

Abstract
Pompe disease is a systemic metabolic disorder characterized by lack of acid-alpha glucosidase (GAA) resulting in ubiquitous lysosomal glycogen accumulation. Respiratory and ambulatory dysfunction are prominent features in patients with Pompe yet the mechanism defining the development of muscle weakness is currently unclear. Transgenic animal models of Pompe disease mirroring the patient phenotype have been invaluable in mechanistic and therapeutic study. Here, we demonstrate significant pathological alterations at neuromuscular junctions (NMJs) of the diaphragm and tibialis anterior muscle as prominent features of disease pathology in Gaa knockout mice. Postsynaptic defects including increased motor endplate area and fragmentation were readily observed in Gaa−/− but not wild-type mice. Presynaptic neuropathic changes were also evident, as demonstrated by significant reduction in the levels of neurofilament proteins, and alterations in axonal fiber diameter and myelin thickness within the sciatic and phrenic nerves. Our data suggest the loss of NMJ integrity is a primary contributor to the decline in respiratory and ambulatory function in Pompe and arises from both pre- and postsynaptic pathology. These observations highlight the importance of systemic phenotype correction, specifically restoration of GAA to skeletal muscle and the nervous system for treatment of Pompe disease.
INTRODUCTION
Pompe disease (glycogen storage disease type II, acid-maltase deficiency) is a neuromuscular disorder characterized by the systemic deficiency of the glycogen metabolizing enzyme acid-alpha glucosidase (GAA). Affecting 1:40 000 individuals, GAA deficiency results in extensive glycogen accumulation within lysosomes resulting in disruption of cellular architecture and function. While the genetic component of Pompe disease is well defined, little is known mechanistically about how systemic glycogen accumulation results in neuromuscular failure. Emerging evidence suggests that neuronal pathology prevails with disease progression. Both human and animal models of Pompe disease support the notion that neuropathology may at least, in part, contribute toward neuromuscular dysfunction in Pompe disease. This is strengthened by several patient case reports documenting glycogen accumulation in the central nervous system (CNS), and in particular motoneurons (1,2), in conjunction with respiratory dysfunction in animal models (1,3–5). The combined neuro- and myopathic events in Pompe suggest that the synaptic interface at the neuromuscular junction (NMJ) may be compromised leading to loss of function.
It is becoming evident that the NMJ can be altered in a spectrum of diseases spanning autoimmune (Myasthenia gravis, Lambert–Eaton myasthenic syndrome), neurodegenerative [spinal muscular atrophy (SMA), Charcot–Marie–Tooth (CMT), amyotrophic lateral sclerosis (ALS)] and myopathic (myotubular myopathy, dynamin-2-related centronuclear myopathy) disorders. In autoimmune disorders, the pathology is well characterized, that is, dysfunction results from the specific targeting of components of the NMJ. In Myasthenia gravis, for example, ∼80% of patients have antibodies against acetylcholine receptors (AChR), while AChR antibody-negative patients develop an immune response to other critical NMJ proteins including muscle-specific kinase (MuSK) and low-density lipoprotein receptor-related protein 4 (6). However, in neurodegenerative and myopathic disorders, the NMJ represents a site of early pathologic vulnerability despite no definitive association of the disease-causing determinant protein with the NMJ (7,8). Recent studies also suggest that NMJ dysfunction contributes to pathogenesis in myopathies, particularly centronuclear myopathies. In myotubular myopathy, which is caused by mutations in the MTM1 gene affecting calcium homeostasis and excitation–contraction coupling (9,10), NMJ abnormalities coincide with the onset of motor pathology (11,12). MTM1 encodes a phosphoinositide phosphatase that regulates endosomal vesicle sorting and provides evidence that NMJ dysfunction can result from defects originating in muscle. Thus, NMJ dysfunction is emerging as a hallmark of early pathology in a wide spectrum of neuropathic and skeletal muscle disorders.
The current therapeutic regimen for Pompe disease is enzyme replace therapy (ERT), a bi-monthly infusion of recombinant GAA enzyme into patients. ERT prolongs survival although complete rescue is not achieved. Shortcomings of ERT appear to result from inefficient cation-independent mannose 6-phosphate receptor (CI-MPR)-mediated GAA uptake (13), impaired intracellular CI-MPR and GAA trafficking (14,15), and the inability of ERT to traverse the blood–brain barrier to target CNS pathology. In a clinical population, therefore, muscle weakness after ERT may result from progressive deterioration of lower motor neuron function (15–17). Another consideration, however, is that ERT may not effectively target NMJ pathology. Currently, there have been no formal attempts to characterize the peripheral nerve and NMJ in Pompe disease, and accordingly we used an established murine model, the Gaa−/− mouse, to characterize NMJ architecture to define pre- and postsynaptic alterations. Our results demonstrate that NMJ pathology is evident in both diaphragm and tibialis anterior (TA) muscles with hallmarks of widespread neuropathology also evident. These data provide evidence that NMJ abnormalities are a contributory factor to muscle weakness and the pathology of Pompe disease.
RESULTS
Pathologic alterations in NMJs of the diaphragm and the phrenic nerve in Pompe mice
To determine the prevalence of peripheral nerve and NMJ abnormalities in Gaa−/− mice, we selected the primary inspiratory muscle (diaphragm) due to the direct association of respiratory insufficiency and Pompe disease (18–21). Access to the phrenic nerve and diaphragm from a Pompe patient is usually only possible at end-stage of disease. Therefore, we utilized the Gaa−/− mouse as a model organism for elucidating the pre- and postsynaptic pathology in Pompe disease.
