

Abstract
Neuromyelitis optica (NMO) pathogenesis involves binding of anti-aquaporin-4 (AQP4) autoantibodies (NMO-IgG) present in serum to AQP4 on astrocytes, which causes complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC). Human immunoglobulin G (hIgG) is effective for treatment of humorally mediated neurological autoimmune diseases and has been reported to improve disease outcome in a limited number of NMO patients. Here, we investigated hIgG actions on NMO-IgG pathogenicity using an in vivo rat model of NMO and in vitro assays. In rats administered NMO-IgG by intracerebral injection, the size of neuroinflammatory demyelinating lesions was reduced by ∼ 50 % when hIgG was administered by intraperitoneal injection to reach levels of 10-25 mg/mL in rat serum, comparable with human therapeutic levels. In vitro, hIgG at 10 mg/mL reduced by 90 % NMO-IgG-mediated CDC following addition of NMO-IgG and human complement to AQP4-expressing cells. The hIgG effect was mainly on the classical complement pathway. hIgG at 10 mg/mL also reduced by up to 90 % NMO-IgG-mediated ADCC as assayed with human natural killer cells as effector cells. However, hIgG at up to 40 mg/mL did not affect AQP4 cell surface expression or its supramolecular assembly in orthogonal arrays of particles, nor did it affect NMO-IgG binding to AQP4. We conclude that hIgG reduces NMO-IgG pathogenicity by inhibition of CDC and ADCC, providing a mechanistic basis to support further clinical evaluation of its therapeutic efficacy in NMO.
Keywords: AQP4, NMO, human immunoglobulin, astrocyte, complement, neuroinflammation
Introduction
Neuromyelitis optica (NMO) is an inflammatory demyelinating disease of the central nervous system (CNS) causing optic neuritis and transverse myelitis. A defining feature of NMO is the presence in serum of anti-aquaporin-4 (AQP4) autoantibodies (NMO-IgG) that bind to water channel AQP4 on astrocyte end-feet (Lennon et al., 2005). In vitro assays and animal models suggests that NMO pathogenesis involves NMO-IgG binding to AQP4 on astrocytes causing complement-dependent cytotoxicity (CDC), which is supported by findings of early loss of AQP4 and GFAP in human NMO lesions with perivascular immunoglobulin and complement deposition (Lucchinetti et al., 2002; Misu et al., 2007). The primary astrocyte cytotoxicity results in blood-brain barrier disruption, recruitment and degranulation of inflammatory cells (granulocytes and macrophages), and secondary oligodendrocyte injury and myelin loss (Papadopoulos and Verkman, 2012). Intracerebral injection in mice of NMO-IgG and human complement (Saadoun et al., 2010), or in rats of NMO-IgG alone (Asavapanumas et al., 2014), produces NMO-like pathology with astrocyte cytotoxicity, complement deposition, inflammation and demyelination. A significant role of antibody-dependent cellular cytotoxicity (ADCC) has also been demonstrated in NMO, as mice administered a mutated NMO-IgG lacking ADCC effector function showed reduced pathology as did mice treated with a Fcγ receptor (FcγR) blocking antibody (Ratelade et al., 2013).
The therapeutic efficacy of human immunoglobulin G (hIgG) administered intravenously was first reported in 1981 in the autoimmune disease idiopathic thrombocytopenic purpura (ITP) (Imbach et al., 1981). hIgG has since been used for the treatment of a broad range of immune-mediated demyelinating diseases of the nervous system including Guillain-Barré syndrome, chronic inflammatory demyelinating polyneuropathy, diabetic polyneuropathy, multifocal motor neuropathy, relapsing-remitting multiple sclerosis and myasthenia gravis (Gelfand, 2012). hIgG has been reported to have pleiotropic actions on the immune system, including accelerated clearance of autoantibodies, inhibition of complement deposition, interference with antigen recognition, and block of Fcγ receptors (Berger et al., 2013; Jacob and Rajabally, 2009). Other possible immunomodulatory actions of hIgG have been reported as well, including cytokine neutralization, inhibition of leukocyte migration, expansion of regulatory T cell populations, and dendritic cell activation (Jacob and Rajabally, 2009).
Limited reported data support the clinical benefit of hIgG in NMO (reviewed in Wingerchuk, 2013). hIgG has shown efficacy in the prevention of relapses in a small cohort of 8 NMO patients, with reduction in mean relapse rate from 1.8/yr in the year before hIgG treatment to 0.006/yr during a mean follow-up of 19.3 months (Magraner et al., 2013). The Expanded Disability Status Scale (EDSS) decreased from 3.3 to 2.6 in the hIgG-treated group. Other case studies also support a beneficial effect of hIgG in preventing relapse in NMO (Bakker and Metz, 2004; Okada et al., 2007). hIgG efficacy has also been suggested for treatment of acute NMO relapses, with clinical improvement seen in five out of 11 relapses in 10 patients reported in a retrospective study, with decreased EDSS from 7 to 6.5 at a median of 2 months after hIgG (Elsone et al., 2013).
Here, we tested the efficacy of hIgG in a rat model of NMO and investigated its potential cellular mechanism(s) of actions. In vitro studies of hIgG effects on each of the major steps in NMO pathogenesis suggested inhibition of CDC and ADCC as the principal mechanisms of hIgG clinical benefit in NMO.
Materials and Methods
Rats
Lewis rats were purchased from Charles River Lab (Wilmington, MA). Experiments were done using weight-matched female rats (100–200 g), age 8 to 12-weeks. Rats were housed and bred in the animal laboratory resource center at the University of California San Francisco. Protocols were approved by the University of California San Francisco Committee on Animal Research.
Antibodies and sera
Recombinant monoclonal NMO antibody rAb-53, which recognizes extracellular epitope(s) on AQP4, was generated from a clonally expanded plasmablast population from cerebrospinal fluid of an NMO patient, as described and characterized previously (Bennett et al., 2009; Crane et al., 2011). A chimeric NMO-IgG (NMO-IgGc), provided by Dr. Jeff Bennett (Univ. Colorado Denver), was generated by cloning the sequence of the variable region of heavy and light chains of rAb-53 upstream of the constant region of mouse IgG2a. NMO serum was obtained from seropositive individuals who met the revised diagnostic criteria for clinical disease, with non-NMO (seronegative) human serum as control. In some studies IgG was purified from pooled NMO serum (NMO-IgGserum) or control serum using a protein A-resin (GenScript, Piscataway, NY) and concentrated using Amicon Ultra Centrifugal Filter Units (Millipore, Billerica, MA). Human immunoglobulin (Privigen®, CSL Behring, Kankakee, IL) was dialysed against phosphate-buffered saline (PBS), which is the buffer used for the in vitro assays.
Rat model of NMO
Rats were anesthetized with intraperitoneal ketamine (75-100 mg/kg) and xylazine (5-10 mg/kg) and mounted in a stereotaxic frame, as described (Asavapanumas et al., 2013). Following a midline scalp incision, a burr hole of diameter 1 mm was made in the skull 3.5 mm to the right of the bregma. A 30-gauge needle attached to 50-μl gas-tight glass syringe (Hamilton, Reno, NV) was inserted 5 mm deep to infuse 750 μg NMO-IgGserum in a total volume of 10 μl (at 1 μl/min). Rats were administered 2g/kg hIgG or bovine serum albumin (BSA) intraperitoneally 1 day before and 1 day after intracerebral injection of NMO-IgG (4 rats per group). Rats were anesthetized at 5 days following NMO-IgG administration and perfused through the left cardiac ventricle with 100 ml PBS and then 25 ml of PBS containing 4% paraformaldehyde (PFA). Brains were collected and processed for immunostaining. hIgG concentration in rat serum was measured with a human IgG Elisa kit according to manufacturers' protocol (Genway, San Diego, CA) and leukocytes were counted using a Hemavet 850 (Drew Scientific, Oxford, CT).
