studies to develop genomic biomarkers of clinical relevance for adjuvant immunotherapy studies. Clearly, evidence of immune activity with clinical implications can be discovered in primary melanoma tumors. Measuring expression of key immune genes in FFPE tissue should be a promising way to make this information available to researchers, clinicians, and patients.
MATERIALS AND METHODS
Patients and samples
This study was approved by the Institutional Review Boards, and patients provided written informed consent when required. The investigation was conducted according to the Declaration of Helsinki Principles. The training set included FFPE primary melanoma tumors from 40 patients with completely resected stage II–III melanoma identified by screening dermatopathology databases between January 2001 and January of 2011 at GMC (32 patients) and MSSM (8 patients). Authorized personnel obtained clinical information at each institution. Patients with incomplete clinical follow-up in the medical record were contacted by mail and telephone under an IRB-approved protocol and included if adequate follow-up was obtained. The test set included additional patients from the GMC (16 patients), MSSM (7 patients), and New York University Medical Center (New York, NY, 25 patients). A complete review of all patient records was performed on December 31, 2011 for the training set and December 31, 2012 for the test set. Data prepared at all three centers were reviewed centrally at MSSM to determine whether patients had recurred, progressed to unresectable stage III or stage IV, or had died, and all living patients were censored on this date or the most recent preceding date of available follow-up.
Analysis of gene expression
RNA was extracted from primary melanoma specimens using the Ambion RecoverAll Total Nucleic Acid Isolation Kit (Life Technologies, Carlsbad, CA, Supplementary Methods page 3 online). A concentration of RNA of at least 20 ngml−1 and a detectable peak on the tracing at 50 bp or above was required for NanoSrting analysis. A total of 446 genes were selected on the basis of a PubMed literature review (Figure 1a and Supplementary Tables S1 and S2 online). The nCounter platform (NanoString Technologies, Seattle, WA) was used to quantify relative mRNA copy number (Supplementary Methods page 4 online; Geiss et al., 2008).
Immunohistochemistry
IHC was performed on 5-μm charged slides using anti-CD2 mAb (MRQ-11, Ventana Medical Systems, Tucson, AZ). Sections were deparaffinized and stained using a Ventana BenchMark XT immunostainer. Slides were evaluated by two of the study authors (SGB & MMM) in a blinded manner in eight random high powered fields using an ocular micrometer with a 1mm2 grid (Nikon Eclipse E40, Tokyo, Japan).
Ensemble classification/regression method and receiver operating characteristic curves
Classification was performed using an ensemble feature selection method encapsulating two standard classifiers: random forest and elastic net, both embedded in data bootstrapping to boost the robustness of the final gene panel. The starting 446 genes from the training experiment were ranked and filtered based on prediction power of melanoma progression in the training cohort, and a subset of 53 genes was selected as a final gene panel. Receiver operating characteristic curves were generated and the area under the curve was calculated on both training and test data sets. Detailed methods are included in the Supplementary appendix online page 6 and in Supplementary Figure S1 online.
Demographic, survival, and multivariable analysis
The two-tailed student’s t-tests generated P-values for continuous variables including age, depth, and mitotic rate. Other noncontinuous characteristics were analyzed using the two-tailed Fisher’s exact test or, in the case of TILs, a χ2-test. Graphpad Prism version 5.0 (GraphPad Software, La Jolla, CA) was used (San Diego, CA) and statistical significance was defined as P<0.05 without correction for multiple comparisons. For RFS and DSS analysis, Kaplan–Meier analysis and log-rank (Mantel–Cox) tests were performed. Standard multivariable logistic and Cox proportional hazards analysis were performed using XLSTAT (Addinsoft, Brooklyn, NY) software.
Coexpression and gene network analysis
From the NIH GEO database, 46 samples of gene expression data identified on the basis of origin in primary melanoma tissue and expression platform (Supplementary Table S6 online) were collected (GEO accession ID: GSE15605)26. Coexpression network analysis was performed using Weighted Gene Coexpression Network Analysis (WGCNA)27 to identify highly correlated gene modules among whole-genome genes in early-stage melanoma patients. To construct a network around the 53-gene panel, a biologically relevant gene set surrounding the 53-gene panel was obtained using the knowledge base tool using VisAnt28,29 (for full details see Supplementary Methods page 9 online).
Pathway and gene ontology enrichment
Gene panels were annotated using the functional database and tool DAVID.30,31 The default list of whole genome was the background set, and each network list was tested for enrichment of the KEGG pathways, or GO term biological process, or GO term molecular function (MF).
Supplementary Material
Acknowledgments
We wish to acknowledge the support of the clinical research education program, under the direction of Dr Gabrilove and CePORTED funded under the auspices of CONDUITS, the Icahn School of Medicine NCATS ULITR4067, an NIH T32HL094283 grant under the direction of Dr Margaret Baron, the Von Hess Grant from Lancaster General Hospital, and a fellow’s grant from Bristol Myers Squibb (Shanthi Sivendran, PI). Funding to support this work was provided by the Tisch Cancer Institute, a career development award from the Dermatology Foundation (Yvonne Saenger PI), the Von Hess grant from Lancaster General Health, and a fellow’s grant from Bristol Myers Squibb (Shanthi Sivendran PI).
Abbreviations
- DSS
disease-specific survival
- FFPE
formalin-fixed and paraffin-embedded
- IHC
immunohistochemistry
- RFS
recurrence-free survival
- TIL
tumor-infiltrating lymphocyte
Footnotes
CONFLICT OF INTEREST
SS, RC, ALD, and YS have filed a patent for the 53-gene panel. The remaining authors state no conflict of interest.
