

Abstract
HCF-1 is a transcriptional cofactor required for activation of herpes simplex virus immediate-early genes by VP16 as well as less clearly defined roles in cell proliferation, cytokinesis, and spliceosome formation. It is expressed as a large precursor that undergoes proteolysis to yield two subunits that remain stably associated. VP16 uses a degenerate 4-amino acid sequence, known as the HCF-binding motif, to bind to a six-bladed β-propeller domain at the N terminus of HCF-1. Functional HCF-binding motifs are also found in LZIP and Zhangfei, two cellular bZIP transcription factors of unknown function. Here we show that the HCF-binding motif occurs in a wide spectrum of DNA-binding proteins and transcriptional cofactors. Three well characterized examples were further analyzed for their ability to use HCF-1 as a coactivator. Krox20, a zinc finger transcription factor required for Schwann cell differentiation, and E2F4, a cell cycle regulator, showed a strong requirement for functional HCF-1 to activate transcription. In contrast, activation by estrogen receptor-α did not display HCF dependence. In Krox20, the HCF-binding motif lies within the N-terminal activation domain and mutation of this sequence diminishes both transactivation and association with the HCF-1 β-propeller. The activation domain in the C-terminal subunit of HCF-1 contributes to activation by Krox20, possibly through recruitment of p300. These results suggest that HCF-1 is recruited by many different classes of cellular transcription factors and is therefore likely to be required for a variety of cellular processes including cell cycle progression and development.
HCF-1 (also known as C1 factor) is a heterodimeric nuclear protein composed of a family of polypeptides generated from a 2035-amino acid precursor by site-specific proteolysis (1–3). After cleavage of the precursor, the N- and C-terminal processing products remain as a stable complex through two matching sets of self-interaction domains (3, 4). HCF-1 is expressed and processed in most cell types and with the exception of unstimulated sensory neurons, is predominantly nuclear and tightly associated with chromatin (3, 5–8). The two HCF-1 subunits are composed of multiple functional domains. The N-terminal subunit includes a six-bladed β-propeller domain characteristic of proteins in the kelch superfamily and an adjacent basic region (9–11). The C-terminal subunit includes a transcriptional activation domain (HCF-1AD), a pair of degenerate fi-bronectin type III repeats, and a bipartite nuclear localization signal (4, 12, 13).
The cellular functions of HCF-1 appear to be multifaceted and are still only partially understood. The most clearly established role is in regulation of viral and cellular transcription. This was first described in the context of the herpes simplex virus (HSV)1 transactivator VP16 (reviewed in Refs. 14 and 15). HCF-1 is necessary for the recruitment of VP16 to HSV immediate-early gene promoters and for transcriptional activation by the assembled VP16-induced complex (13). In addition, both subunits of HCF-1 have been shown to interact with cellular transcriptional activators and repressors (7, 16–21). Analysis of a temperature-sensitive hamster cell line (tsBN67) carrying a missense mutation in the HCF-1 β-propeller has revealed a role in cell cycle progression and cytokinesis (22, 23). Last, HCF-1 is incorporated into spliceosome complexes and may participate in mRNA splicing as well as transcription (24).
The N-terminal β-propeller domain is central to most HCF-1 functions. The 380-amino acid domain encompasses six kelch (HCFKEL) repeats, which are predicted to form the six blades of the propeller structure. The domain is sufficient to bind VP16 and assemble a VP16-induced complex on DNA in association with the cellular POU protein Oct-1 (11). In the HCF-1 protein encoded by the tsBN67 cell line, there is a missense mutation in the β-propeller that changes proline 134 to serine (11, 22). At the non-permissive temperature, HCF-1 is unable to support transactivation by VP16 and is released from the cellular chromatin (8, 22). Yeast interaction screens using the β-propeller domain identified two ATF/cAMP-response element-binding protein-like basic leucine zipper proteins known as LZIP/Luman and Zhangfei (7, 16, 18). Sequence comparison with VP16 revealed a 4-amino acid sequence motif, termed the HCF-binding motif (HBM), that is used by all three proteins to recognize the HCF-1 β-propeller (16, 25). This motif ((D/E)HXY) consists of an acidic residue (aspartic acid or glutamic acid) followed by an invariant histidine, any residue (X), and then an invariant tyrosine. The HBM is an integral part of the LZIP transactivation domain, and recruitment of HCF-1 is required for activation by LZIP (26). Recently, functional HBM sequences have been identified in the peroxisome proliferator-activated receptor γ-coactivator PGC-1β and a novel nuclear export factor termed HPIP (27, 28).
As a starting point for study of defined cellular processes regulated by HCF-1, we searched the protein sequence data bases to identify known transcription factors that contain potential HBMs. Many candidates were identified in this way, and in several the HBM lies in a region of the protein already known to be involved in gene activation. Three candidates, Krox20 (also known as EGR2, NGF1-B), E2F4, and estrogen receptor α (ERα), representing well studied members of unrelated protein families, were selected for further analysis. Krox20 is a member of the Krüpple-like zinc finger protein family and plays a critical role in hindbrain development and myelination of the peripheral nervous system (29). E2F4 plays a key role in regulating the cell cycle as well as differentiation processes such as adipogenesis (30). ERα is a steroid hormone receptor that stimulates gene expression in a ligand-dependent manner (31). We found that transactivation by Krox20 and E2F4 but not ERα was dependent on the presence of functional HCF-1. Mutation of the HBM or deletion of the activation domain of HCF-1 significantly reduced the ability of Krox20 to stimulate transcription from a synthetic promoter composed of a GC-rich Krox20 binding site or a natural promoter from the human bFGF-2 gene. The results of these experiments suggest that HCF-1 plays multiple roles in the regulation of cellular transcription. In addition to ubiquitous processes such as promoting cell proliferation, HCF-1 is likely to be important for tissue-specific events including myelination by Schwann cells and development of the central nervous system.
Experimental Procedures
Data Base Analysis
The SWISS-PROT and Translated EMBL (TrEMBL) data bases were searched using ScanProSite, a web-based motif search tool provided by the Expert Protein Analysis System (ExPASy)2 proteomics server of the Swiss Institute of Bioinformatics (32). Using the query “[DE]HxY,” searches were restricted to mammalian proteins and individual entry annotations were used to identify transcription factors.