Immunostaining
Brains were post-fixed for 2 h in 4% PFA. Five micrometer-thick paraffin sections were immunostained at room temperature for 1 h with antibodies against AQP4 (1:200, Santa Cruz Biotechnology, Santa Cruz, CA), GFAP (1:100, Millipore), myelin basic protein (MBP) (1:200, Santa Cruz Biotechnology), ionized calcium-binding adaptor molecule-1 (Iba1; 1:1,000; Wako, Richmond, VA), albumin (1:200, Santa Cruz Biotechnology), neurofilament (1:200; Millipore), C5b-9 (1:50, Hycult Biotech, Uden, The Netherlands) or CD45 (1:10, BD Biosciences, San Jose, CA), followed by the appropriate fluorescent secondary antibody (1:200, Invitrogen, Carlsbad, CA) or biotinylated secondary antibody (1:500, Vector Laboratories, Burlingame, CA). Tissue sections were examined with a Leica (Wetzlar, Germany) DM 4000 B microscope. AQP4, GFAP and MBP immunonegative areas were defined by hand and quantified blindly by two different observers using ImageJ (http://rsbweb.nih.gov/ij/). Data are presented as area (mm2) of immunonegative area.
Cell culture
Chinese hamster ovary (CHO) cells stably expressing human AQP4-M23 (named CHO-AQP4 cells) were generated as described (Crane et al., 2011) and cultured at 37 °C in 5% CO2 95% air in F-12 Ham's Nutrient Mixture medium supplemented with 10% fetal bovine serum, 200 μg/ml geneticin (selection marker), 100 U/ml penicillin and 100 μg/ml streptomycin. Human natural killer cells (NK-cells) were purchased from Fox Chase Cancer Center (Philadelphia, PA) in which parental NK-92 cells (ATCC CRL-2407) were retrovirally-transduced to express the high-affinity 176V variant of the Fcγ receptor CD16 in pBMN-NoGFP (Yusa et al., 2002). Primary astrocyte cultures from rat neonatal brain were generated as described (Asavapanumas et al., 2013).
Quantification of cell surface AQP4
CHO-AQP4 cells or rat astrocytes were grown on coverglasses until confluence and incubated for 3 h with specified concentrations of hIgG or BSA at 37 °C. Cells were then washed extensively in cold PBS and blocked for 20 min in 1% BSA at 4 °C. Remaining AQP4 at the cell surface was labeled with 50 μg/ml NMO-IgGc at 4 °C for 1 h. Subsequently, NMO-IgGc was labeled by incubation for 1 h at 4 °C with goat anti-mouse IgG-conjugated Alexa Fluor 555 (1:200, Invitrogen), and the plasma membrane was stained using a fluorescent lectin, wheat germ agglutinin (WGA)-conjugated Alexa Fluor 488 (1:200; Invitrogen). Cells were then washed in cold PBS and fixed for 15 min in 4% PFA. Cell surface AQP4 was quantified as the (background-subtracted) ratio of red (surface AQP4) to green (plasma membrane) fluorescence using ImageJ, as described (Ratelade et al., 2011).
Super-resolution imaging
The size of plasma membrane AQP4 orthogonal array of particles (OAP) was measured by super-resolution (dSTORM) imaging, as described (Rossi et al., 2012; Smith et al., 2013). Briefly, CHO-AQP4 cells or rat astrocytes were treated with 40 mg/mL hIgG or BSA for 3 h at 37 °C and then fixed, permeabilized and stained sequentially with primary anti C-terminal AQP4 antibody (Santa Cruz Biotechnology) and goat anti-rabbit IgG-conjugated Alexa Fluor 647 secondary antibody (Invitrogen) at 1 μg/ml each in PBS containing 1% BSA. Single images of AQP4 distribution were acquired under TIRF conditions then Alexa Fluor 647 was switched to dark state by incubation of fixed cells with 100 mM β-mercaptoethylamine in PBS at pH 9.0 and increase of laser power (van de Linde et al., 2011). Spontaneous reactivation of individual fluorophores was measured over 10-20,000 frames. Detection and localization of individual fluorophores, drift correction and image reconstruction was performed with the QuickPALM plugin for Image J (Henriques et al., 2010). Image segmentation and analysis of array size was performed as described (Smith et al., 2013).
NMO-IgG and C1q binding
CHO-AQP4 cells were incubated for 1 h with specified concentrations of NMO-IgGc or NMO-IgGserum with hIgG or BSA (40 mg/mL) at 23 °C. Cells were then rinsed with PBS, fixed in 4% PFA for 15 min and permeabilized with 0.1% Triton X-100. Cells were blocked in 1% BSA and incubated for 30 min with 0.4 μg/mL polyclonal, C-terminal-specific rabbit anti-AQP4 antibody (Santa Cruz Biotechnology), then rinsed with PBS. Cells were then incubated for 30 min with 4 μg/mL goat anti-mouse IgG-conjugated Alexa Fluor 555 (to detect NMO-IgGc) or anti-human IgG-conjugated Alexa Fluor 555 (to detect NMO-IgGserum) and goat anti-rabbit IgG-conjugated Alexa Fluor 488 (to detect AQP4). NMO-IgG binding to AQP4 was quantified as described (Crane et al., 2011). To assay C1q binding CHO-AQP4 cells were incubated with 20 μg/mL rAb-53 for 1 h at 23 °C then with 120 μg/mL human C1q with or without hIgG (40 mg/mL) for 1 h at 23 °C, as described (Phuan et al., 2012). Cells were washed, fixed and C1q was stained with a rabbit FITC-conjugated anti-C1q antibody (GeneTex, Irvine, CA).
CDC and ADCC assays
CHO-AQP4 cells were grown in 96-well plates until confluence and incubated with 20 μg/mL rAb-53 for 1 h at 23 °C, as described (Ratelade et al., 2013). Cells were then washed extensively. For assay of CDC, human or rat complement was pre-incubated for 1 h at 4 °C with specified concentrations of hIgG or BSA and then added to the NMO-IgG-coated CHO-AQP4 cells for 1 h at 23 °C. For assay of ADCC, NK-cells were pre-incubated with hIgG or BSA for 1 h at 37 °C and then added to target cells for 2 h at 37 °C. Target cells were then washed extensively in PBS and viability was measured by addition of 20% AlamarBlue (Invitrogen) for 1 h at 37 °C. Fluorescence was measured with a plate reader at excitation/emission wavelengths of 560/590 nm. Percentage cell viability was computed as: [(sample – 100% lysis)/(no lysis – 100% lysis)] × 100 where ‘100% lysis’ is fluorescence of cells incubated in 1% Triton X-100 and ‘no lysis’ is fluorescence of cells incubated with human complement or NK-cells but no NMO-IgG. Hemolysis assays using IgM-coated sheep red blood cells (for assay of classical complement pathway) and uncoated rabbit red blood cells (for assay of alternative pathway) were performed according to manufacturers' instructions (Complement Technology Inc., Tyler, TX).
Statistics
Data are presented as mean ± S.E.M. Statistical comparisons were made using the non-parametric Mann-Whitney test when comparing two groups.