Our methodological approach was to label three distinct structures that comprise the NMJ, namely the motor neuron axon, the presynaptic terminal and the postsynaptic motor endplate (Fig. 1). Under high magnification, we noted fragmentation and expansion of AChR area in samples from affected mice (Fig. 1 A and B). The low magnification view shows denervation and lack of overlap between the presynaptic synaptotagmin and postsynaptic α-bungarotoxin-labeled AChR clusters (Fig. 1C and D). To quantify these alterations, we measured the distribution of AChR clusters and counted axon(s) in contact with postsynaptic sites and for the presence of synaptotagmin at the synapse (Fig. 2). Motor endplates in the diaphragm of Gaa−/− mice were significantly larger than those of wild type (WT) displaying a mean area of 316.8 μm2 ± 6.284 (standard error of the mean, SEM), n = 581, compared with 275.8 μm2 ± 7.233 (SEM), n = 527 (P ≤ 0.0001; two-tailed, unpaired t-test) (Fig. 2A). In agreement, frequency distribution in WT and Gaa−/− mice revealed that the endplate size is right-shifted between the WT and Gaa−/− groups (Fig. 2B). Postsynaptic abnormalities were further characterized quantifying fragmentation of motor endplates. Fragmentation was defined according to (22) as the presence of five or more AChR island and/or an area of the postsynapse with severe and/or irregularly shaped AChR clusters. Gaa−/− endplates displayed an approximate 3.3-fold higher frequency of fragmentation [8.3% ± 2.5%, n = 120 compared with WT 2.5% ± 1.4% n = 120 (P = 0.047; two-tailed, Mann–Whitney test)] (Fig. 2C). Together these results indicate that AChR clusters are distinct and dispersed in 9-month-old Pompe mice compared with WT.
Figure 1.
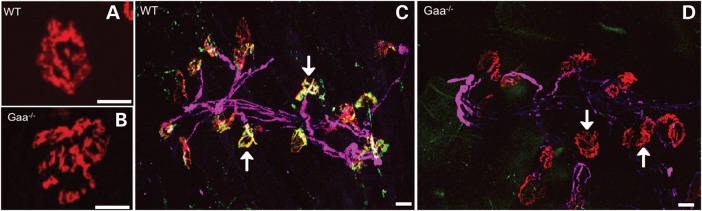
Widespread disruption of diaphragmatic NMJs in Pompe animals. Confocal images of AChR distribution in the diaphragm of representative (A and C) 9-month-old old WT 129SVE and (B and D) age-matched Gaa−/− mice. Lower magnification images from the same tissue samples show triple immunolabeling with anti-NFH (magenta), anti-synaptotagmin (green) and alpha-bungarotoxin (red). The white arrow in (C) indicates colocalization of presynaptic synaptotagmin and α-bungarotoxin labeling. (D) Representative area of NMJs in Gaa−/− diaphragm show marked reduction in colocalization of pre- and postsynaptic labels. White arrows indicate loss of presynaptic staining in motor endplates. Scale bars represent 10 μm.
Figure 2.

Synaptic pathology in the diaphragm of Pompe animals. (A) Mean diaphragm endplate size is significantly larger in Gaa−/− (gray column) mice compared with age-matched WT controls (white column; ****P ≤ 0.0001, Mann–Whitney t-test, two-tailed; n = 4 biological replicates per group). (B) Histogram of endplate size showing frequency distribution of endplate area across the diaphragm of WT and Gaa−/−animals (n = 4). (C) The mean percentage of fragmented diaphragm endplates in Gaa−/− (gray) and WT (white) mice (*P ≤ 0.05, Mann–Whitney t-test, two-tailed). (D) Quantification of multi- (gray) or single- (white) innervated endplates within the diaphragm of Gaa−/− compared with age-matched control mice (two-way repeated measures ANOVA, n = 4 biological replicates per group). (E) Quantification of NMJ innervation in the diaphragm of 9-month-old Gaa−/− mice relative to age-matched controls (two-way ANOVA with Sidak's multiple comparison test, n = 4 animals per group).
To determine whether this increase in endplate size was a physiological response to presynaptic abnormalities, the number of motor axons that innervated individual endplates was quantified using neurofilament staining. No significant differences in endplates that were innervated by multiple or single axons were observed between WT or Gaa−/− mice (Fig. 2D). However, quantification of a presynaptic marker (synaptotagmin II) enabled assessment of nerve occupancy within the endplate and revealed dramatic differences between the groups (Fig. 2E). In WT mice, 55.56% of endplates were fully occupied, that is, complete overlap occurred between pre- and postsynaptic markers in the NMJs. In contrast, only 9.78% of endplates in Gaa−/− mice were fully occupied by presynaptic inputs (Fig. 2E). This trend was reciprocated in the number of endplates displaying no staining with the presynaptic marker. In WT mice, 17.43% of endplates did not display any presynaptic staining while this fraction increased significantly to 56.79% in diaphragm NMJs from Gaa−/− mice (P ≤ 0.01, two-way ANOVA, Fig. 2E).
In WT and Gaa−/− mice, light microscopy of transverse sections of phrenic nerves stained with toluidine blue were performed to assess myelin and axon diameters (Fig. 3). Transverse sections from Gaa−/− mice were atypical in appearance, with higher frequency of large and hypermyelinated fibers, tomacula and myelin infoldings (Fig. 3B). Morphometric analyses of nerve fiber diameters (axon and myelin) revealed an overall increase in nerve fiber size in affected animals (Fig. 3C). Measurements of axon to fiber (myelin and axon) diameters (referred to as G-ratios) were significantly reduced in Gaa−/− samples with a mean ratio of 0.4490 ± 0.0080 compared with 0.6961 ± 0.0037 in WT (P ≤ 0.0001, two-tailed, unpaired t-test) (Fig. 3C and D).
