

Abstract
Macrocyclic bis-carbocyanines are efficacious inhibitors of tau aggregation. To extend the structure activity relationship of this inhibitor class, N,N′-alkylene bis-thiacarbocyanines linked by three to nine carbon alkyl chains were synthesized and examined for inhibitory activity against recombinant human tau aggregation in vitro. At 10 micromolar concentration, inhibitory activity varied with linker length, with four methylene units being most efficacious. On the basis of absorbance spectroscopy measurements, linker length also affected compound folding and aggregation propensity, with a linker length of four methylene units being optimal for preserving open monomer conformation. These data suggest that inhibitory potency can be optimized through control of linker length, and that a contributory mechanism involves modulation of compound folding and aggregation.
Keywords: Alzheimer’s disease, tau, neurofibrillary tangle, cyanine dye, aggregation
Aggregation and accumulation of the microtubule-associated protein tau into neurofibrillary lesions accompanies several neurodegenerative disorders, including Alzheimer’s disease (AD) and certain forms of frontotemporal lobar degeneration.1 Because lesion formation correlates with neurodegeneration and cognitive decline, inhibitors of tau aggregation are being investigated as potential therapeutic agents for slowing progression of these disorders.2 Although small-molecule tau aggregation inhibitors have been reported, including acridine, phenothiazine, carbocyanine, and many other scaffold derivatives,3–7 the structural features responsible for inhibitory potency are not fully understood. To clarify the mechanism of tau aggregation inhibition, we have been characterizing the structure activity relationship (SAR) of carbocyanine derivatives, which are capable of inhibiting tau aggregation in vitro and in ex vivo brain slices prepared from transgenic animal models of tauopathy.5, 8–10 We found that inhibitory potency was influenced by the polarizability of constituent cyanine heterocycles and by the length of the polymethine bridge that connected them.5 Potency could be further augmented by linking carbocyanine units with alkyl chains to create macrocyclic, bis-carbocyanines.11 Although a previous report on bis-acridine inhibitors of prion conversion found that linker length represents an additional variable that can influence inhibitory potency,12 the mechanism underlying the observation was not examined. Moreover, whether such an approach may be effective against tau protein has not been reported. Here we address these issues by characterizing the SAR of bis-thiacarbocyanines in an in vitro assay for tau aggregation inhibition.
To create a novel library of inhibitors, N,N′-alkylene bis-thiacarbocyanines were synthesized from 2-methylbenzothiazole in two steps as described previously11 except that dibromo alkanes (Sigma-Aldrich Co) varying in length from 3 to 9 carbon atoms were used to prepare intermediate bis-quaternary salts 1a – 1g (Scheme 1). The final bis-carbocyanines 2a – 2g, which contained linkers of length 3 to 9 methylene units (Scheme 1), were purple/pink solids except 2g which was a brown oil with poor solubility in aqueous solution. Therefore, only compounds 2a – 2f were analyzed further as described below.
Scheme 1.
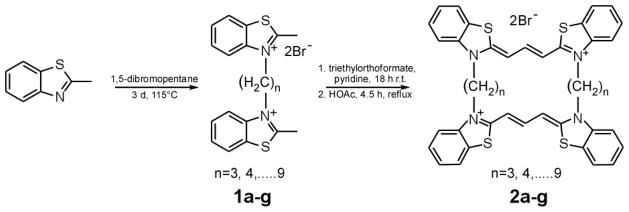
General Synthetic Scheme for the cyclic cyanines reported herein.11
To quantify their tau aggregation inhibitory activity, compounds 2a – 2f were incubated in the presence of full-length human tau isoform 2N4R and octadecyl sulfate (ODS) inducer under near-physiological conditions of pH, ionic strength, bulk tau concentration (4 μM), and reducing environment. The ODS inducer was included to greatly accelerate aggregation of full-length tau without the need for agitation.13 Near complete inhibition of aggregation was found for 2b at 1 μM final concentration (Fig. 1), whereas at least partial inhibition was found for all bis-carbocyanines at 10 μM (Fig. 2). Compounds 2b and 2d, which contained even-numbered methylene units in their linkers, were the most efficacious inhibitors at this concentration. These data reveal that linker length can in fact influence inhibitory potency of bis-carbocyanines.