Results
hIgG reduces NMO pathology in a rat model of NMO
Recognizing the limited clinical data on hIgG in human NMO, we first investigated the potential beneficial effect of hIgG in a robust animal model of NMO produced by intracerebral injection of NMO-IgG in rats (Asavapanumas et al., 2013). At 5 days after NMO-IgG injection rats show astrocyte damage with loss of AQP4 and GFAP, blood-brain barrier leakage, perivascular deposition of activated complement, inflammatory cell infiltration and myelin loss. Surrounding this central, complement-dependent lesion is a ‘penumbra’ lesion with selective AQP4 loss. Importantly, the rat model does not require addition of exogenous human complement or pre-existing inflammation. In pilot studies, we determined the dose and frequency of hIgG administration to give serum levels comparable to therapeutic levels in hIgG-treated humans of 15-30 mg/mL. Fig. 1A shows the experimental design: rats were administered 2 g/kg hIgG (or BSA as control) by intraperitoneal injections at days 0 and 2, and NMO-IgG by intracerebral injection at day 1. Rats were sacrificed at day 6.
Figure 1. hIgG reduces NMO-IgG pathogenicity in rat brain in vivo.
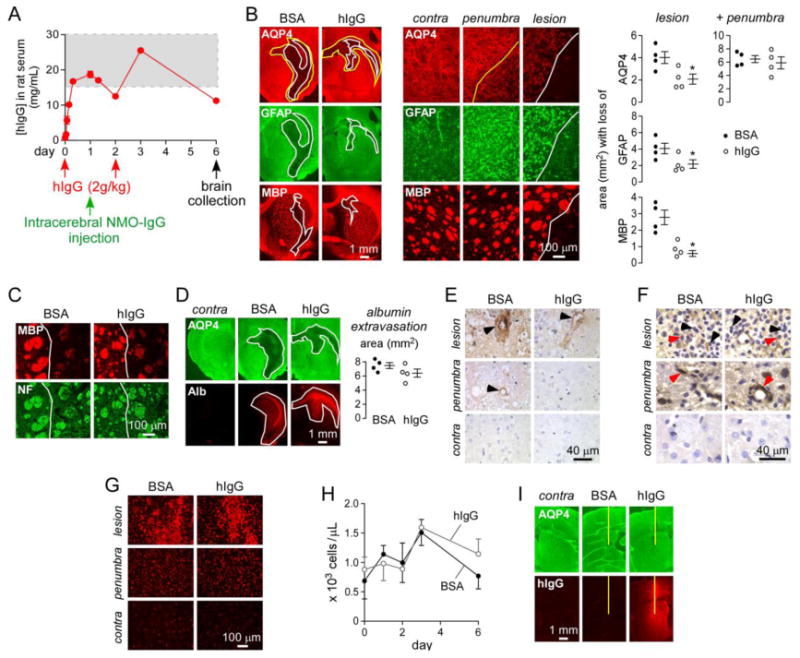
A. Rats were administered hIgG or BSA intraperitoneally at day 0 and day 2, and 750 μg NMO-IgGserum was injected intracerebrally at day 1. Brains were collected at day 6. Graph shows serum hIgG concentration as measured by human IgG ELISA (mean ± S.E., n=4). Dashed rectangle indicates therapeutic levels of hIgG in human serum. B. (left) AQP4, GFAP and myelin (MBP) immunofluorescence in BSA- and hIgG-treated rats. White line delimits the central lesion with loss of AQP4, GFAP and MBP. Yellow line delimits the penumbra area with selective loss of AQP4. (middle) Higher magnification of the contralateral (non-injected) hemisphere, penumbra and lesion. (right) Area with loss of AQP4, GFAP and MBP in the central and total (central + penumbra) lesion (mean ± S.E., n=4, * P < 0.05). C. MBP and neurofilament (NF) immunofluorescence in the central lesion (white line) of rats studied in A. D. Staining for AQP4 and albumin (Alb) in BSA- and hIgG-treated rats. Contra: contralateral non-injected brain hemisphere. White line delimits the area of AQP4 loss and albumin extravasation. (right) Area of albumin extravasation (mean ± S.E., n=4). E. C5b-9 staining in lesion, penumbra and contralateral hemisphere. Arrowheads denote perivascular complement deposition. F. CD45 immunohistochemistry. Black arrowhead shows granulocytes and red arrowhead macrophages. G. Iba1 immunofluorescence for microglia/macrophages. H. Neutrophil blood counts in BSA- and hIgG-treated rats (mean ± S.E., n=4). I. Anti-human IgG staining of brains from rats administered intraperitoneally with hIgG or BSA followed by intracerebral injection of PBS. Contra: contralateral non-injected brain hemisphere. Yellow line represents needle tract.
Serum concentrations of hIgG in rats measured using a human IgG Elisa were between 10 and 25 mg/mL during the course of the study (Fig. 1A). Brain immunofluorescence in rats administered NMO-IgG (with BSA as control) showed a central lesion with loss of AQP4, GFAP and myelin (MBP) (Fig. 1B, left and central panels, white line), surrounded by a penumbra with selective loss of AQP4 (yellow line). Rats injected with control (non-NMO) IgG did not develop central or penumbra pathology (not shown). The size of the central lesion was reduced by ∼ 50% in hIgG-treated rats compared to BSA-treated controls, as seen by immunofluorescence and quantified by lesion size (Fig. 1B, right panel). However, the total area (central lesion + penumbra) with loss of AQP4 was not reduced by hIgG.
Staining with MBP and a neurofilament antibody showed demyelinating pathology in the central lesion with minor axonal damage (Fig. 1C). Blood-brain barrier leakage, as assessed by albumin extravasation, was similar in brains of hIgG- and BSA-treated rats (Fig. 1D) and corresponded to the area of AQP4 loss (central lesion + penumbra). Fig. 1E shows perivascular deposition of activated complement in the central lesion, as assessed by immunohistochemistry using an antibody against the complement membrane attack complex (MAC, C5b-9). C5b-9 antibody staining was reduced in brains of hIgG-treated rats. Very limited or no deposition of activated complement was seen in the penumbra or contralateral hemisphere. Inflammatory cell infiltration was similar in hIgG- and BSA-treated rats in the central lesion and penumbra as seen by CD45 staining showing granulocytes and macrophages (Fig. 1F). Fig. 1G shows similar microglial activation/macrophage recruitment in penumbra and central lesion of hIgG- and BSA-treated rats. Blood counts for neutrophils (Fig. 1H), monocytes and lymphocytes (not shown) were not affected by hIgG administration over the time course of the experiment.
We studied the extravasation of hIgG in brain by staining with an anti-human IgG secondary antibody. For this experiment rats were administered 2 g/kg hIgG (or BSA as control) by intraperitoneal injection. After 6 hours, rats were injected intracerebrally with PBS (NMO-IgG was not injected as it would be detected by the anti-human IgG antibody). Rats were sacrificed after 24 h and brains were processed for staining. Fig. 1I shows that the needle insertion allows hIgG entry in the brain. No staining was seen in BSA-injected rats. Protection by hIgG in this model is thus likely due to its extravasation to the site of NMO-IgG deposition.
hIgG does not affect AQP4 surface expression or organization in orthogonal arrays of particles
In vitro experiments were done to investigate potential mechanisms of reduced NMO-IgG pathogenicity in the NMO rat model. The amount of cell surface AQP4 expression is an important determinant of NMO-IgG binding and pathogenicity. We investigated the effect of hIgG on AQP4 cell surface expression in primary cultures of rat astrocytes and in AQP4-transfected cells (CHO-AQP4 cells), testing the possibility that hIgG might contain AQP4-binding antibodies that induce AQP4 internalization, as found previously for NMO-IgG (Ratelade et al., 2011). Astrocytes and CHO-AQP4 cells were incubated for 3 h at 37 °C (temperature allowing for internalization) with hIgG or BSA as a control (Fig. 2A). Cells were washed extensively, cooled to 4 °C to block endocytosis, and cell surface AQP4 was labeled using a saturated amount of NMO-IgGc (antibody with mouse constant region, allowing distinction from non-specific deposition of hIgG on cells) and a red fluorescent anti-mouse IgG secondary antibody (Fig. 2A). Cell surface was labeled green with the lectin WGA. Fig. 2B shows no significant reduction in cell surface AQP4 in response to hIgG in CHO-AQP4 cells or rat astrocytes as quantified by the red-to-green fluorescence ratio, even at a concentration of 40 mg/mL, higher than human therapeutic levels of 15-30 mg/mL. A positive control for AQP4 internalization was obtained by incubation of CHO-AQP4 cells with NMO-IgG.