Figure 3.

Alterations of nerve morphology in the phrenic nerve of Pompe mice. Representative cross section of the phrenic nerve from (A) WT mice and (B) Gaa−/− mice. (C, D) Significantly lower average G-ratio in phrenic nerves from 9-month-old Gaa−/− mice (0.4490 ± 0.0080, n = 447 fibers from four independent animals) compared with age-matched WT control mice (mean ratio 0.6961 ± 0.0037, n = 300 fibers from three independent animals), indicating thicker myelin sheaths relative to the axon diameter (C; ****P ≤ 0.0001; two-tailed, unpaired t-test), with lower G-ratios across a range of axon calibers (D). Scale bars represent 50 μm.
Neuromuscular abnormalities in distal regions of the peripheral nerve and skeletal muscle in Pompe mice
To determine whether NMJ pathology was limited to respiratory muscle or was more widespread in Gaa−/− mice, we conducted a similar analysis of a locomotor muscle, the TA (Figs 4 and 5). Whole mount preparations of the TA were reacted with antibodies to neurofilament heavy (NFH) polypeptide, synaptotagmin and α-bungarotoxin and analyzed by confocal microscopy (Fig. 4). When individual synaptic junctions were viewed, expansion of AChR distribution was noted, as seen previously in the diaphragm (Figs 1 and 2). While innervation of the NMJ was comparable between the samples, a decrease in the colocalization of the presynaptic marker and postsynaptic AChRs were notable (Fig. 4C and D). Quantification of endplate size within the TA revealed significant enlargements in Gaa−/− mice (633.3 μm2 ± 15.19, n = 498) compared with WT (558.0 μm2 ± 19.01, n = 220; P = 0.0040, two-tailed, t-test) (Fig. 5A). We qualitatively observed an increased prevalence of endplates per field of view in the Gaa−/− samples (data not shown). Despite this, distribution of endplate size approximates those of WT (Fig. 5B). Fragmented endplates were also more frequent in the TA of Gaa−/− mice compared with those of WT (19.29 ± 3.34%, n = 140, Gaa−/− compared with 10.20 ± 2.51%, n = 147, WT, P = 0.03; two-tailed, Mann–Whitney test) (Fig. 5C). In keeping with NMJ innervation within the diaphragm (Fig. 2D), no significant difference was observed between partially innervated endplates between groups (Fig. 4D). Within TA isolated from Gaa−/− mice, there was a significant increase in the number of denervated endplates void of presynaptic terminal staining (64.50% Gaa−/− versus 15.90% WT, P = ≤ 0.01; two-way ANOVA, Sidak's multiple comparison test) (Fig. 5E). Concordantly, there was a significant decrease in endplates with complete occupancy by the presynaptic terminal between groups (8.95% Gaa−/− compared with 47.54% WT, P = ≤ 0.05) (Fig. 5E).
Figure 4.

Widespread NMJ disruption in the TA of 9-month-old Pompe animals. Confocal micrographs of individual postsynaptic sites in in the TA of (A, C) 9-month-old WT and (B, D) 9-month-old Gaa−/− mice, as visualized by the binding of α-bungarotoxin. Lower magnification images of the distribution of NFH (magenta), synaptotagmin (green) and alpha-bungarotoxin (red). The white arrow in (C) indicates colocalization of presynaptic synaptotagmin and α-bungarotoxin labeling in AChR clusters. (D) Representative area of NMJs in Gaa−/− diaphragm show marked reduction in colocalization of pre- and postsynaptic labels. Scale bars represent 10 μm.
Figure 5.

Synaptic pathology in hindlimb muscle of 9-month-old Pompe animals. (A) The mean TA endplate size is significantly larger in Gaa−/− (gray column) mice compared with age-matched WT controls (white column; P ≤ 0.05, t-test, two-tailed; n = 4 biological replicates per group). (B) Histogram of endplate size showing frequency distribution of endplate area across the TA of WT and Gaa−/− animals (n = 4). (C) The mean percentage of all measured TA endplates that were fragmented in Gaa−/− (gray) and WT (white) mice (P ≤ 0.05, Mann–Whitney t-test, two-tailed). (D) Quantification of multi- (gray) or single- (white) innervated endplates within the diaphragm of Gaa−/− compared with age-matched control mice (two-way repeated measures ANOVA, n = 4 biological replicates per group). (E) Quantification of NMJ innervation in the TA of 9-month-old Gaa−/− mice relative to age-matched controls (two-way ANOVA with Sidak's multiple comparison test, n = 4 animals per group).
The marked endplate expansion and denervation of both respiratory and locomotor muscles during progression of Pompe disease prompted us to analyze the endplate area and innervation status in early-stage disease animals (Supplementary Material, Fig. S1). At 6 weeks of age, we noted a significant increase in endplate area, the diaphragm of Gaa−/− mice (396.8 ± 9.056 μm2) relative to age-matched WT mice (294.1 ± 6.68 μm2, P ≤ 0.0001) (Supplementary Material, Fig. S1A). However, we did not observe any difference in the endplate area within the TA muscle isolated from either Gaa−/− (464.1 ± 10.3 μm2) relative to age-matched WT mice (458.5 ± 11.55 μm2). In addition, we did not detect any difference in the innervation status of NMJs from either muscle group at 6 weeks of age (Supplementary Material, Fig. S1E and F).