Fig. 1.

Cyclic cyanines inhibit tau aggregation. Tau (2N4R isoform; 4 μM) was incubated (37°C for 22 h) without agitation in assembly buffer (10 mM HEPES, pH 7.4, 100 mM NaCl, 5 mM dithiothreitol) in the presence or absence of fibrillization inducer ODS (50 μM) and macrocyclic inhibitors. Control reactions contained DMSO vehicle (2% (v/v) final concentration). Reactions were stopped with glutaraldehyde, stained with uranyl acetate, and subjected to electron microscopy as described previously.15 (A) In the presence of DMSO vehicle control, 2N4R tau formed abundant filaments. (B) In the presence of 1 μM 2b, 2N4R tau aggregation was inhibited almost completely.
Fig. 2.
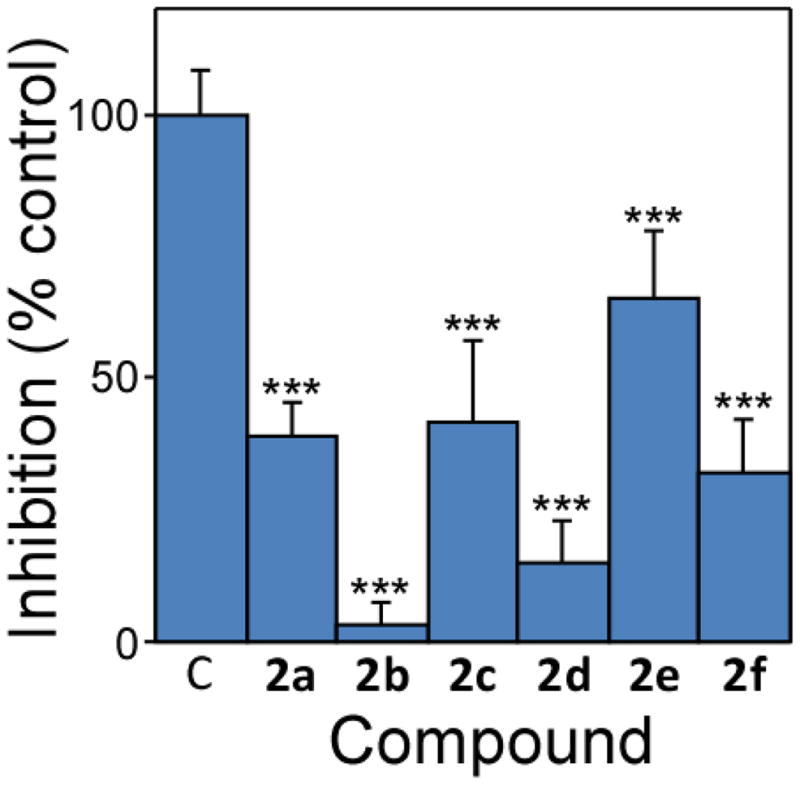
Compound efficacy varies with linker length. Tau (2N4R; 4 μM) was incubated without agitation in assembly buffer containing 50 μM ODS inducer and 10 μM 2a–f. Reaction products were then stained with uranyl acetate and viewed by electron microscopy. Each bar represents total filament length expressed as the normalized percentage of filament length measured in DMSO vehicle alone (quadruplicate determinations ± SD) as described previously.5 Data were analyzed by one-way ANOVA and Dunnett’s multiple comparison test (***, p < 0.001 compared to DMSO control reaction).
Cyclic carbocyanines undergo complex folding and aggregation reactions14 that may influence their tau aggregation antagonist activity.10, 11 Therefore, the effects of linker length on 2a–f folding were investigated using absorption spectroscopy over visible wavelengths 400–650 nm.10 In methanol solvent, which depresses compound aggregation,11, 14 all compounds showed two absorption optima: a sharp peak at 560 nm corresponding to the open monomer conformation, and a broader peak centered at 520 nm corresponding to the closed conformation (Fig. 3A).11, 14 Quantitatively, however, the compounds differed in relative peak heights, with some favoring the 560 nm (open) monomer conformation relative to the 520 nm (closed) conformation. Compound 2b, the most efficacious inhibitor at 1 and 10 μM concentrations, adopted primarily open monomer conformation under these conditions (Fig. 3A). In contrast, 2a, a compound that was inactive at 1 μM and weakly inhibitory at 10 μM adopted primarily closed conformation (Fig. 3A). Compounds with longer linkers (2c–f) populated both closed and open conformations that varied over a more narrow range (Fig. 3A). These data show that linker length influences compound folding, with the shortest linkers favoring discrete conformations.