Figure 2. hIgG does not affect AQP4 cell surface expression or assembly in OAPs.
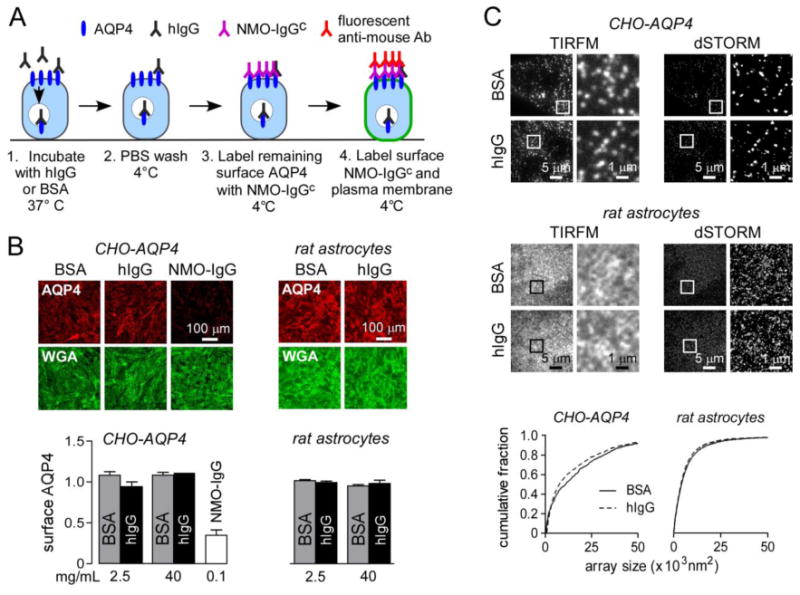
A. Experimental protocol to quantify cell surface AQP4. B. Fluorescence micrographs showing surface AQP4 (red) and plasma membrane staining (wheat germ agglutinin, WGA, green) in CHO-AQP4 cells and rat astrocytes incubated for 3 h at 37 °C with 40 mg/mL BSA or hIgG (or 100 μg/mL NMO-IgG as positive control). Graphs show cell surface AQP4 quantified as red to green fluorescence ratio (1.0 corresponds to cell surface AQP4 without BSA or hIgG) for indicated concentrations of BSA or hIgG (mean ± S.E., n=5). C. TIRFM and dSTORM imaging of AQP4 OAPs in CHO-AQP4 cells and rat primary astrocytes following incubation with 40 mg/mL BSA or hIgG for 3 h at 37 °C. Graphs represent OAP size distribution.
In addition to the level of AQP4 cell surface expression, the supramolecular assembly of AQP4 tetramers into orthogonal arrays of particles (OAP) is a major determinant of NMO-IgG binding (Crane et al., 2011) and CDC (Phuan et al., 2012). NMO-IgGs generally have relatively low affinity for individual tetramers consisting the long AQP4-M1 isoform and a higher affinity for OAPs containing many tetramers enriched in the short isoform AQP4-M23 (Crane et al., 2011). We also showed that, independent of NMO-IgG binding to AQP4, NMO-IgG-mediated CDC requires AQP4 OAP assembly to allow multivalent binding of complement protein C1q on clustered NMO-IgG (Phuan et al., 2012). Super-resolution (dSTORM) microscopy was used to investigate whether hIgG affected AQP4 OAP assembly. Astrocytes (expressing both AQP4 isoforms) or CHO-AQP4 cells (expressing only AQP4-M23) were incubated for 3 h at 37 °C with hIgG or BSA. Fig. 2C shows labeled OAPs stained with an anti-AQP4 antibody and a fluorescently labeled secondary antibody visualized by TIRFM and dSTORM. OAPs were smaller in primary rat astrocytes than in CHO-AQP4 cells because of the presence of the AQP4-M1 isoform. The OAP size distributions were not significantly affected by incubation with hIgG compared to BSA, indicating that hIgG does not alter AQP4 cell membrane organization.
hIgG does not interfere with NMO-IgG binding to AQP4
Anti-idiotype antibodies in hIgG could bind NMO-IgG, preventing its binding to AQP4. We studied the effect of hIgG on binding of the recombinant monoclonal chimeric NMO-IgGc and on polyclonal NMO-IgGserum purified from NMO serum. Binding of NMO-IgGc was detected with an anti-mouse secondary antibody, avoiding non-specific detection of background human immunoglobulins contained in hIgG. Fig. 3A shows the procedure to quantify NMO-IgGc binding to AQP4 in CHO-AQP4 cells. NMO-IgGc was added to live cells at room temperature with hIgG or BSA, cells are then washed and fixed, and AQP4 was stained using a C-terminus antibody. Binding of NMO-IgGc was quantified by a red (NMO-IgGc) to green (AQP4) fluorescence ratio, as described (Crane et al., 2011). Fig. 3B shows no effect of hIgG (40 mg/mL) on binding of the high affinity NMO-IgGc at non-saturating (2.5 μg/mL) and saturating (20 μg/mL) concentrations. In the experiment using human NMO-IgGserum, we used rat IgG instead of hIgG for selective detection of NMO-IgGserum with an anti-human IgG secondary antibody. Fig. 3C shows no inhibition of rat IgG (40 mg/mL) on polyclonal NMO-IgGserum binding to AQP4 at non-saturating (25 μg/mL) and saturating (100 μg/mL) concentrations of NMO-IgGserum.
Figure 3. hIgG does not affect NMO-IgG binding to AQP4.
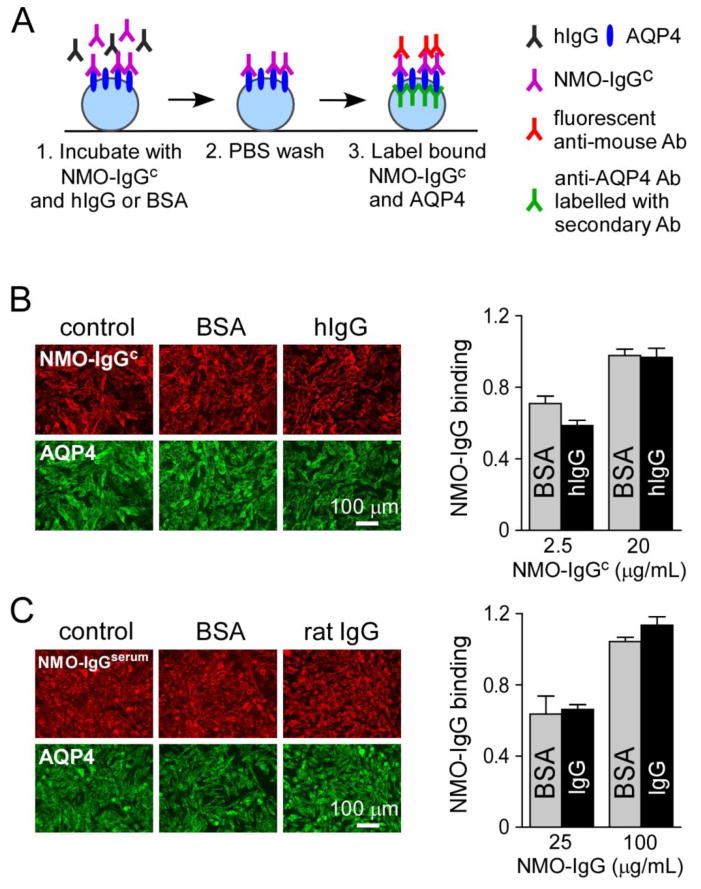
A. Experimental protocol to quantify NMO-IgGc binding to AQP4 in CHO-AQP4 cells. Cells were incubated with NMO-IgGc and 40 mg/mL hIgG or BSA for 1 h at 23 °C. Cells were then washed, fixed and permeabilized. Bound NMO-IgGc was labeled with a red fluorescent secondary antibody and AQP4 with a C-terminus primary antibody and green fluorescent secondary antibody. B. Fluorescence micrographs showing bound NMO-IgGc (red) to AQP4 (green) in CHO-AQP4 cells after 1-h incubation with 20 μg/mL NMO-IgGc and 40 mg/mL hIgG or BSA. (right) NMO-IgGc binding quantified as red to green fluorescence ratio at 2.5 and 20 μg/mL NMO-IgGc and 40 mg/mL BSA or hIgG (mean ± S.E., n=5). C. Fluorescence micrographs showing bound NMO-IgGserum (red) and AQP4 (green) in CHO-AQP4 cells after 1-h incubation with 100 μg/mL NMO-IgGserum and 40 mg/mL BSA or rat IgG. (right) NMO-IgGserum binding quantified as in B (mean ± S.E., n=5).