Expression Plasmids
The full-length murine Krox20 open reading frame was amplified by PCR from a cDNA clone provided by Dr. Jeffrey Milbrandt (Washington University, St. Louis, MO), and subcloned into the cytomegalovirus enhancer-driven mammalian expression vectors pCGT and pCGN, thereby adding N-terminal T7 and hemagglutinin-epitope tags (11). Note that the mouse Krox20 protein was used in these experiments. Mouse and human proteins are highly homologous (overall 89% identical and 95% similar), especially within the activation and DNA-binding domains. In addition, Krox20 sequences (full-length or the first 170 residues) were subcloned into pCGNGal4, creating a hem-agglutinin-tagged fusion to the yeast Gal4 DNA-binding domain (residues 1 to 94). All PCR amplified fragments were confirmed by DNA sequencing. Alanine substitution mutagenesis of the Krox20 HBM (residues 162 to 165) was performed by QuikChange™ (Stratagene) and verified by DNA sequencing. A diagnostic PstI site was incorporated into the changes to identify mutants.
The plasmid encoding Gal4-E2F4-(240–412) was kindly provided by Dr. David Johnson (University of Texas M. D. Anderson Cancer Center, Houston, TX) (33). The Gal4-responsive luciferase reporter plasmid used in this study was p5xGal4-E1B-luc and contains five tandem Gal4-binding sites (CGGAGTACTGTCCTCCG) (34). The plasmid encoding the estrogen receptor (CMV-ER) and estrogen response element-luc reporter were kind gifts of Dr. Michael Garabedian (NYU School of Medicine) (35). The bFGF-2 promoter and GC-luciferase reporter plasmid (GCGGGGGCG-luc) were provided by Drs. Jeffrey Milbrandt and John Svaren (University of Washington, St. Louis, MO) (36, 37).
Cell Culture and Protein Expression Assays
Maintenance and electroporation of 293T, HeLa, and tsBN67-derived cells, luciferase reporter assays, extract preparation, and immunoblotting have been described previously (2, 13, 26). For temperature shift experiments, tsBN67 cells were grown at 33.5 °C. After electroporation, the transfected cells were plated and allowed to recover at 33.5 °C for 2 h before transfer to 39.5 °C.
Results
Candidate HCF-binding Motifs Are Found in Many Cellular Transcription Factors
To identify cellular proteins that can interact with HCF-1, we searched the SWISS-PROT and EMBL protein data bases for sequences matching the HBM consensus ((D/E)HXY) using the web-based ScanProsite search tool. As anticipated, this short and degenerate motif was present in a large number of mammalian proteins of different functions and cellular localization. SWISS-PROT alone yielded more than 370 unique hits. Given the previously established role of HCF-1 in regulation of gene expression, and the predominantly nuclear localization of the protein, we focused on known or suspected DNA-binding proteins and transcriptional co-regulators (see Fig. 1 for a listing). As expected, the search identified previously known HBM-containing proteins. These include HSV VP16 and its homologues from related α-herpesviruses as well as the cellular proteins LZIP, Zhangfei, HPIP, and PGC-1α and -β (7, 16, 18, 27, 28). The sequences flanking the core motif show no obvious homology, although there is evidence that flanking sequences do influence HBM function (38).
Fig. 1. Candidate HCF-binding motifs are found in many human transcription factors.

Comparison of known or candidate HBM sequences. For each, the core motif ((E/D)HXY, where X can be any residue), is highlighted. The first 8 sequences correspond to experimentally verified HBM proteins (see text for details). SWISS-PROT or EMBL accession numbers and amino acid coordinates are given.
In terms of the candidate HBM-containing proteins, we noted that in several examples, the HBM-like sequence is conserved in non-human counterparts and/or falls within a region of the protein known to contain an activation domain. A clear example is the Krox20/EGR2 protein (Fig. 2A). Orthologs from other vertebrates preserve the putative HBM, although in birds, fish, and amphibians the first position is changed from an aspartic acid to a glutamic acid. In fish and amphibians the variable third position is also changed from leucine to isoleucine that preserves the hydrophobic nature of the side group. The activation domain of Krox20 has been mapped to the N terminus of the protein (residues 1–184) and encompasses the HBM-like sequence (residues 162–165) (39). This arrangement is strongly reminiscent of LZIP in which the N-terminal activation domain includes the HBM (26).
Fig. 2. Temperature-dependent transactivation by Krox20 and E2F4 in tsBN67 cells.
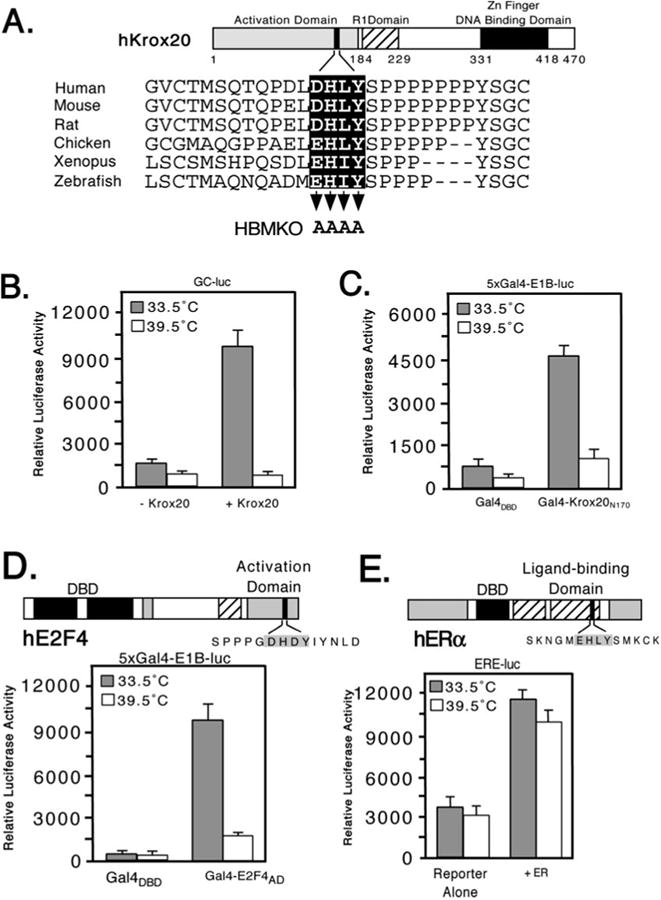
A, schematic showing structure of murine Krox20 with sequences of the HBM region from the following orthologs: Zebrafish (SWISS-PROT accession Q05159), Xenopus (Q08427), chicken (Q98T82), rat (P51774), mouse (P08152), and human (P11161). Three functional domains have been defined: an N-terminal activation domain (residues 1–184), a repression domain (193–229), and a DNA-binding domain composed of three zinc fingers (331–418). Residues mutated to alanine in Krox20 HBMKO are indicated by arrowheads. B, hamster tsBN67 cells were cotransfected with an expression plasmid (500 ng) encoding full-length mouse Krox20 together with a Krox20 responsive reporter (GC-luc, 5 μg) and incubated at 33.5 or 39.5 °C for 40 h. Each assay was performed in triplicate and the mean ± S.D. are shown. C, as in panel B, except that tsBN67 cells were cotransfected with 500 ng of expression plasmid encoding Gal4DBD or Gal4-Krox20N170 together with a Gal4 reporter (5xGal4-E1B-luc, 500 ng). D, domain structure of human E2F4. The HBM (filled box) lies within the transactivation domain (33). For the reporter assay tsBN67 cells were cotransfected with 250 ng of Gal4DBD or Gal4-E2F4-(240–412) expression plasmid and 500 ng of Gal4-luciferase reporter. E, in the estrogen receptor (ERα) the putative HBM (filled box) lies in the ligand-binding domain. 500 ng of ERα expression plasmid were cotransfected into tsBN67 cells together with an ER-responsive reporter plasmid (ERE-luc, 500 ng). Note that the calf serum used to culture the cells contains sufficient levels of natural ligands to activate the transfected receptors.