The observations of dramatic postsynaptic terminal abnormalities in the TA at 9-months of age suggest that distal axon pathology is a prominent feature of Pompe disease progression. To expand our understanding of Pompe-associated neuropathology, we focused on the morphological and biochemical properties of the sciatic nerve. Morphometric analysis of sciatic nerve revealed obvious abnormalities of Pompe nerve fibers, including wider separation of individual nerve fibers and alteration of normal axonal profiles by the presence of increased frequency of large and hypermyelinated fibers and tomacula (Fig. 6). Morphological irregularity in myelinated axon fibers was frequently observed in addition to an increase in extracellular matrix (Fig. 6B asterisk) of the sciatic nerve of Gaa−/− mice (Fig. 6A and B). G-ratio analysis from sciatic nerve sections exhibited a significantly reduced ratio in the Pompe nerve (Fig. 6C and D), reflecting thicker myelin around individual axons. The mean G-ratio in Pompe nerves was 0.4035 ± 0.005730 as compared with 0.5261 ± 0.004489 in WT (Fig. 6 D; P < 0.0001; two-tailed, unpaired t-test).
Figure 6.

Alterations of tissue morphology in the sciatic nerve of Pompe mice. Morphological analysis of the sciatic nerve from (A) WT and (B) Pompe (Gaa−/−) mice highlights altered nerve fiber distribution and increased extracellular space between individual nerve fibers (asterisks). (C, D) Significantly lower average G-ratio in sciatic nerves from 9-month-old Gaa−/− mice (0.4111 ± 0.0055, n = 850 fibers from four independent animals) compared with age-matched WT control mice (mean ratio 0.5431 ± 0.0048, n = 869 fibers from three independent animals), indicating thicker myelin sheaths relative to the axon diameter (C; ****P ≤ 0.0001; two-tailed, unpaired t-test), with lower G-ratios across a range of axon calibers (D). Scale bars represent 50 μm.
To determine if NMJ deterioration impaired physiological function in 9-month-old Gaa−/− mice, we performed in situ isometric twitch and torque analysis from TA muscles of WT and Gaa−/− mice (23–26) (Fig. 7). At 9 months of age, we noted impaired torque production across all frequencies tested with peak torque significantly impaired in Gaa−/− mice relative to WT controls (Gaa−/−, 8.83 ± 0.71 mN/g bodyweight; WT, 16.43 ± 0.67 mN/g bodyweight; P = 8.26 × 10−6). No significant difference was observed in mean the bodyweight of Gaa−/− and WT mice (WT 28.7 ± 0.65 g (n = 8); Gaa−/− 27.5 ± 1.54 g (n = 6), P = 0.34).
Figure 7.

Loss of Gaa impairs neuromuscular function in 9-month-old mice. In situ analysis of muscle torque production revealed a significant decrease in performance of Gaa−/− mice relative to age-matched WT mice across all frequencies tested (*P ≤ 0.05).
Previous reports have detected an abundance of lysosomes in skeletal muscle of Pompe mice (27–29). On the basis of the observed morphological abnormalities in affected sciatic nerves, we asked if there was an accumulation of lysosomes within the axons or the Schwann cells of sciatic nerve isolated from Gaa−/− mice (Fig. 8). Confocal laser microscopy revealed intense lysosomal-associated membrane protein 1 (LAMP1) staining in S100-labelled Schwann cells from Pompe mice (Fig. 8D–F). This is in contrast to low abundance of LAMP1-reactive cell profiles in WT sciatic nerve sections co-labeled with S100 (Fig. 8A–C).
Figure 8.

Accumulation of LAMP1 in Schwann cells in the sciatic nerve of affected mice. Sciatic nerves from WT (A, C and E) and Gaa−/− (B, D and F) mice were doubled, labeled with anti-S100 (red) and anti-LAMP1 (green) antibodies. Nuclei were stained with DAPI (blue). Arrows indicate the perinuclear area of S100-positive Schwann cells, while arrowheads indicate the paranodal region of myelin internodes. The majority of LAMP1 in Gaa−/− is detected as perinuclear or paranodal puncta in S100-positive Schwann cells, with pronounced abnormal accumulation in a subset of the Schwann cells. Paranodal LAMP1 vesicles in Gaa−/− also appear less punctate. Scale bars = 10 μm. Blue = DAPI, green = S100, red =LAMP1.
The increase in LAMP1 detection prompted us to perform western blot analysis of sciatic nerves isolated from Gaa−/− and WT animals (Fig. 9). Immunoblotting of individual nerve lysates from each group showed a significant increase in lysosomal-associated membrane protein-2 (LAMP2, Fig. 9B, P = 0.012; two-way, unpaired t-test) with suggestive evidence albeit non-statistically significant of an increase in LAMP1 (Fig. 9A, P = 0.069; two-way, unpaired t-test). No statistically significant difference was observed in myelin basic protein (MBP) (P = 0.340; two-way, unpaired t-test) (Fig. 9C) or growth-associated protein (43 kD; GAP43; P = 0.070; two-way, unpaired t-test) (Fig. 9D). Analysis of non-phosphorylated heavy and medium neurofilament polypeptides revealed a significant reduction in Gaa−/− mice (Fig. 9E, P = 0.047; two-way, unpaired t-test). Together these data suggest that reduced axon caliber manifests with Pompe progression.
Figure 9.

Neuropathic changes in the sciatic nerve of Pompe animals. Representative western blot images and quantification normalized to α-tubulin for select proteins within the sciatic nerve of 9-month-old WT and Gaa−/− mice. (A) LAMP1 (P ≥ 0.05; two-tailed, unpaired t-test). (B) LAMP2 (P ≤ 0.05; two-tailed, unpaired t-test). (C) MBP (P ≥ 0.05; two-tailed, unpaired t-test). (D) Growth-associated protein 43 kDa (P ≥ 0.05; two-tailed, unpaired t-test). (E) Non-phosphorylated neurofilament heavy and medium polypeptide (P ≤ 0.05; two-tailed, unpaired t-test).