hIgG reduces NMO-IgG-mediated CDC by acting on the classical pathway
To study effects of NMO-IgG-dependent CDC, CHO-AQP4 cells were coated with NMO-IgG and washed. Separately, human complement was preincubated with hIgG or BSA and then added to the NMO-IgG-coated cells for 1 h at 23 °C (Fig. 4A). Cell viability was assayed using AlamarBlue. Fig. 4B (left) shows cytotoxicity for different amounts of human complement as a function of hIgG concentration. hIgG strongly inhibited NMO-IgG-mediated CDC, with 90 % inhibition at 10 mg/mL in the presence of 0.6 % complement. Fig. 4B (right) shows similar data using rat complement.
Figure 4. hIgG strongly inhibits NMO-IgG-mediated CDC and the classical complement pathway.
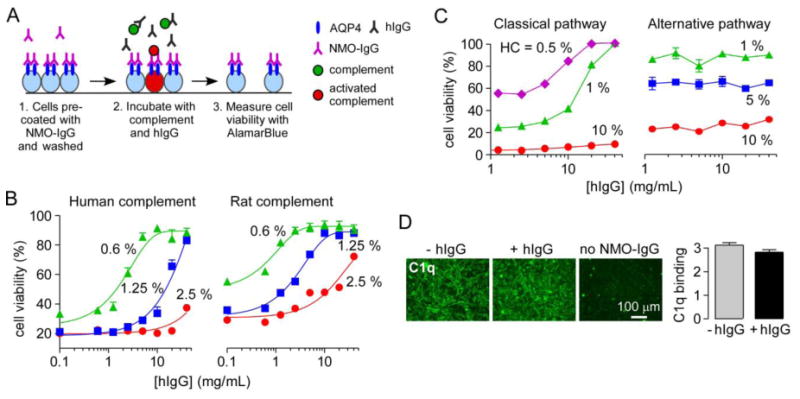
A. Experimental protocol to measure complement-dependent cytotoxicity in NMO-IgG-coated CHO-AQP4 cells. CHO-AQP4 cells were incubated with 20 μg/mL rAb-53 for 1 h at 23 °C and washed extensively. Complement was pre-incubated for 1 h at 4 °C with different concentrations of hIgG and then added to the NMO-IgG-coated CHO-AQP4 cells for 1 h at 23 °C. Target cells were then washed extensively in PBS and viability was measured by addition of 20% AlamarBlue for 1 h at 37 °C. B. Cell viability as a function of hIgG concentration for indicated concentrations of human or rat complement (mean ± S.E., n=3). C. Cell viability in IgM-coated sheep red blood cells (assay of classical pathway) and uncoated rabbit red blood cells (assay of alternative pathway) for different concentrations of human complement (HC) (mean ± S.E., n=3). D. C1q binding to NMO-IgG-coated CHO-AQP4 cells with or without 40 mg/mL hIgG. Graph shows C1q binding as fluorescence intensity (mean ± S.E., n=5).
To investigate whether hIgG action involves the classical and/or alternative complement pathways, hemolysis assays were done with sheep red blood cells coated with IgM for the classical pathway and rabbit red blood cells in calcium and magnesium-free conditions (to inhibit assembly of the C1 complex) for the alternative pathway. Fig. 4C shows that 20 mg/mL hIgG inhibited hemolysis of sheep red blood cells by nearly 100 % in presence of 0.5 % human complement, but did not inhibit lysis of rabbit red blood cells. We conclude that hIgG inhibits mainly the classical complement pathway. We investigated the possibility that hIgG might inhibit C1q binding to cell-bound IgG, which is the initiating event in the classical pathway. Purified human C1q protein was added to CHO-AQP4 cells coated with NMO-IgG in absence or presence of hIgG. Cells were then washed and bound C1q was detected with a FITC-conjugated antibody. No significant difference in C1q binding to NMO-IgG was seen in presence of a high concentration (40 mg/mL) of hIgG (Fig. 4D). No C1q binding was seen in absence of NMO-IgG.
hIgG reduces NMO-IgG-mediated ADCC in vitro
Because of the involvement of ADCC in NMO, we also studied the inhibitory effect of hIgG on ADCC. An in vitro assay of ADCC was performed in which human NK-cells were used as effector cells, as described previously (Ratelade et al., 2012). NK-cells were pre-incubated with hIgG or BSA and then added to NMO-IgG-coated CHO-AQP4 cells (Fig. 5A). Cell viability was measured with Alamar Blue. Fig. 5B shows a 90 % reduction of NMO-IgG-dependent cytotoxicity with 10 mg/mL hIgG in presence of 5x104 NK-cells per well.
Figure 5. hIgG strongly inhibits NMO-IgG-mediated ADCC.
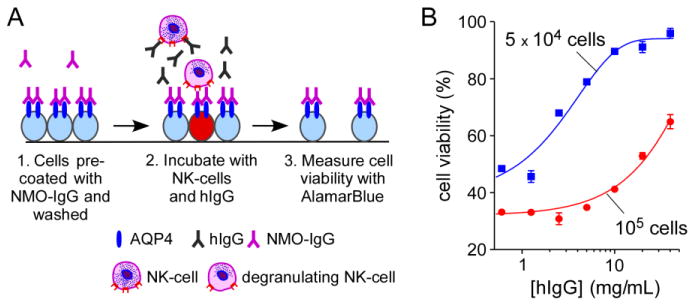
A. Experimental protocol to measure cytotoxicity produced by NK-cell addition to NMO-IgG-coated CHO-AQP4 cells. CHO-AQP4 cells were incubated with 20 μg/mL rAb-53 for 1 h at 23 °C and washed extensively. NK-cells were pre-incubated with hIgG for 1 h at 37 °C and then added to target cells for 2 h at 37 °C. Target cells were then washed extensively in PBS and viability was measured by addition of 20% AlamarBlue for 1 h at 37 °C. B. Cell viability as a function of hIgG concentration for different numbers of NK-cells added per well (mean ± S.E., n=3).