Transcriptional Activation by Krox20 and E2F4 Is Temperature-sensitive in tsBN67 Cells
To determine whether any of these sequence matches represent functional HBMs, we selected three candidates, Krox20, E2F4, and ERα, and tested them for HCF dependence. Each activator was analyzed by transient transfection of tsBN67, a derivative of BHK21 that carries a temperature-sensitive version of HCF-1. At the permissive temperature of 33.5 °C, HCF-1 is functional and the cells proliferate normally, whereas at the non-permissive temperature (39.5 °C), the HCF-1 β-propeller domain is inactivated and the cells asynchronously arrest (11, 22).
Transactivation by Krox20 was examined in two contexts (Fig. 2, B and C). In the first (Fig. 2B), full-length Krox20 (Krox20FL) was cotransfected into tsBN67 cells together with a Krox20-responsive luciferase reporter (GC-luc) composed of two Krox20-binding sites (5′-GCGGGGGCG-3′) placed upstream of the prolactin core promoter. Pools of transfected cells were split into halves and incubated for 40 h at either the permissive or non-permissive temperature, and then assayed for luciferase activity. Expression of Krox20 gave a 7-fold stimulation at the permissive temperature but this was abolished when cells were cultured at 39.5 °C. The reporter alone showed a similar reduction reflecting the activity of low levels of endogenous Krox20 protein. The result of this experiment suggests that Krox20 requires HCF-1 to activate transcription. In the second approach (Fig. 2B), we tested an N-terminal fragment of Krox20 (residues 2–170) corresponding to the activation domain (39). This fragment was fused to the Gal4 DNA-binding domain (Gal4DBD) and cotransfected with a luciferase reporter gene (5xGal4-E1B-luc) containing five Gal4-binding sites upstream of the adenovirus E1B TATA box. Gal4-Krox20N170 activated transcription 5-fold at the permissive temperature and this activation was reduced to only 2-fold at the non-permissive temperature. This result substantiates the temperature-dependent activation observed using full-length Krox20 and argues that HCF-1 contributes to transcriptional activation rather than recognition of the GC-rich Krox20-bind-ing site.
We also tested a C-terminal fragment from human E2F4 (residues 240–412) fused to the Gal4DBD (Gal4-E2F4-(240– 412)) (Fig. 2D). This fragment includes the activation and pocket protein-binding domains but not the DNA-binding or dimerization domains (33). Expression of Gal4-E2F4-(240– 412) at the permissive temperature increased transcription of the report by ∼8-fold and this was significantly reduced in cells maintained at the non-permissive temperature. In contrast to Krox20 and E2F4, activation by the full-length estrogen receptor (ERα) using a reporter linked to an estrogen response element showed little difference at the two temperatures (Fig. 2E). Under these conditions, ERα is activated by ligand present in the calf sera. Addition of the ligand estradiol (100 nM) to the culture media gave similar results (data not shown). These results indicate that not all of the activators containing HBM-like sequences require HCF-1 to activate transcription, thus highlighting the role of HCF-1 in specific programs of gene expression.
Mutation of the HBM Prevents Transactivation by Krox20
To determine whether the HBM-like sequence in Krox20 contributes to activation domain function, we generated a substitution mutant (Krox20 HBMKO, Fig. 2A) simultaneously changing all four residues of the motif to alanine. HeLa cells were cotransfected with the wild type and mutant versions of full-length Krox20 together with the GC-luc reporter (Fig. 3A). Wild type Krox20 (WT) activated transcription 6-fold, whereas the mutant (HBMKO) activated transcription only 1.5-fold. This indicates that the HBM identified through a sequence data base search is an important component of the Krox20 activation domain.
Fig. 3. Mutation of the HBM prevents transactivation by Krox-20.