DISCUSSION
Although traditionally defined and treated as a myopathy, the ubiquitous depletion of GAA and systemic glycogen accumulation in Pompe disease suggest pathological involvement of non-muscle tissue (30). Supporting this argument, recent evidence has expanded our understanding of pathogenesis in non-muscle tissues (30,31). Additionally, glycogen accumulation within the CNS (1,2,4,32–35) and skeletal muscle (36) are likely to contribute to both pre- and postsynaptic pathology at the NMJ and ultimately lead to muscle dysfunction. To elucidate potential mechanisms that lead to incomplete muscle activation, we investigated the contribution of pre- and postsynaptic pathology in Pompe disease. For the first time, we report the presence of pathological changes in respiratory and limb muscle NMJs in a murine Pompe model, which recapitulates many of the clinical phenotypes of the disease (37). Moreover, these changes can be detected in young diseased animals and become more prominent with disease status. Susceptibility of NMJ pathology is accompanied by severe alterations in the phrenic and sciatic nerves in Pompe animals. The combination of peripheral nerve and NMJ pathology results in a severe loss of neuromuscular function (in situ force measures). Overall, the data suggest the respiratory and ambulatory impairments associated with Pompe disease are likely to reflect mechanisms, which extend beyond skeletal muscle pathology.
Pompe disease is characterized by a wide variation of mutations and by multisystem involvement. Respiratory failure has long been recognized as a primary feature in Pompe patients and considered the major factor contributing to mortality. Numerous clinical and animal studies have shown respiratory dysfunction in patients and models of Pompe disease; however, the pathological mechanisms that link progressive lysosomal glycogen accumulation to skeletal muscle weakness are unclear. Possible mechanisms have recently been supported by evidence revealing disruption of autophagy regulation, loss of proper lysosomal trafficking and mitochondrial function (38).
The data presented here indicate that NMJ pathology is a potential underlying mechanism contributing to progressive muscle weakness in Pompe disease. In agreement with other myopathies (11,39), we observed a significant increase in motor endplate size in both the TA and diaphragm accompanying the progression of Pompe pathology. Our detection of moderate abnormalities within the diaphragm at 6 weeks of age suggests progressive deterioration of NMJ components accompanies Pompe disease progression. Further evaluation will be required to characterize the physiological effect of these early histological abnormalities and evaluate how NMJ deterioration contributes to Pompe disease pathogenesis. The endplate expansion observed in this study contrasts with aging (40) and neuropathic-induced NMJ alterations, which result in reduced motor endplate area (41,42), and could potentially highlight subtle differences between neuropathic- or myopathic-induced NMJ dysfunction. Our observed differences in the frequency of fragmentation based on the muscle in question (either TA or diaphragm) could result from muscle or nerve specific susceptibility to glycogen accumulation. Degeneration of axon terminals has been proposed to be a crucial event in the materialization of motoneuron disease and neurodegenerative disease in general. The codependence of motor neurons and muscle for the trophic, electrical and chemical signals required for maintenance of proper innervation (43) are severely affected. Although Pompe disease has been traditionally classified as a neuromuscular disease, the primary focus has been placed on defining disease manifestation and therapeutic efficacy in skeletal and cardiac muscle. Indeed, our data from the Gaa−/− mouse model suggest that Pompe disease displays a classic neuromuscular disease phenotype. We speculate that this denervation results primarily from glycogen accumulation within the neuronal cell body. In support of this, biochemical analysis revealed significant reduction in neurofilament and neuronal plasticity proteins and only a minor difference in MBP between Pompe and WT nerves.
To our knowledge, this is the first study to describe myelin abnormalities in Pompe mice. Transverse section analysis of sciatic and phrenic nerve bundles demonstrated gross morphological disruption to individual nerve fibers and increased myelin occupation of individual fibers indicating neuropathology. Perturbation of Schwann cells is known to induce switching between a non-proliferative myelinating phase to a non-myelinating proliferative phase in axons (44,45) and terminal Schwann cells have been shown to participate in NMJ development and dissociation (46–51). Periodic Acid Schiff staining detected observable accumulation of glycogen in a subset of axons in the sciatic nerve (Supplementary Material, Fig. S2B). LAMP1 accumulation near Schwann cell nuclei support the possibility of enhanced susceptibility of glial cells in Pompe disease, however, whether disruption of normal lysosomal trafficking or prolonged systemic disease status is ultimately responsible warrants further study.
The NMJ serves as a site of pathological interest in a spectrum of neuropathic and myopathic disorders and more recently in aging models. Inherent to its malleable nature, NMJ structural abnormalities range from alterations in endplate size, fragmentation, pre-terminal sprouting, neurofilament infiltration and swelling (22,52). The mechanism of enhanced susceptibility of motoneurons to glycogen accumulation and how this leads to denervation of the NMJ remains unknown. Our data suggest that perturbations of lysosomal trafficking within the nerve may be partly accountable for Pompe-associated neuropathology. Regardless of the mechanism, replacement of GAA to the entire motor unit is likely to be essential in the treatment of Pompe disease and may account for the failure to reverse functional decline with the current human recombinant GAA enzyme replacement strategy.
In summary, this study demonstrates that significant peripheral nerve and NMJ pathology develop as a result of Pompe disease. We have found that these events occur both in respiratory and locomotor muscles. While therapeutic strategies are available to address skeletal muscle GAA deficiency, correction of neuronal deficits is severely limited utilizing an ERT approach. For this reason, Pompe disease represents an attractive candidate for gene therapy. Correction of both the CNS and skeletal muscle may ultimately be required to reverse or attenuate disease progression. Finally, determining whether an accelerated temporal pattern exists for the development of the specific molecular mechanisms that impact NMJ pathogenesis in Pompe disease will be critical in defining the impact of therapies.