Discussion
We found that hIgG at therapeutic concentrations (when used in humans) reduced NMO pathology in a rat model of NMO produced by intracerebral injection of NMO-IgG. hIgG inhibited NMO-IgG-dependent astrocyte damage, complement activation and myelin loss, but did not affect the penumbra area around the central lesion with selective loss of AQP4. hIgG in vitro did not reduce cell surface AQP4 expression or inhibit its organization in OAPs, nor did it inhibit the binding of NMO-IgG to AQP4. Remarkably, however, hIgG strongly inhibited NMO-IgG-dependent CDC and ADCC in AQP4-expressing cell cultures.
hIgG was first used as IgG replacement therapy in patients with hypogammaglobulinemia. In 1981, its use in children with ITP gave a reproducible increase in platelet count (Imbach et al., 1981). The current FDA-approved indications for hIgG are limited to primary immunodeficiency disease, chronic lymphocytic leukemia, Kawasaki's disease, chronic inflammatory demyelinating polyneuropathy and multifocal motor neuropathy; however, hIgG is often used for off-label indications in the United States (Gelfand, 2012). The immunomodulatory actions of hIgG have produced benefit in several autoimmune neuromuscular and demyelinating disorders (Winkelmann and Zettl, 2012). Some of these disorders have similar features to those of NMO such as a humorally mediated mechanism with autoantibodies against a target expressed in the nervous system, and involvement of complement activation. hIgG is first-line therapy for Guillain-Barré syndrome, a cause of acute neuromuscular paralysis, in which serum autoantibodies against gangliosides expressed on peripheral nerves are found in ∼50% of patients (van Doorn et al., 2008). Although the presence of anti-ganglioside antibodies correlates with clinical symptoms, their pathogenicity has not been proven in humans. In mice, anti-ganglioside antibodies are toxic to peripheral nerves by a mechanism involving complement activation (Halstead et al., 2008). hIgG is also a first-line therapy in multifocal motor neuropathy in which anti-ganglioside antibodies targeting motor neurons (anti-GM1-IgM) are found in up to 43 % of the patients (Winkelmann and Zettl, 2012). hIgG has been shown to block complement activation mediated by anti-GM1 antibodies (Piepers et al., 2010; Yuki et al., 2011). In experimental animals, hIgG reduces the severity of experimental autoimmune encephalomyelitis by several potential mechanisms, including expansion of a specific regulatory T cell population, neutralization of the pro-inflammatory cytokine TNFα, and inhibition of metalloproteinase activity (Ephrem et al., 2008; Niimi et al., 2011; Weishaupt et al., 2002). Finally, hIgG has also shown efficacy in myasthenia gravis (Kim et al., 2011), an autoimmune disease that sometimes accompanies NMO (Jarius et al., 2012; Leite et al., 2012), in which autoantibodies against acetylcholine receptors (AChRs) at the neuromuscular junction cause complement activation (Kim et al., 2011).
As mentioned in the Introduction, the clinical evince for hIgG efficacy in NMO is currently limited, but hIgG seems to be beneficial for prevention and treatment of relapses. Fig. 6 diagrams the major mechanisms by which hIgG could reduce NMO-IgG pathogenicity in NMO, as based upon human NMO pathology as well as results in NMO rodent models and the study here. We found here that hIgG did not affect AQP4 cell surface expression or its organization in OAPs in primary astrocyte cultures in vitro. Though not studied here because of the limitations of current animal models, hIgG could accelerate NMO-IgG catabolism or suppress its secretion by plasmablasts. Previous work suggested that hIgG could saturate the recycling receptor FcRn on endothelial cells and decrease the half-life of pathogenic antibodies in blood (Li et al., 2005). A previous study demonstrated that hIgG reduced blood-brain barrier disruption in a cecal puncture model of sepsis (Esen et al., 2012), though it is difficult to establish whether this was a direct effect or a secondary effect due to a global antiinflammatory response. Here, we found comparable albumin extravasation, a measure of blood-brain barrier integrity, in control and hIgG-treated rats.
Figure 6. Potential mechanisms of hIgG benefit in NMO.
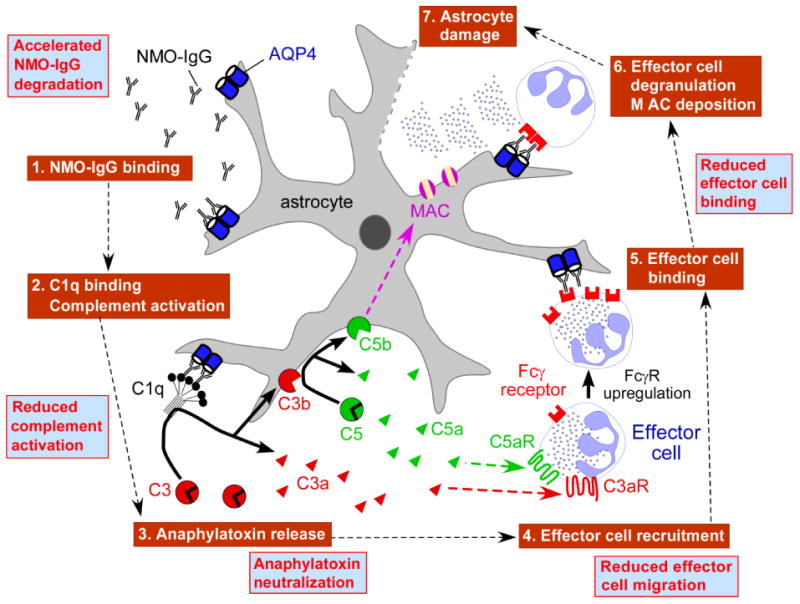
Schematic showing NMO pathogenesis mechanism. Boxes in blue show potential beneficial effects of hIgG. MAC: membrane attack complex. FcγR: Fcγ receptor.
We found that hIgG did not affect NMO-IgG binding to AQP4. It has been suggested that antiidiotype antibodies against pathogenic autoantibodies may be present in hIgG and suppress their binding to target antigens (Gelfand, 2012). The lack of effect of hIgG on NMO-IgG binding to AQP4 suggests that anti-idiotype antibodies are not involved in the protective effect of hIgG, which is not unexpected, as NMO serum probably contains many pathogenic anti-AQP4 antibodies targeting various 3-dimensional epitopes on the AQP4 extracellular surface (Nicchia et al., 2009).
A major effect of hIgG is inhibition of complement activation (Berger et al., 2013). hIgG has been reported to decrease binding/activation of C1q, C3a, C3b and C4 and thus prevent the formation of the membrane attack complex (Gelfand, 2012; Piepers et al., 2010). We found here that hIgG reduced the size of the central lesion in the rat intracerebral injection model, characterized by loss of AQP4, GFAP and MBP. We previously reported that this central lesion is largely complement-dependent, as inhibition of endogenous complement with cobra venom factor or injection of a modified NMO-IgG lacking CDC effector function produced very little pathology (Asavapanumas et al., 2014). Inhibition of complement activation by hIgG is supported by reduced MAC deposition in hIgG-treated rats, and the CDC assays in vitro. By using complementary hemolysis assays we concluded that hIgG inhibits mainly the complement classical pathway, which is consistent with a report showing decreased serum concentrations of C1q and C4 in hIgG-treated patients (Piepers et al., 2010).
hIgG is also reported to modulate pro-inflammatory cytokine signaling and neutralize anaphylatoxins C3a and C5a (Schwab and Nimmerjahn, 2013), which may be relevant to its beneficial effect in NMO. hIgG decreased IL-1β and IL6 production in a spinal cord injury model (Nguyen et al., 2012). IL6 is elevated in NMO and promotes NMO-IgG secretion by plasmablasts, and IL6 receptor block by the monoclonal antibody tocilizumab has been shown to be beneficial in NMO (Chihara et al., 2011).