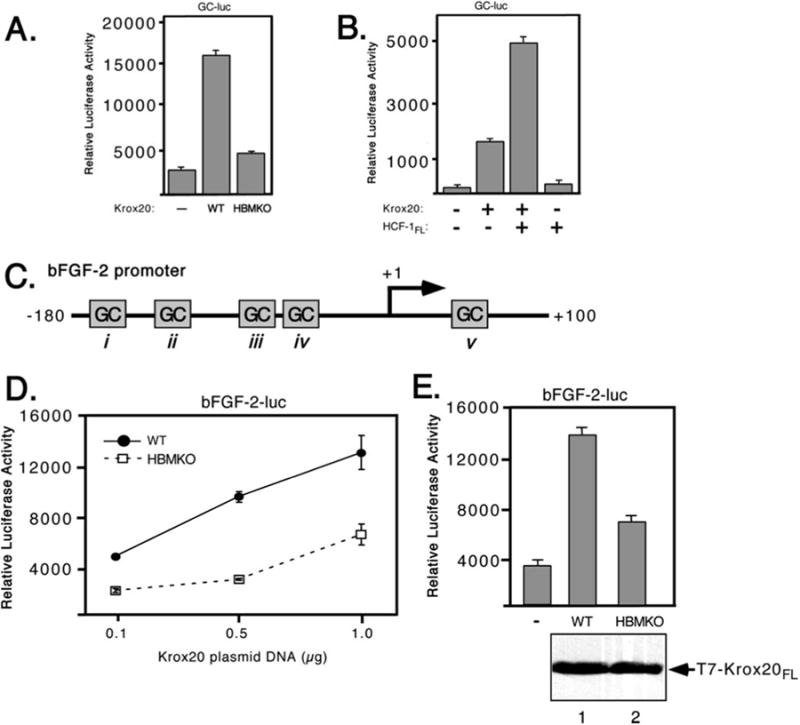
A, GC-luc reporter; B, HCF-dependent activation of the bFGF-2 promoter. C, schematic showing the human bFGF-2 promoter that lacks a TATA box but contains multiple GC-rich boxes (i–v) that serve as binding sites for Sp1 and Krox24/Egr-1. Egr-1 was shown to bind to human bFGF-2 promoter at two sites (sites i and iii) (36). D, 293T cells were cotransfected with increasing amounts of wild type (filled circles) or HBMKO (open squares) Krox20 expression plasmids (0.1, 0.5, and 1 μg) and a bFGF-2 promoter reporter plasmid (bFGF-2-luc, 100 ng). Luciferase activity was measured after 40 h. E, in a separate experiment, protein extracts were prepared from cells transfected with 0.5 μg of expression plasmid, resolved by 10% SDS-PAGE, and immunoblotted with αT7 antibody.
We also examined the consequence of overexpressing HCF-1 (Fig. 3B). HeLa cells were transfected with expression plasmids encoding full-length Krox20 and HCF-1. Coexpression of both proteins resulted in a greater level of activation than with Krox20 alone. This stimulation was not observed when HCF-1 was expressed on its own. This suggests that HCF-1 can only contribute to activation when brought to the promoter via a DNA-binding protein such as Krox20.
HCF-1 Is Required for Activation of the bFGF-2 Promoter
The bFGF-2 promoter (shown schematically in Fig. 3C) contains multiple GC-rich elements and at least two of these (boxes i and iii) have been shown to bind Krox20 (or the related Krox24/EGR1) protein in vitro, leading to promoter activation (36, 40). To address the role of HCF-1 in regulation of the bFGF-2 promoter, we cotransfected 293T cells with increasing amounts of expression plasmids encoding wild type and HB-MKO Krox20 together with the full-length bFGF-2 promoter driving the luciferase reporter gene (41). The promoter was activated in a dose-dependent manner by wild type Krox20, but showed a significantly reduced response to Krox20 HBMKO (Fig. 3D). As shown in Fig. 3E, the experiment was repeated using 500 ng of each Krox20 expression plasmid and extracts were assayed for protein expression by immunoblotting with an antibody against the T7 epitope-tagged Krox20 proteins as well as for luciferase activity. Activation of the bFGF-2 promoter was significantly reduced with the HBMKO mutant compared with wild type, although there was little detectable difference in protein expression. These results show that mutation of the candidate HBM in Krox20 reduces its ability to activate transcription from a natural promoter.
The HCF-1 β-Propeller Associates with the Activation Domain of Krox20
By analogy to VP16, LZIP, and HPIP, it is possible that the HCF-1 β-propeller alone is sufficient for recognition of the HBM in Krox20 (11, 16, 26, 28). To date, we have been unable to detect a stable interaction between Krox20 and HCF-1 using gel mobility shift or in vitro coimmunoprecipitation assays, suggesting the interaction is weak or stabilized by other factors present in vivo (data not shown). Instead, we have used a mammalian one-hybrid recruitment assay in transfected 293T cells to characterize the interaction between the β-propeller of HCF-1 and Krox20. Gal4-HCF-1N380WT was co-expressed with Krox20FL and the Gal4-responsive reporter (Fig. 4A). The β-propeller of HCF-1 did not activate the reporter gene unless coexpressed with Krox20FL. The specificity of this interaction was verified using the tsBN67 version of the HCF-1 β-propeller. This is identical to wild type except that proline 134 has been changed to serine (P134S). Previous studies have shown that this single substitution is sufficient to prevent interaction with VP16 and LZIP (11, 16, 25). As expected, activation was not observed using the P134S mutant, indicating that recruitment of Krox20FL to the promoter was dependent on a functional β-propeller domain.
Fig. 4. The Krox-20 activation domain interacts with the β-propeller of HCF-1.
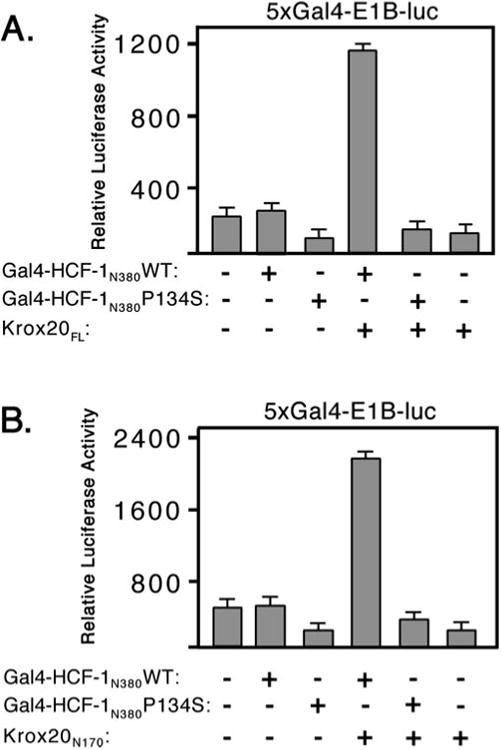
A, 293T cells were cotransfected with 5xGal4- E1B-luc reporter (100 ng) together with expression plasmids (500 ng) encoding Gal4-HCF-1N380 WT or P134S and full-length Krox20 (Krox20FL). Luciferase activity was measured after 40 h. B, as in the previous panel except that only the N terminus of Krox20 (Krox20N170) was used.