Supplementary material is linked to the online version of the paper at http://www.nature.com/jid
References
- Ascierto PA, Capone M, Urba WJ, et al. The additional facet of immunoscore: immunoprofiling as a possible predictive tool for cancer treatment. J Transl Med. 2013;11:54. doi: 10.1186/1479-5876-11-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azimi F, Scolyer RA, Rumcheva P, et al. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J Clin Oncol. 2012;30:2678–83. doi: 10.1200/JCO.2011.37.8539. [DOI] [PubMed] [Google Scholar]
- Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch CM, Gershenwald JE, Soong SJ, et al. Multivariate analysis of prognostic factors among 2,313 patients with stage III melanoma: comparison of nodal micrometastases versus macrometastases. J Clin Oncol. 2010;28:2452–9. doi: 10.1200/JCO.2009.27.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch CM, Soong SJ, Gershenwald JE, et al. Prognostic factors analysis of 17,600 melanoma patients: validation of the American Joint Committee on cancer melanoma staging system. J Clin Oncol. 2001;19:3622–34. doi: 10.1200/JCO.2001.19.16.3622. [DOI] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Fridman WH, et al. The prognostic impact of anticancer immune response: a novel classification of cancer patients. Semin Immunopathol. 2011;33:335–40. doi: 10.1007/s00281-011-0264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogunovic D, O’Neill DW, Belitskaya-Levy I, et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc Natl Acad Sci USA. 2009;106:20429–34. doi: 10.1073/pnas.0905139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busam KJ, Antonescu CR, Marghoob AA, et al. Histologic classification of tumor-infiltrating lymphocytes in primary cutaneous malignant melanoma. A study of interobserver agreement. Am J Clin Pathol. 2001;115:856–60. doi: 10.1309/G6EK-Y6EH-0LGY-6D6P. [DOI] [PubMed] [Google Scholar]
- Clemente CG, Mihm MC, Jr, Bufalino R, et al. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77:1303–10. doi: 10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Fortina P, Surrey S. Digital mRNA profiling. Nat Biotechnol. 2008;26:293–4. doi: 10.1038/nbt0308-293. [DOI] [PubMed] [Google Scholar]
- Francken AB, Accortt NA, Shaw HM, et al. Prognosis and determinants of outcome following locoregional or distant recurrence in patients with cutaneous melanoma. Ann Surg Oncol. 2008;15:1476–84. doi: 10.1245/s10434-007-9717-9. [DOI] [PubMed] [Google Scholar]
- Fridman WH, Galon J, Dieu-Nosjean MC, et al. Immune infiltration in human cancer: prognostic significance and disease control. Curr Top Microbiol Immunol. 2011;344:1–24. doi: 10.1007/82_2010_46. [DOI] [PubMed] [Google Scholar]
- Galon J, Angell HK, Bedognetti D, et al. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39:11–26. doi: 10.1016/j.immuni.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Geiss GK, Bumgarner RE, Birditt B, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–25. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- Harbst K, Staaf J, Lauss M, et al. Molecular profiling reveals lowand high-grade forms of primary melanoma. Clin Cancer Res. 2012;18:4026–36. doi: 10.1158/1078-0432.CCR-12-0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruit WH, Suciu S, Dreno B, et al. Selection of immunostimulant AS15 for active immunization with MAGE-A3 protein: results of a randomized phase II study of the European Organisation for research and treatment of cancer melanoma group in metastatic melanoma. J Clin Oncol. 2013;31:2413–20. doi: 10.1200/JCO.2012.43.7111. [DOI] [PubMed] [Google Scholar]
- Loetscher P, Uguccioni M, Bordoli L, et al. CCR5 is characteristic of Th1 lymphocytes. Nature. 1998;391:344–5. doi: 10.1038/34814. [DOI] [PubMed] [Google Scholar]
- Mann GJ, Pupo GM, Campain AE, et al. BRAF mutation, NRAS mutation, and the absence of an immune-related expressed gene profile predict poor outcome in patients with stage III melanoma. J Invest Dermatol. 2013;133:509–17. doi: 10.1038/jid.2012.283. [DOI] [PubMed] [Google Scholar]
- Manolio TA, Chisholm RL, Ozenberger B, et al. Implementing genomic medicine in the clinic: the future is here. Genet Med. 2013;15:258–67. doi: 10.1038/gim.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton DL, Thompson JF, Cochran AJ, et al. Sentinel-node biopsy or nodal observation in melanoma. N Engl J Med. 2006;355:1307–17. doi: 10.1056/NEJMoa060992. [DOI] [PubMed] [Google Scholar]
- Ott PA, Carvajal RD, Pandit-Taskar N, et al. Phase I/II study of pegylated arginine deiminase (ADI-PEG 20) in patients with advanced melanoma. Invest New Drugs. 2013;31:425–34. doi: 10.1007/s10637-012-9862-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnepenninckx V, Lazar V, Michiels S, et al. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J Natl Cancer Inst. 2006;98:472–82. doi: 10.1093/jnci/djj103. [DOI] [PubMed] [Google Scholar]
- Wong SL, Balch CM, Hurley P, et al. Sentinel lymph node biopsy for melanoma: American Society of Clinical Oncology and Society of Surgical Oncology joint clinical practice guideline. J Clin Oncol. 2012;30:2912–8. doi: 10.1200/JCO.2011.40.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.