Another effect of hIgG is saturation and inhibition of activating Fcγ receptors (Schwab and Nimmerjahn, 2013), which are expressed on immune cells and bind the Fc portion of IgG resulting in effector cell activation with ADCC and/or phagocytosis. The involvement of neutrophils, eosinophils and macrophages in NMO pathogenesis has been demonstrated in animal models of NMO (Asavapanumas et al., 2013; Saadoun et al., 2012; Zhang and Verkman, 2013), and they represent the major leukocyte types present in human NMO lesions (Lucchinetti et al., 2002; Misu et al., 2007). Also, we previously showed that ADCC mediated by NMO-IgG is required for development of a full NMO lesion (Ratelade et al., 2013). hIgG inhibition of NMO-IgG-dependent ADCC, as shown here in vitro, may thus account, in part, for its beneficial effect in NMO. However, the in vivo data here showed that the penumbra region in the rat intracerebral model was not reduced by hIgG, suggesting that hIgG action on ADCC is not the primary mechanism of its beneficial effect in this model, as we previously showed that penumbra pathology is ADCC-dependent (Asavapanumas et al., 2014).
hIgG thus has pleiotropic immunomodulatory effects, with the evidence here supporting a major effect on complement activation in accounting for its beneficial effect in NMO. Of note, several immune mechanisms targeted by potential therapies under consideration for NMO are also targeted by hIgG, including eculizumab and alternative monoclonal antibodies for complement inhibition (Phuan et al., 2013), rituximab for B cell depletion (Kim et al., 2013), and tocilizumab for IL6 receptor block (Chihara et al., 2011). As hIgG targets the major immune mechanisms in NMO, its multiple actions may produce more benefit than individual therapeutics targeting a single mechanism in NMO, and with fewer side effects. The limited anecdotal evidence for hIgG efficacy in NMO, the documented immunomodulatory actions of hIgG, and the mechanistic studies here support controlled clinical studies of hIgG in NMO.
Highlights.
hIgG reduces lesion severity in a rat model of neuromyelitis optica
hIgG does not affect AQP4 cell surface expression or NMO-IgG binding to AQP4
hIgG reduces NMO-IgG-mediated complement and cellular cytotoxicity
Acknowledgments
This work was supported by grants EY13574, DK35124, EB00415 and DK72517 from the National Institutes of Health, a grant from the Guthy-Jackson Charitable Foundation, and a sponsored research agreement from CSL Behring (Kankakee, IL). We thank Dr. Jeffrey Bennett (Univ. Colorado Denver, Aurora, CO) for providing recombinant monoclonal NMO antibody, Dr. Mel Berger (CSL Behring) for advice, and Accelerated Cure (Waltham, MA) for providing human NMO sera.
Abbreviations
- ADCC
antibody-dependent cellular cytotoxicity
- AQP4
aquaporin-4
- BSA
bovine serum albumin
- CDC
complement-dependent cytotoxicity
- CNS
central nervous system
- dSTORM
direct stochastic reconstruction microscopy
- EDSS
expanded disability status scale
- FcγR
Fcγ receptor
- GFAP
glial fibrillary acidic protein
- hIgG
human immunoglobulin G
- ITP
idiopathic thrombocytopenic purpura
- MAC
membrane attack complex
- MBP
myelin basic protein
- NK-cell
natural killer cell
- NMO
neuromyelitis optica
- NMO-IgG
neuromyelitis optica immunoglobulin G antibody
- OAP
orthogonal array of particles
- PFA
paraformaldehyde
- TIRF
total internal reflection fluorescence
- WGA
wheat germ agglutinin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Asavapanumas N, Ratelade J, Verkman AS. Unique neuromyelitis optica pathology produced in naive rats by intracerebral administration of NMO-IgG. Acta Neuropathol. 2014 doi: 10.1007/s00401-013-1204-8. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker J, Metz L. Devic's neuromyelitis optica treated with intravenous gamma globulin (IVIG) Can J Neurol Sci. 2004;31:265–267. doi: 10.1017/s0317167100053932. [DOI] [PubMed] [Google Scholar]
- Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, Glogowska M, Case D, Antel JP, Owens GP, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66:617–629. doi: 10.1002/ana.21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M, McCallus DE, Lin CS. Rapid and reversible responses to IVIG in autoimmune neuromuscular diseases suggest mechanisms of action involving competition with functionally important autoantibodies. J Peripher Nerv Syst. 2013 doi: 10.1111/jns5.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chihara N, Aranami T, Sato W, Miyazaki Y, Miyake S, Okamoto T, Ogawa M, Toda T, Yamamura T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci U S A. 2011;108:3701–3706. doi: 10.1073/pnas.1017385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JM, Lam C, Rossi A, Gupta T, Bennett JL, Verkman AS. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin-4 m1/m23 isoforms and orthogonal arrays. J Biol Chem. 2011;286:16516–16524. doi: 10.1074/jbc.M111.227298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsone L, Panicker J, Mutch K, Boggild M, Appleton R, Jacob A. Role of intravenous immunoglobulin in the treatment of acute relapses of neuromyelitis optica: experience in 10 patients. Mult Scler. 2013 doi: 10.1177/1352458513495938. [DOI] [PubMed] [Google Scholar]
- Ephrem A, Chamat S, Miquel C, Fisson S, Mouthon L, Caligiuri G, Delignat S, Elluru S, Bayry J, Lacroix-Desmazes S, et al. Expansion of CD4+CD25+ regulatory T cells by intravenous immunoglobulin: a critical factor in controlling experimental autoimmune encephalomyelitis. Blood. 2008;111:715–722. doi: 10.1182/blood-2007-03-079947. [DOI] [PubMed] [Google Scholar]
- Esen F, Senturk E, Ozcan PE, Ahishali B, Arican N, Orhan N, Ekizoglu O, Kucuk M, Kaya M. Intravenous immunoglobulins prevent the breakdown of the blood-brain barrier in experimentally induced sepsis. Crit Care Med. 2012;40:1214–1220. doi: 10.1097/CCM.0b013e31823779ca. [DOI] [PubMed] [Google Scholar]
- Gelfand EW. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med. 2012;367:2015–2025. doi: 10.1056/NEJMra1009433. [DOI] [PubMed] [Google Scholar]
- Halstead SK, Zitman FM, Humphreys PD, Greenshields K, Verschuuren JJ, Jacobs BC, Rother RP, Plomp JJ, Willison HJ. Eculizumab prevents anti-ganglioside antibody-mediated neuropathy in a murine model. Brain. 2008;131:1197–1208. doi: 10.1093/brain/awm316. [DOI] [PubMed] [Google Scholar]
- Henriques R, Lelek M, Fornasiero EF, Valtorta F, Zimmer C, Mhlanga MM. QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat Methods. 2010;7:339–340. doi: 10.1038/nmeth0510-339. [DOI] [PubMed] [Google Scholar]
- Imbach P, Barandun S, d'Apuzzo V, Baumgartner C, Hirt A, Morell A, Rossi E, Schoni M, Vest M, Wagner HP. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet. 1981;1:1228–1231. doi: 10.1016/s0140-6736(81)92400-4. [DOI] [PubMed] [Google Scholar]
- Jacob S, Rajabally YA. Current proposed mechanisms of action of intravenous immunoglobulins in inflammatory neuropathies. Curr Neuropharmacol. 2009;7:337–342. doi: 10.2174/157015909790031166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarius S, Paul F, Franciotta D, de Seze J, Munch C, Salvetti M, Ruprecht K, Liebetrau M, Wandinger KP, Akman-Demir G, et al. Neuromyelitis optica spectrum disorders in patients with myasthenia gravis: ten new aquaporin-4 antibody positive cases and a review of the literature. Mult Scler. 2012;18:1135–1143. doi: 10.1177/1352458511431728. [DOI] [PubMed] [Google Scholar]