To better define sequences in Krox20 required for this interaction, the recruitment assay was repeated using Krox20N170 in place of Krox20FL (Fig. 4B). Again Krox20N170 was able to activate the reporter in the presence of Gal4-HCF-1N380WT, but not the P134S mutant. This result shows that the Krox20 activation domain was sufficient for interaction with the HCF-1 β-propeller domain.
The HCF-1 C-terminal Activation Domain Is Important for Krox20 Function
HCF-1 contains a potent activation domain (HCF-1AD) located in a domain termed the “acidic region” that forms part of the C-terminal subunit (Fig. 5A) (13). The HCF-1AD activates transcription when tethered to a promoter by a heterologous DNA-binding domain and is required for efficient transactivation by the VP16-induced complex and LZIP. We next asked whether the HCF-1AD contributes to Krox20 coactivator function by using two cell lines derived from hamster tsBN67 cells. In these lines, the temperature-sensitive defect has been complemented by stable expression of human HCF-1 (HCF-1Δrep) with (WT) or without (ΔAD) the HCF-1AD peptide (residues 1613 to 1654). To prevent exchange of subunits with the endogenous hamster protein that is stable at both temperatures, we used a version of human HCF-1 lacking the central repeat region (designated Δrep). Cells were maintained at 39.5 °C to inactivate the endogenous tsBN67 HCF-1 protein and ensure that cell proliferation and transcription were dependent on the recombinant HCF-1Δrep protein. As shown in Fig. 5B, activation by the N terminus of Krox20 (Gal4-Krox20N170) was reduced ∼2-fold in the absence of the HCF-1AD. This reduction was specific to activated transcription because background promoter activity (Gal4DBD alone) was equivalent in the two cell lines. The results obtained using full-length Krox20 were more important (Fig. 5, C and D). Using the GC reporter (Fig. 5C), wild type Krox20 stimulated the promoter ∼5-fold in WT cells but was essentially inactive in the absence of the HCF-1AD. Likewise, the bFGF-2 promoter showed a markedly different response in the two cell lines, although there was some stimulation in the ΔAD cells expressing the highest level of Krox20 protein. This context dependence may reflect the contribution of other elements of the bFGF-2 promoter and suggests that the coactivator requirements for the Krox20 activation domain to function vary from gene to gene.
Fig. 5. HCF-1AD is required for transactivation by Krox-20.
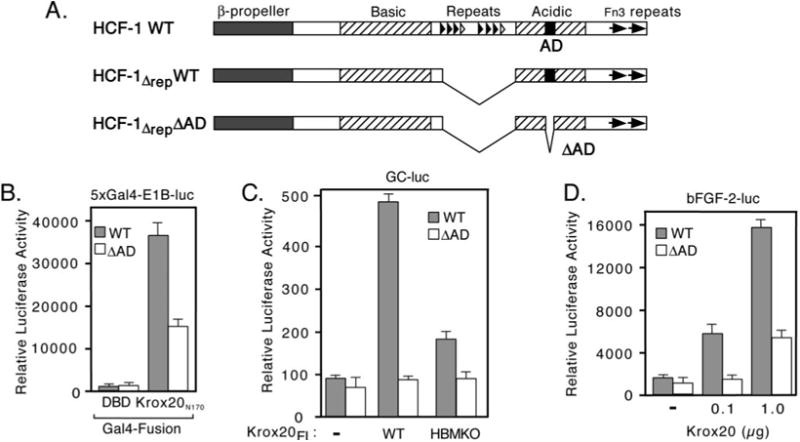
A, structure of wild type (WT) and activation domain deleted (ΔAD) versions of HCF-1 used to complement tsBN67 cells (13). Major structural features of HCF-1 are indicated. Note the central HCF-1PRO repeats were removed (HCF-1Δrep) to prevent processing and subunit exchange. B, equivalent cultures of tsBN67 cells that have been complemented with a derivative of human HCF-1 (HCF-1Δrep) with (R-tsBN67-HCF-1ΔrepWT) or without (R-tsBN67-HCF-1ΔrepΔAD) the C-terminal activation domain (HCF-1AD) were transiently transfected with 5xGal4-E1B-luc (500 ng) together with expression plasmids (500 ng) encoding Gal4DBD or Gal4-Krox20N170 and assayed for luciferase activity after incubation at 39.5 °C for 40 h. C, equal numbers of WT and ΔAD cells were transfected with the GC-luc reporter (500 ng) together with expression plasmids (500 ng) encoding WT or HBMKO full-length Krox20 and assayed after incubation for 40 h at 39.5 °C. D, R-tsBN67-HCF-1ΔrepWT and R-tsBN67-HCF-1ΔrepΔAD cells were transfected with bFGF-2-luc reporter (500 ng) with or without cotransfected wild type Krox20 expression plasmid (100 ng and 1 μg).
Activation by Krox20 Can Be Stimulated by Expression of p300
How HCF-1 functions as a coactivator is not well understood. Many coactivators contain acetyltransferase activity and are thought to act through covalent modification of other transcription factors or the N-terminal tails of the core histones (42). HCF-1 is not known to possess any enzymatic activities, however, the HCF-1AD has been shown to act synergistically with the coactivator p300 to stimulate transcription (13). This well characterized coactivator has been shown to acetylate a variety of transcription factors as well as histones and may thus indirectly provide this function to HCF-1 (43). We therefore asked whether transactivation by Krox20 could be stimulated by coexpression of p300 (Fig. 6A). Full-length Krox20 was cotransfected into 293T cells with or without a p300 expression plasmid and levels of Krox-dependent transcription was measured using the GC-luc reporter. On its own, Krox20FL activated transcription 2.5-fold but this was increased to ∼20-fold in the presence of overexpressed p300. Expression of p300 alone did not stimulate the reporter beyond that of Krox20 alone. This result suggests that the coactivator p300 can cooperate with the HCF-1AD to activate Krox20-regulated promoters.
Fig. 6. Expression of p300 stimulates activation by Krox-20.