MATERIALS AND METHODS
Experimental animals
The Gaa−/− mouse (Taconic, Hudson, NY) originally developed by Raben et al. (53) was outbred to a 129SVE background (5). All Gaa−/− and 129SVE mice were age and gender-matched via timed mating. All animals were obtained from Taconic and were maintained on a 12 h light/dark cycle with water and food provided ad libitum. Gaa−/− male and female mice were compared with the background strain 129SVE (WT). All animal studies were approved in accordance with the guidelines set forth by the University of Florida Institutional Animal Care and Use Committee. All animals were sacrificed at 6 weeks for histologic or 9 months of age for physiologic, histologic and biochemical assays.
NMJ staining
Gaa−/− and WT mice were anesthetized with 2% isofluorane and sacrificed by thoracotomy. Diaphragm, TA, phrenic and sciatic nerves were immediately dissected and processed according to standard protocols. For neuromuscular immunostaining, dissected diaphragm and TA muscles were finely teased in 4°C saline solution for a maximum of 10 min to maximize surface area while maintaining neuromuscular connectivity. Muscles were then fixed in 4% paraformaldehyde (PFA) in 0.1 M phosphate buffered saline (PBS) for 15 min. Samples were washed for three times for 5 min each prior to permeabilization with 2% triton X-100 in PBS for 30 min. Samples were then blocked overnight at 4°C in blocking buffer (1% Triton X-100, 4% BSA in PBS). Following blocking, samples were incubated in primary antibody solution containing anti-ZNP1 (Zebrafish International Research Center 1:200) and anti-NFH polypeptide (EnCor Biotechnology Inc., 1:400) for a minimum of 24 h to label neuronal and presynaptic structures. Samples were washed six times for 30 min each prior to incubation with secondary antibodies anti-mouse Alexa 488 (1:200 dilution, Jackson Immunolabs), Anti-Chicken Alexa 647 (1:1000 dilution, Life Technologies) and Alexa 594-conjugated α-bungarotoxin. Samples were washed as described above, blotted dry and mounted onto Superfrost glass slides in Prolong Gold anti-fade with DAPI (Life Technologies).
Morphological studies of the phrenic and sciatic nerves
Nerves were processed as previously described (54). Animals were sacrificed and a ∼5 mm piece of the proximal end of each left sciatic nerve and the left phrenic nerve immediately distal to the heart was obtained for morphological analyses. Samples were fixed immediately in 3% glutaraldehyde in 0.2 mol/l Na Cacodylate buffer (pH 7.3) before embedding in epon as described previously (55) with minor modifications. Thick sections (0.5 micron) were stained with toluidine blue and surveyed by light microscopy. For the morphometric studies, three WT and four Gaa−/− mice were evaluated for distribution of fiber diameter (axon with myelin, ∼300 fibers/animal), myelin sheath thickness (300 fibers/animal) and G-ratio (300 fibers/animal) on light level images. We analyzed the data using the public domain NIH Image J program (52).
Sciatic nerve immunostaining
Freshly dissected sciatic nerves were quickly frozen in OCT using isopentane chilled with liquid nitrogen. Samples were stored at −80°C prior to cryosectioning. Longitudinal nerve cryosections (6 µm thickness) were mounted onto glass slides and were subsequently dried at room temperature for 60 min, fixed with 4% PFA in 0.1 M PBS for 10 min at room temperature and then rinsed twice in fresh 0.1 M PBS. Sections were permeabilized in −20°C methanol for a period of 5 min, rinsed in 0.1 M PBS and blocked in 10% normal goat serum (NGS) for 30 min. Subsequently, samples were incubated with anti-CD107a, LAMP1 (1:500, BD Pharmigen 553792) and S100 (1:500, Dako) primary antibodies overnight at 4°C. Slides were rinsed three times in 0.1 M PBS and incubated with anti-rat Alexa 488 and anti-rabbit Alexa 594 secondary antibodies (Life Technologies) (1:500) for 2 h at room temperature. Following further rinses in 0.1 M PBS, samples were mounted with Prolong Gold anti-fade and the nuclear dye DAPI (Life Technologies).
Confocal microscopy
Fluorescent micrographs were acquired using a LEICA TCS AOBS spectral confocal microscope. The following wavelengths were specified for each fluorophore; DAPI Excitation (Ex) 430 nm, Emission (Em) 480 nm; Alexa 488, Ex 500 nm, Em 545 nm; Alexa 594, Ex 605 nm, Em 655 nm and Alexa 647, Ex 660 nm, Em 730 nm. Images for NMJs were acquired using a z-stack from 1 μm-thick slice sections throughout whole muscle preparations. Four randomly chosen fields of view were used to acquire images in each TA muscle analyzed (n = 4/group). Three randomly chosen fields of view were used to acquire images in each diaphragm muscle analyzed (n = 4/group). Maximum intensity projections of confocal stacks were used for quantification (described below). Images for sciatic nerve were acquired using a z-stack from 6 μm-thick sections, and average intensity projections of confocal stacks were presented within the manuscript.
Image quantification
All measurements were performed using ImageJ. Endplate area was calculated using single channel (Alexa 594) maximum intensity projections and manually tracing the circumference of individual α-bungarotoxin-labeled structures. Innervation was performed as described in Murray et al. and Valdez et al. (22,42). Briefly, innervation was determined by the extent of overlap or co-localization between α-bungarotoxin and synaptotagmin labeling. NMJ fragmentation was quantified as described by Valdez et al. (22). Sciatic nerve G-ratios were assessed by measuring both the outer diameter of myelinated axons and the inner membrane representing the axonal diameter alone as described by Notterpek et al. (56). Statistical analysis was performed using GraphPad Prism6. All data are presented as means and standard errors.