Fig. 3.

Linker length influences cyclic cyanine conformation and aggregation state. Absorbance spectra for all compounds at 10 μM final concentration in (A) 100% MeOH, and (B) tau aggregation conditions (4 μM 2N4R tau, 50 mM ODS, assembly buffer; 22 h incubation). Absorbance peaks at 560 nm correspond to open monomer (O) whereas shorter wavelengths correspond to closed (C) and H-aggregate (H) conformations.11
To examine the effects of linker length on aggregation propensity, the experiment was repeated in aqueous solution containing 2N4R tau and assembly buffer. Under these conditions, the most efficacious compounds (2b, 2d) continued to favor open monomer conformation, whereas less active inhibitors (e.g., 2a, 2c) populated greater amounts of hypsochromically shifted species corresponding to closed and H-aggregate conformations (Fig. 3B). These data are consistent with the inhibitory potency of bis-carbocyanines reflecting the distribution of compound conformations and aggregation states rather than simple bulk concentration, with those compounds that favor the open monomer conformation having the greatest efficacy in the low micromolar concentration regime.
To gain insight into the mechanism through which linker length modulated compound conformation and aggregation propensity, low energy conformations (i.e., the ground states) of 2a–f monomer cations were calculated semi-empirically via molecular dynamics in gas phase assuming symmetry and a 2+ charge. Each conformer simulation ran for 5,000 steps at a target temperature of 300K, with step intervals of 2.0 fs and frame intervals of 10 fs. Under these conditions, all compounds were predicted to fluctuate between open and closed states, with the relative population of conformers dependent on linker length. 2a, which had the shortest linker (3-methylene units), was predicted to selectively populate a fully closed “clamshell” conformation (Fig. 4A). This was because the odd number of methylene units in its linker positioned the heterocyclic rings adjacent to each other where they could efficiently interact. The simulation was consistent with 2a conformation determined in methanol solvent (Fig. 3A), and with the high aggregation propensity of 2a observed in aqueous solution (Fig. 3B). The latter behavior reflected the large flat surface area available for supporting H-aggregate formation in the closed conformation. In contrast, 2b formed a stable conformer that was open and nonplanar (Fig. 4B). This was because the even number of methylene units in its linker positioned the heterocyclic rings on opposite sides of the compound where they could not efficiently interact. This simulation also was consistent with absorbance measurements, which detected primarily open conformation in methanol solvent (Fig. 3A) and low aggregation propensity in aqueous solvent (Fig. 3B). The latter behavior was consistent with the predicted non-planar geometry of the open conformation, which presented only limited flat surface areas to support H-aggregate formation. As linker length increased beyond four methylene units, the simulations predicted complex mixtures of open and closed conformations, perhaps reflecting the increasing entropy associated with longer linkers. In general, compounds with odd numbers of methylene units in the linker (2c, 2e) populated open conformers that interconverted with closed conformers similar to that of 2a, whereas compounds with even numbers of methylene units in the linker (2d, 2f) transiently populated open conformers resembling 2b. Together the simulations suggested that the superior efficacy of 2b at low micromolar concentrations derived from its relatively short linker length, which favored a discrete conformational ensemble, and the even number of methylene units in its linker, which stabilized an open monomer conformation that was aggregation resistant.
Fig 4.