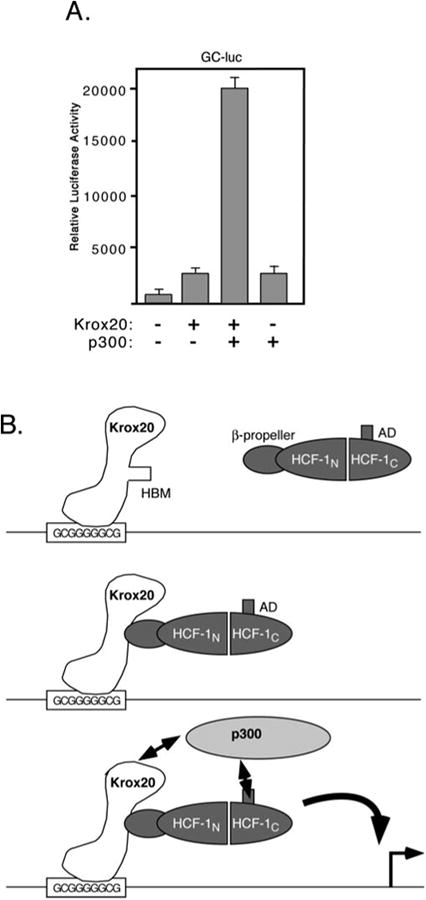
A, 293T cells were cotransfected with the GC-luc reporter (5 μg) together with expression plasmids encoding full-length Krox20 (500 ng) and p300 (6 μg). Luciferase activity was assayed 40 h post-transfection. B, model showing recruitment of HCF-1 and p300 to the activation domain of Krox20.
Discussion
The characterization of cellular transcription factors that associate with HCF-1 should provide important clues to its function during development and differentiation. As one means to achieve this, we searched the protein sequence data bases for transcription factors that contain a putative core HCF-binding motif sequence. This search identified a broad spectrum of sequence specific transcription factors and transcriptional co-factors, including proteins that act as activators or repressors. In a few cases, multiple members of a single family were represented including seven different zinc finger DNA-binding domain proteins.
While this article was in preparation, Wysocka and colleagues (44) described association of the HCF-1 N terminus with the human Set1/Ash2 histone methyltransferase complex. The enzymatic activity associated with this complex is likely to play a critical role in the coactivator function of HCF-1. Most significantly, the complex interacts with the β-propeller domain and both the human and nematode Set1 proteins contain HBM-like sequences (EHNY and EHCY, respectively). Revisiting our data base search results, we identified human Set1 (accession number O15047) in the collection of expressed sequence tags that lacked annotation. The fact that we identified all known interaction partners for the β-propeller domain suggests that our screen achieved relatively complete coverage of the human proteome.
The degenerate nature of the HBM consensus sequence makes it difficult to distinguish functional examples of the motif from fortuitous matches and it seems reasonable to assume that the list of candidates includes examples of both classes. Of the three candidates selected for further analysis, transactivation by two (Krox20 and E2F4) showed a strong dependence on the presence of functional HCF-1 when tested in tsBN67 cells but the third (ERα) did not. The non-responsiveness of ERα to inactivation of HCF-1 may reflect redundancy in the activation mechanism of this receptor in tsBN67 cells or represents an example of a fortuitous sequence match. Despite the lack of obvious sequence conservation for residues flanking known HBMs, mutagenesis and peptide competition studies using VP16 have clearly demonstrated the importance of sequence context (38, 45). For example, mutation of valine 358 to alanine in VP16, which lies at the –3 position relative to the acidic residue of the HBM, resulted in a 6-fold reduction in ability of an HBM peptide to compete for HCF-1 binding (38). Hydrophobic residues (valine or leucine) occur at this position in VZV ORF10, LZIP, and HPIP but are replaced by proline or serine in EHV VP16, BHV BTIF, Zhanfei, and PGC-1(α,β), suggesting some flexibility. It may be significant that Krox20 and E2F4 also have proline at this position, whereas ERα has a lysine.
The important but poorly understood role of the sequences that flank the 4-amino acid HBM is reminiscent of the LXXLL motif (also known as the NR box), another short and degenerate sequence motif involved in activator-coactivator interactions (46, 47). In the coactivator SRC1, an 8-amino acid peptide containing a single LXXLL motif is sufficient to bind ERα (48). Likewise, an octapeptide from VP16, which includes the HBM, can compete effectively with the complete VP16 protein for binding to HCF-1 (38). For LXXLL-mediated interactions, selectivity is determined by three parameters: the sequence of the core LXXLL motif itself, the sequence of the flanking residues, and in some cases, the relative spacing of two or more LXXLL motifs (49). Sequence context is undoubtedly important for HBM function, but there is no evidence that spacing between HBMs will play a significant role. Of the candidate HBM-containing proteins identified in this study, only T-box protein 5 (TBX5) and chromodomain-helicase-DNA binding protein 2 (CHD-2) contained more than one HBM consensus sequence and furthermore, these sequences are separated by more than 200 amino acids. What defines a genuine HBM from a fortuitous match is not yet known but detailed mutagenesis and structural modeling studies may be sufficient to decipher the code.
The placement of the HBM within the N-terminal activation domain of Krox20 is analogous to the bZIP transactivator LZIP where it has been shown that the N-terminal activation domain is composed of the HBM and two LXXLL motifs (26). Presumably in Krox20 and E2F4, the activation domains include additional protein-protein interaction modules that act cooperatively with the HBM through recruitment of other coactivator activities. Accordingly, we show that expression of the coactivator p300 leads to an increase in activation by Krox20.
Although we and others have shown that the C terminus of E2F4 can activate transcription as a Gal4 DNA-binding domain fusion, it is not known where or when the full-length E2F4 protein functions as an activator. The putative HBM of E2F4 is conserved in the mouse and rat orthologs, implying an important and evolutionarily conserved function, however, chromatin immunoprecipitation studies in cultured cells indicate that E2F4 acts predominantly as a repressor of E2F-regulated genes through recruitment of the p107 and p130 pocket proteins (50). The HBM overlaps with the pocket protein-binding domain (residues 382–413) raising the possibility that pocket proteins and HCF-1 might compete for binding. Promoter context or cell cycle status may determine which coregulator is recruited to a given molecule and thus allow E2F4 to switch between activator and repressor modes. E2F1 also contains a potential HBM but it is located in the N terminus of the protein, within a region involved in the interaction of E2F1 with cyclin A/CDK2, again creating an opportunity for regulation (51). Clearly the functional significance of these interactions needs to be explored. Interestingly, antagonism of pRb family members through the expression of SV40 large T antigen or adenovirus E1A has been shown to suppress the tsBN67 cell proliferation defect (52). This suggests that HCF-1 plays a role in countering the cell cycle checkpoint imposed by the pocket proteins.