In situ isometric torque analysis
Isometric torque analysis was performed on the TA muscle with modifications to procedures described elsewhere (23–26). Under anesthesia (2–2.5% isofluorane, 97.5–98% O2, flow rate 1 l/min), the skin and fascia surrounding the distal hindlimb were surgically removed exposing the TA. 4-0 braided silk surgical suture (Teleflex medical) was tied just distal to the myotendinous junction, and then the tendon cut distal to the suture knot. In addition, to prevent movement of the paw during stimulation, the extensor digitorum longus and peroneus longus tendons near the ankle were cut. Mice were positioned in dorsal recumbency on a heated platform to maintain body temperature at 37°C. A clamp was used to secure the hindlimb at 90° at the knee and the paw secured to the physiology table using transpore surgical tape (3M). The TA tendon was secured to a 300C-LR-FP muscle lever (Aurora Scientific). Cathode and anode electrodes were inserted distal to the fibular head to stimulate the peroneal nerve. Under control of the Dynamic Muscle Control (DMC) and Analysis (DMA) Software suite (Aurora Scientific), optimal electrode placement was determined by repositioning the electrodes and stimulating the nerve at 1 Hz until maximum twitch amplitude was recorded indicating bracketing of the peroneal nerve. Optimal length-tension was determined by performing isometric twitch stimulation at an increasing range of amplitude and varying tensions until maximum twitch amplitude was observed. Three successive tetanic stimulations (200 Hz, 100 pulses per train, 60 s rest between) were performed and the muscle allowed to rest for 5 min. Single stimulations at 15, 30, 60, 100, 120, 160 and 200 Hz were then performed with 30 s between each. Data were processed with the DMA software, and a torque-frequency curve derived. Significance was determined using the multiple t-test with Holm-Sidak multiple comparison between groups and frequencies. All values are reported as mean and SEM.
Western blot
Sciatic nerves were frozen in liquid nitrogen and homogenized using a pestle and mortar in sample application buffer (62.5 mm Tris, 10% glycerol, 3.0% SDS, pH 6.8; 100 μl per nerve). Homogenized samples were quantified using the BioRad DC protein assay and diluted in laemmli buffer. For cytoskeletal/structural targets, 9 μg of lysate was resolved by acrylamide gel electrophoresis, and for cytosolic proteins, 18 μg was resolved. The following antibodies were used to probe blotted membranes overnight at 4°C; LAMP1 (1:300 dilution, Abcam ab24170), LAMP2 (1:300 dilution, Abcam GL2A7), MBP (1:1000 dilution, Millipore MAB386), GAP43 (1:1000 dilution, Abcam 12274), SMI33 (1:1000 dilution) and α-tubulin (1:10 000 dilution, Sigma Aldrich T9026). Blots were imaged using the LI-COR imaging system with appropriate labeled secondary antibodies. Image studio lite (LI-COR) was used to quantify blots and statistical significance determined using GraphPad Prism 6.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants from the NIH K01AR066077 and MDA 216676 (D.J.F.), NHLBI PO1 HL59412-06 (B.J.B.), 1R01HD052682-01A1 (B.J.B. and D.D.F.), 15T32HL083810-04 (M.S.S.) and Parker B. Francis (M.K.E.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Arthritis and Musculoskeletal and Skin Diseases or National Heart, Lung, and Blood Institute of the National Institutes of Health.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Robin Yoon for morphometric assessment of endplate receptors and nerve fibers detailed in this paper.
Conflict of Interest statement. None declared.
REFERENCES
- 1.DeRuisseau L.R., Fuller D.D., Qiu K., DeRuisseau K.C., Donnelly W.H., Jr, Mah C., Reier P.J., Byrne B.J. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc. Natl. Acad. Sci. USA. 2009;106:9419–9424. doi: 10.1073/pnas.0902534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gambetti P., DiMauro S., Baker L. Nervous system in Pompe's disease. Ultrastructure and biochemistry. J. Neuropathol. Exp. Neurol. 1971;30:412–430. doi: 10.1097/00005072-197107000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Mah C.S., Falk D.J., Germain S.A., Kelley J.S., Lewis M.A., Cloutier D.A., DeRuisseau L.R., Conlon T.J., Cresawn K.O., Fraites T.J., Jr, et al. Gel-mediated delivery of AAV1 vectors corrects ventilatory function in Pompe mice with established disease. Mol. Ther. 2010;18:502–510. doi: 10.1038/mt.2009.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee K.Z., Qiu K., Sandhu M.S., Elmallah M.K., Falk D.J., Lane M.A., Reier P.J., Byrne B.J., Fuller D.D. Hypoglossal neuropathology and respiratory activity in Pompe mice. Front. Physiol. 2011;2:31. doi: 10.3389/fphys.2011.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falk D.J., Mah C.S., Soustek M.S., Lee K.Z., Elmallah M.K., Cloutier D.A., Fuller D.D., Byrne B.J. Intrapleural administration of AAV9 improves neural and cardiorespiratory function in Pompe disease. Mol. Ther. 2013;21:1661–1667. doi: 10.1038/mt.2013.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takamori M. Structure of the neuromuscular junction: function and cooperative mechanisms in the synapse. Ann. N Y Acad. Sci. 2012;1274:14–23. doi: 10.1111/j.1749-6632.2012.06784.x. [DOI] [PubMed] [Google Scholar]