- Kim JY, Park KD, Richman DP. Treatment of myasthenia gravis based on its immunopathogenesis. J Clin Neurol. 2011;7:173–183. doi: 10.3988/jcn.2011.7.4.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Huh SY, Lee SJ, Joung A, Kim HJ. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013;70:1110–1117. doi: 10.1001/jamaneurol.2013.3071. [DOI] [PubMed] [Google Scholar]
- Leite MI, Coutinho E, Lana-Peixoto M, Apostolos S, Waters P, Sato D, Melamud L, Marta M, Graham A, Spillane J, et al. Myasthenia gravis and neuromyelitis optica spectrum disorder: a multicenter study of 16 patients. Neurology. 2012;78:1601–1607. doi: 10.1212/WNL.0b013e31825644ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Zhao M, Hilario-Vargas J, Prisayanh P, Warren S, Diaz LA, Roopenian DC, Liu Z. Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. J Clin Invest. 2005;115:3440–3450. doi: 10.1172/JCI24394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, Trebst C, Weinshenker B, Wingerchuk D, Parisi JE, et al. A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain. 2002;125:1450–1461. doi: 10.1093/brain/awf151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magraner MJ, Coret F, Casanova B. The effect of intravenous immunoglobulin on neuromyelitis optica. Neurologia. 2013;28:65–72. doi: 10.1016/j.nrl.2012.03.014. [DOI] [PubMed] [Google Scholar]
- Misu T, Fujihara K, Kakita A, Konno H, Nakamura M, Watanabe S, Takahashi T, Nakashima I, Takahashi H, Itoyama Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain. 2007;130:1224–1234. doi: 10.1093/brain/awm047. [DOI] [PubMed] [Google Scholar]
- Nguyen DH, Cho N, Satkunendrarajah K, Austin JW, Wang J, Fehlings MG. Immunoglobulin G (IgG) attenuates neuroinflammation and improves neurobehavioral recovery after cervical spinal cord injury. J Neuroinflammation. 2012;9:224. doi: 10.1186/1742-2094-9-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicchia GP, Mastrototaro M, Rossi A, Pisani F, Tortorella C, Ruggieri M, Lia A, Trojano M, Frigeri A, Svelto M. Aquaporin-4 orthogonal arrays of particles are the target for neuromyelitis optica autoantibodies. Glia. 2009;57:1363–1373. doi: 10.1002/glia.20855. [DOI] [PubMed] [Google Scholar]
- Niimi N, Kohyama K, Kamei S, Matsumoto Y. Intravenous immunoglobulin therapy prevents development of autoimmune encephalomyelitis and suppresses activation of matrix metalloproteinases. Neuropathology. 2011;31:392–400. doi: 10.1111/j.1440-1789.2010.01183.x. [DOI] [PubMed] [Google Scholar]
- Okada K, Tsuji S, Tanaka K. Intermittent intravenous immunoglobulin successfully prevents relapses of neuromyelitis optica. Intern Med. 2007;46:1671–1672. doi: 10.2169/internalmedicine.46.0217. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012;11:535–544. doi: 10.1016/S1474-4422(12)70133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuan PW, Ratelade J, Rossi A, Tradtrantip L, Verkman AS. Complement-dependent cytotoxicity in neuromyelitis optica requires aquaporin-4 protein assembly in orthogonal arrays. J Biol Chem. 2012;287:13829–13839. doi: 10.1074/jbc.M112.344325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuan PW, Zhang H, Asavapanumas N, Leviten M, Rosenthal A, Tradtrantip L, Verkman AS. C1q-targeted monoclonal antibody prevents complement-dependent cytotoxicity and neuropathology in in vitro and mouse models of neuromyelitis optica. Acta Neuropathol. 2013;125:829–840. doi: 10.1007/s00401-013-1128-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepers S, Jansen MD, Cats EA, van Sorge NM, van den Berg LH, van der Pol WL. IVIg inhibits classical pathway activity and anti-GM1 IgM-mediated complement deposition in MMN. J Neuroimmunol. 2010;229:256–262. doi: 10.1016/j.jneuroim.2010.08.023. [DOI] [PubMed] [Google Scholar]
- Ratelade J, Asavapanumas N, Ritchie AM, Wemlinger S, Bennett JL, Verkman AS. Involvement of antibody-dependent cell-mediated cytotoxicity in inflammatory demyelination in a mouse model of neuromyelitis optica. Acta Neuropathol. 2013;126:699–709. doi: 10.1007/s00401-013-1172-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratelade J, Bennett JL, Verkman AS. Evidence against Cellular Internalization in Vivo of NMO-IgG, Aquaporin-4, and Excitatory Amino Acid Transporter 2 in Neuromyelitis Optica. J Biol Chem. 2011;286:45156–45164. doi: 10.1074/jbc.M111.297275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratelade J, Zhang H, Saadoun S, Bennett JL, Papadopoulos MC, Verkman AS. Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathol. 2012;123:861–872. doi: 10.1007/s00401-012-0986-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Moritz TJ, Ratelade J, Verkman AS. Super-resolution imaging of aquaporin-4 orthogonal arrays of particles in cell membranes. J Cell Sci. 2012;125:4405–4412. doi: 10.1242/jcs.109603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–361. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S, Waters P, Macdonald C, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain. Ann Neurol. 2012;71:323–333. doi: 10.1002/ana.22686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol. 2013;13:176–189. doi: 10.1038/nri3401. [DOI] [PubMed] [Google Scholar]
- Smith AJ, Jin BJ, Ratelade J, Verkman AS. Aggregation state determines the localization and function of M1- and M23-aquaporin-4 in astrocytes. J Cell Biol. 2013 doi: 10.1083/jcb.201308118. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Linde S, Loschberger A, Klein T, Heidbreder M, Wolter S, Heilemann M, Sauer M. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat Protoc. 2011;6:991–1009. doi: 10.1038/nprot.2011.336. [DOI] [PubMed] [Google Scholar]
- van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barre syndrome. Lancet Neurol. 2008;7:939–950. doi: 10.1016/S1474-4422(08)70215-1. [DOI] [PubMed] [Google Scholar]
- Weishaupt A, Kuhlmann T, Schonrock LM, Toyka KV, Bruck W, Gold R. Effects of intravenous immunoglobulins on T cell and oligodendrocyte apoptosis in high-dose antigen therapy in experimental autoimmune encephalomyelitis. Acta Neuropathol. 2002;104:385–390. doi: 10.1007/s00401-002-0568-y. [DOI] [PubMed] [Google Scholar]
- Wingerchuk DM. Neuromyelitis optica: potential roles for intravenous immunoglobulin. J Clin Immunol. 2013;33(Suppl 1):S33–37. doi: 10.1007/s10875-012-9796-7. [DOI] [PubMed] [Google Scholar]
- Winkelmann A, Zettl UK. Use of intravenous immunoglobulin in the treatment of immune-mediated demyelinating diseases of the nervous system. Curr Pharm Des. 2012;18:4570–4582. doi: 10.2174/138161212802502314. [DOI] [PubMed] [Google Scholar]
- Yuki N, Watanabe H, Nakajima T, Spath PJ. IVIG blocks complement deposition mediated by anti-GM1 antibodies in multifocal motor neuropathy. J Neurol Neurosurg Psychiatry. 2011;82:87–91. doi: 10.1136/jnnp.2010.205856. [DOI] [PubMed] [Google Scholar]
- Yusa S, Catina TL, Campbell KS. SHP-1- and phosphotyrosine-independent inhibitory signaling by a killer cell Ig-like receptor cytoplasmic domain in human NK cells. J Immunol. 2002;168:5047–5057. doi: 10.4049/jimmunol.168.10.5047. [DOI] [PubMed] [Google Scholar]
- Zhang H, Verkman AS. Eosinophil pathogenicity mechanisms and therapeutics in neuromyelitis optica. J Clin Invest. 2013;123:2306–2316. doi: 10.1172/JCI67554. [DOI] [PMC free article] [PubMed] [Google Scholar]