The addition of Krox20 to the growing roster of HCF-dependent activators offers a unique opportunity to study HCF-1 function in a developmental and cell-specific context. Disruption of the murine Krox20 gene blocks differentiation of Schwann cells within the peripheral nervous system and also leads to loss of rhombomeres 3 and 5, resulting in abnormal development of the hindbrain and associated cranial sensory ganglia (53, 54). Both the hypomyelination and segmentation defects are thought to reflect insufficient expression of Krox20 target genes. This hypothesis is supported by the finding that ectopic expression of Krox20 in primary Schwann cells is sufficient to induce several myelin-associated genes (29, 55).
In addition to the gene disruption studies in mice, Krox20 has also been implicated in myelination through analysis of congenital hypomyelinating neuropathies. Eight missense mutations in the Krox20 protein have been identified in patients presenting syndromes of this type (reviewed in Ref. 56). Seven of the eight mutations map to the DNA-binding domain of Krox20 and presumably these prevent recognition of key target genes. The eighth mutant prevents binding of the NAB1 and NAB2 corepressors, suggesting that repression by Krox20 might also be important for proper execution of the myelination program or that transactivation must be modulated through the concerted action of coactivators and corepressors (57, 58). Whether HCF-1 plays a similar modulatory role is now testable.
Acknowledgments
We thank Jeffrey Milbrandt, John Svaren, David Johnson, and Michael Garabedian for plasmids used in this study. We also thank Richard Freiman, Naoko Tanese, and Ravi Tikoo for thoughtful comments that strengthened the manuscript.
Footnotes
This work was supported by National Science Foundation Grant MCB-98-16856 and National Institutes of Health Grants GM61139 and AI07180-21. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: HSV, herpes simplex virus; HBM, HCFbinding motif; ERα, estrogen receptor α bFGF-2, basic fibroblast growth factor 2; AD, activation domain; luc, luciferase; WT, wild type.
us.expasy.org.
References
- 1.Kristie TM, Pomerantz JL, Twomey TC, Parent SA, Sharp PA. J Biol Chem. 1995;270:4387–4394. doi: 10.1074/jbc.270.9.4387. [DOI] [PubMed] [Google Scholar]
- 2.Wilson AC, LaMarco K, Peterson MG, Herr W. Cell. 1993;74:115–125. doi: 10.1016/0092-8674(93)90299-6. [DOI] [PubMed] [Google Scholar]
- 3.Wilson AC, Peterson MG, Herr W. Genes Dev. 1995;9:2445–2458. doi: 10.1101/gad.9.20.2445. [DOI] [PubMed] [Google Scholar]
- 4.Wilson AC, Boutros M, Johnson KM, Herr W. Mol Cell Biol. 2000;20:6721–6730. doi: 10.1128/mcb.20.18.6721-6730.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frattini A, Faranda S, Redolfi E, Zucchi I, Villa A, Patrosso MC, Strina D, Susani L, Vezzoni P. Genomics. 1994;23:30–35. doi: 10.1006/geno.1994.1455. [DOI] [PubMed] [Google Scholar]
- 6.Kristie TM, Vogel JL, Sears AE. Proc Natl Acad Sci U S A. 1999;96:1229–1233. doi: 10.1073/pnas.96.4.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu R, Yang P, O'Hare P, Misra V. Mol Cell Biol. 1997;17:5117–5126. doi: 10.1128/mcb.17.9.5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wysocka J, Reilly PT, Herr W. Mol Cell Biol. 2001;21:3820–3829. doi: 10.1128/MCB.21.11.3820-3829.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LaBoissière S, Walker S, O'Hare P. Mol Cell Biol. 1997;17:7108–7118. doi: 10.1128/mcb.17.12.7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simmen KA, Newell A, Robinson M, Mills JS, Canning G, Handa R, Parkes K, Borkakoti N, Jupp R. J Virol. 1997;71:3886–3894. doi: 10.1128/jvi.71.5.3886-3894.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson AC, Freiman RN, Goto H, Nishimoto T, Herr W. Mol Cell Biol. 1997;17:6139–6146. doi: 10.1128/mcb.17.10.6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LaBoissière S, Hughes T, O'Hare P. EMBO J. 1999;18:480–489. doi: 10.1093/emboj/18.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luciano RL, Wilson AC. Proc Natl Acad Sci U S A. 2002;99:13403–13408. doi: 10.1073/pnas.202200399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Hare P. Semin Virol. 1993;4:145–155. [Google Scholar]
- 15.Wysocka J, Herr W. Trends Biochem Sci. 2003;28:294–304. doi: 10.1016/S0968-0004(03)00088-4. [DOI] [PubMed] [Google Scholar]
- 16.Freiman RN, Herr W. Genes Dev. 1997;11:3122–3127. doi: 10.1101/gad.11.23.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunther M, Laithier M, Brison O. Mol Cell Biochem. 2000;210:131–142. doi: 10.1023/a:1007177623283. [DOI] [PubMed] [Google Scholar]
- 18.Lu R, Misra V. Nucleic Acids Res. 2000;28:2446–2454. doi: 10.1093/nar/28.12.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piluso D, Bilan P, Capone JP. J Biol Chem. 2002;277:46799–46808. doi: 10.1074/jbc.M206226200. [DOI] [PubMed] [Google Scholar]
- 20.Scarr RB, Sharp PA. Oncogene. 2002;21:5245–5254. doi: 10.1038/sj.onc.1205647. [DOI] [PubMed] [Google Scholar]
- 21.Vogel JL, Kristie TM. EMBO J. 2000;19:683–690. doi: 10.1093/emboj/19.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goto H, Motomura S, Wilson AC, Freiman RN, Nakabeppu Y, Fukushima K, Fujishima M, Herr W, Nishimoto T. Genes Dev. 1997;11:726–737. doi: 10.1101/gad.11.6.726. [DOI] [PubMed] [Google Scholar]