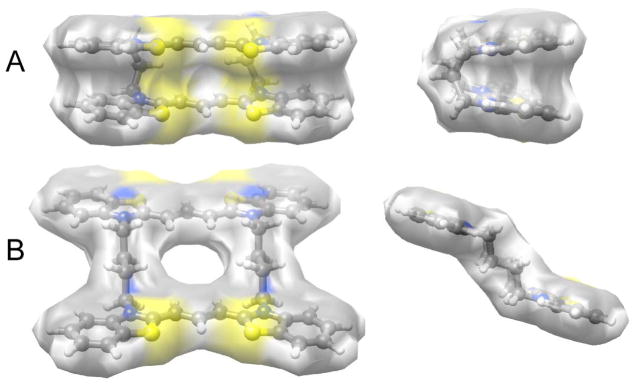
Molecular dynamics simulations predict that alkyl linker length affects monomer conformation. (A) Van der Waals hydrophobic surfaces calculated from energy minimized geometries of 2a and 2b, viewed from same perspective as in Scheme 1 (left), as well as a 90° rotated view (right). Molecular dynamic calculations were performed in Chem3D Pro 12.0.
In conclusion, these data reveal that the potency of macrocyclic cyanine aggregation inhibitors can be optimized through control of linker length. The results further support the hypothesis11 that multivalency arising from delivery of cyanine pharmacophores in an open monomer conformation is a key driver of macrocyclic cyanine potency, and that the observed linker length effects on potency arise at least in part through effects on monomer conformation. These design consideration can be used to maximize potency of tau aggregation inhibitors of this structural class.
Acknowledgments
Work at the Ohio State University was supported by a grant from the Public Health Service (AG14452; JK)
References
- 1.Sergeant N, Bretteville A, Hamdane M, Caillet-Boudin ML, Grognet P, Bombois S, Blum D, Delacourte A, Pasquier F, Vanmechelen E, Schraen-Maschke S, Buee L. Expert Rev Proteomics. 2008;5:207. doi: 10.1586/14789450.5.2.207. [DOI] [PubMed] [Google Scholar]
- 2.Kuret J. In: Protein Folding Diseases: Enzyme Inhibitors and Other Agents as Prospective Therapies; Smith HJ, Sewell RDE, Simons C, editors. Vol. 5. CRC Press, Taylor & Francis Books; Boca Raton, FL: 2007. p. 287. [Google Scholar]
- 3.Ballatore C, Brunden KR, Piscitelli F, James MJ, Crowe A, Yao Y, Hyde E, Trojanowski JQ, Lee VM, Smith AB., 3rd J Med Chem. 2010;53:3739. doi: 10.1021/jm100138f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bulic B, Pickhardt M, Khlistunova I, Biernat J, Mandelkow EM, Mandelkow E, Waldmann H. Angew Chem Int Ed Engl. 2007;46:9215. doi: 10.1002/anie.200704051. [DOI] [PubMed] [Google Scholar]
- 5.Chang E, Congdon EE, Honson NS, Duff KE, Kuret J. J Med Chem. 2009;52:3539. doi: 10.1021/jm900116d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M, Hasegawa M. J Biol Chem. 2005;280:7614. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- 7.Wischik CM, Edwards PC, Harrington CR, Roth M, Klug A. 6,953,794. US Patent. 2005
- 8.Chirita CN, Necula M, Kuret J. Biochemistry. 2004;43:2879. doi: 10.1021/bi036094h. [DOI] [PubMed] [Google Scholar]
- 9.Congdon EE, Figueroa YH, Wang L, Toneva G, Chang E, Kuret J, Conrad C, Duff KE. J Biol Chem. 2009;284:20830. doi: 10.1074/jbc.M109.016089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Congdon EE, Necula M, Blackstone RD, Kuret J. Arch Biochem Biophys. 2007;465:127. doi: 10.1016/j.abb.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Honson NS, Jensen JR, Darby MV, Kuret J. Biochem Biophys Res Commun. 2007;363:229. doi: 10.1016/j.bbrc.2007.08.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.May BC, Fafarman AT, Hong SB, Rogers M, Deady LW, Prusiner SB, Cohen FE. Proc Natl Acad Sci USA. 2003;100:3416. doi: 10.1073/pnas.2627988100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chirita CN, Necula M, Kuret J. J Biol Chem. 2003;278:25644. doi: 10.1074/jbc.M301663200. [DOI] [PubMed] [Google Scholar]
- 14.Herz AH. Photogr Sci Eng. 1974;18:323. [Google Scholar]
- 15.Necula M, Kuret J. Analytical Biochemistry. 2004;329:238. doi: 10.1016/j.ab.2004.02.023. [DOI] [PubMed] [Google Scholar]