- 23.Reilly PT, Herr W. Exp Cell Res. 2002;277:119–130. doi: 10.1006/excr.2002.5551. [DOI] [PubMed] [Google Scholar]
- 24.Ajuh P, Chusainow J, Ryder U, Lamond AI. EMBO J. 2002;21:6590–6602. doi: 10.1093/emboj/cdf652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu R, Yang P, Padmakumar S, Misra V. J Virol. 1998;72:6291–6297. doi: 10.1128/jvi.72.8.6291-6297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luciano RL, Wilson AC. Proc Natl Acad Sci U S A. 2000;97:10757–10762. doi: 10.1073/pnas.190062797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. J Biol Chem. 2002;277:1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]
- 28.Mahajan SS, Little MM, Vazquez R, Wilson AC. J Biol Chem. 2002;277:44292–44299. doi: 10.1074/jbc.M205440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagarajan R, Svaren J, Le N, Araki T, Watson M, Milbrandt J. Neuron. 2001;30:355–368. doi: 10.1016/s0896-6273(01)00282-3. [DOI] [PubMed] [Google Scholar]
- 30.Trimarchi JM, Lees JA. Nat Rev Mol Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 31.McDonnell DP, Norris JD. Science. 2002;296:1642–1644. doi: 10.1126/science.1071884. [DOI] [PubMed] [Google Scholar]
- 32.Gattiker A, Bienvenut W, Bairoch A, Gasteiger E. Proteomics. 2002;10:1435–1444. doi: 10.1002/1615-9861(200210)2:10<1435::AID-PROT1435>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 33.Wang D, Russell JL, Johnson DG. Mol Cell Biol. 2000;20:3417–3424. doi: 10.1128/mcb.20.10.3417-3424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun P, Enslen H, Myung PS, Maurer RA. Genes Dev. 1994;8:2527–2539. doi: 10.1101/gad.8.21.2527. [DOI] [PubMed] [Google Scholar]
- 35.Su LF, Knoblauch R, Garabedian MJ. J Biol Chem. 2001;276:3231–3237. doi: 10.1074/jbc.M005547200. [DOI] [PubMed] [Google Scholar]
- 36.Biesiada E, Razandi M, Levine ER. J Biol Chem. 1996;271:18576–18581. doi: 10.1074/jbc.271.31.18576. [DOI] [PubMed] [Google Scholar]
- 37.Russo MW, Matheny C, Milbrandt J. Mol Cell Biol. 1993;13:6858–6865. doi: 10.1128/mcb.13.11.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu TJ, Monokian G, Mark DF, Wobbe CR. Mol Cell Biol. 1994;14:3484–3493. doi: 10.1128/mcb.14.5.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vesque C, Charnay P. Nucleic Acids Res. 1992;20:2485–2492. doi: 10.1093/nar/20.10.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Svaren J, Sevetson BR, Golda T, Stanton JJ, Swirnoff AH, Milbrandt J. EMBO J. 1998;17:6010–6019. doi: 10.1093/emboj/17.20.6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shibata F, Baird A, Florkiewicz RZ. Growth Factors. 1991;4:277–287. doi: 10.3109/08977199109043913. [DOI] [PubMed] [Google Scholar]
- 42.Näär AM, Lemon BD, Tjian R. Annu Rev Biochem. 2001;70:475–501. doi: 10.1146/annurev.biochem.70.1.475. [DOI] [PubMed] [Google Scholar]
- 43.Chan HM, La Thangue NB. J Cell Sci. 2001;114:2363–2373. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- 44.Wysocka J, Myers MP, Laherty CD, Eisenman RN, Herr W. Genes Dev. 2003;17:896–911. doi: 10.1101/gad.252103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lai JS, Herr W. Mol Cell Biol. 1997;17:3937–3946. doi: 10.1128/mcb.17.7.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heery DM, Kalkhoven E, Hoare S, Parker MG. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 47.Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG. Nature. 1997;387:677–684. doi: 10.1038/42652. [DOI] [PubMed] [Google Scholar]
- 48.Heery DM, Hoare S, Hussain S, Parker MG, Sheppard H. J Biol Chem. 2001;276:6695–6702. doi: 10.1074/jbc.M009404200. [DOI] [PubMed] [Google Scholar]
- 49.Coulthard VH, Matsuda S, Heery D. J Biol Chem. 2002;278:10942–10951. doi: 10.1074/jbc.M212950200. [DOI] [PubMed] [Google Scholar]
- 50.Rayman JB, Takahashi Y, Indjeian VB, Dannenberg JH, Catchpole S, Watson RJ, Riele H, Dynlacht BD. Genes Dev. 2002;16:933–947. doi: 10.1101/gad.969202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu M, Sheppard KA, Peng CY, Yee AS, Piwnica-Worms H. Mol Cell Biol. 1994;14:8420–8431. doi: 10.1128/mcb.14.12.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reilly PT, Wysocka J, Herr W. Mol Cell Biol. 2002;22:6767–6778. doi: 10.1128/MCB.22.19.6767-6778.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneider-Maunoury S, Topilko P, Seitandou T, Levi G, Cohen-Tannoudji M, Pournin S, Babinet C, Charnay P. Cell. 1993;75:1199–1214. doi: 10.1016/0092-8674(93)90329-o. [DOI] [PubMed] [Google Scholar]
- 54.Swiatek PJ, Gridley T. Genes Dev. 1993;7:2071–2084. doi: 10.1101/gad.7.11.2071. [DOI] [PubMed] [Google Scholar]
- 55.Jessen KR, Mirsky R. J Anat. 2002;200:367–376. doi: 10.1046/j.1469-7580.2002.00046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Venken K, Di Maria E, Bellone E, Balestra P, Cassandrini D, Mandich P, De Jonghe P, Timmerman V, Svaren J. Neurogenetics. 2002;4:37–41. doi: 10.1007/s10048-001-0124-2. [DOI] [PubMed] [Google Scholar]
- 57.Sevetson BR, Svaren J, Milbrandt J. J Biol Chem. 2000;275:9749–9757. doi: 10.1074/jbc.275.13.9749. [DOI] [PubMed] [Google Scholar]
- 58.Warner LE, Svaren J, Milbrandt J, Lupski JR. Hum Mol Genet. 1999;8:1245–1251. doi: 10.1093/hmg/8.7.1245. [DOI] [PubMed] [Google Scholar]