- 7.Bowerman M., Murray L.M., Beauvais A., Pinheiro B., Kothary R. A critical smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscul. Disord. 2012;22:263–276. doi: 10.1016/j.nmd.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 8.Goulet B.B., Kothary R., Parks R.J. At the ‘junction’ of spinal muscular atrophy pathogenesis: the role of neuromuscular junction dysfunction in SMA disease progression. Curr. Mol. Med. 2013;13:1160–1174. doi: 10.2174/15665240113139990044. [DOI] [PubMed] [Google Scholar]
- 9.Jungbluth H., Wallgren-Pettersson C., Laporte J. Centronuclear (myotubular) myopathy. Orphanet. J. Rare Dis. 2008;3:26. doi: 10.1186/1750-1172-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dowling J.J., Vreede A.P., Low S.E., Gibbs E.M., Kuwada J.Y., Bonnemann C.G., Feldman E.L. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. 2009;5:e1000372. doi: 10.1371/journal.pgen.1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dowling J.J., Joubert R., Low S.E., Durban A.N., Messaddeq N., Li X., Dulin-Smith A.N., Snyder A.D., Marshall M.L., Marshall J.T., et al. Myotubular myopathy and the neuromuscular junction: a novel therapeutic approach from mouse models. Dis. Model. Mech. 2012;5:852–859. doi: 10.1242/dmm.009746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robb S.A., Sewry C.A., Dowling J.J., Feng L., Cullup T., Lillis S., Abbs S., Lees M.M., Laporte J., Manzur A.Y., et al. Impaired neuromuscular transmission and response to acetylcholinesterase inhibitors in centronuclear myopathies. Neuromuscul. Disord. 2011;21:379–386. doi: 10.1016/j.nmd.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 13.Farah B.L., Madden L., Li S., Nance S., Bird A., Bursac N., Yen P.M., Young S.P., Koeberl D.D. Adjunctive beta2-agonist treatment reduces glycogen independently of receptor-mediated acid alpha-glucosidase uptake in the limb muscles of mice with Pompe disease. FASEB J. 2014;28:2272–2280. doi: 10.1096/fj.13-244202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cardone M., Porto C., Tarallo A., Vicinanza M., Rossi B., Polishchuk E., Donaudy F., Andria G., De Matteis M.A., Parenti G. Abnormal mannose-6-phosphate receptor trafficking impairs recombinant alpha-glucosidase uptake in Pompe disease fibroblasts. PathoGenetics. 2008;1:6. doi: 10.1186/1755-8417-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raben N., Danon M., Gilbert A.L., Dwivedi S., Collins B., Thurberg B.L., Mattaliano R.J., Nagaraju K., Plotz P.H. Enzyme replacement therapy in the mouse model of Pompe disease. Mol. Genet. Metab. 2003;80:159–169. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- 16.Kikuchi T., Yang H.W., Pennybacker M., Ichihara N., Mizutani M., Van Hove J.L., Chen Y.T. Clinical and metabolic correction of Pompe disease by enzyme therapy in acid maltase-deficient quail. J. Clin. Invest. 1998;101:827–833. doi: 10.1172/JCI1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuller D.D., ElMallah M.K., Smith B.K., Corti M., Lawson L.A., Falk D.J., Byrne B.J. The respiratory neuromuscular system in Pompe disease. Respir. Physiol. Neurobiol. 2013;189:241–249. doi: 10.1016/j.resp.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mellies U., Lofaso F. Pompe disease: a neuromuscular disease with respiratory muscle involvement. Respir. Med. 2009;103:477–484. doi: 10.1016/j.rmed.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 19.Mellies U., Ragette R., Schwake C., Baethmann M., Voit T., Teschler H. Sleep-disordered breathing and respiratory failure in acid maltase deficiency. Neurology. 2001;57:1290–1295. doi: 10.1212/wnl.57.7.1290. [DOI] [PubMed] [Google Scholar]
- 20.Prigent H., Orlikowski D., Laforet P., Letilly N., Falaize L., Pellegrini N., Annane D., Raphael J.C., Lofaso F. Supine volume drop and diaphragmatic function in adults with Pompe disease. Eur. Respir. J. 2012;39:1545–1546. doi: 10.1183/09031936.00169011. [DOI] [PubMed] [Google Scholar]
- 21.Byrne B.J., Falk D.J., Pacak C.A., Nayak S., Herzog R.W., Elder M.E., Collins S.W., Conlon T.J., Clement N., Cleaver B.D., et al. Pompe disease gene therapy. Hum. Mol. Genet. 2011;20:R61–R68. doi: 10.1093/hmg/ddr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valdez G., Tapia J.C., Kang H., Clemenson G.D., Jr, Gage F.H., Lichtman J.W., Sanes J.R. Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc. Natl. Acad. Sci. USA. 2010;107:14863–14868. doi: 10.1073/pnas.1002220107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dellorusso C., Crawford R.W., Chamberlain J.S., Brooks S.V. Tibialis anterior muscles in mdx mice are highly susceptible to contraction-induced injury. J. Muscle. Res. Cell. Motil. 2001;22:467–475. doi: 10.1023/a:1014587918367. [DOI] [PubMed] [Google Scholar]
- 24.Huguet A., Medja F., Nicole A., Vignaud A., Guiraud-Dogan C., Ferry A., Decostre V., Hogrel J.Y., Metzger F., Hoeflich A., et al. Molecular, physiological, and motor performance defects in DMSXL mice carrying >1,000 CTG repeats from the human DM1 locus. PLoS Genet. 2012;8:e1003043. doi: 10.1371/journal.pgen.1003043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Metzger F., Sajid W., Saenger S., Staudenmaier C., van der Poel C., Sobottka B., Schuler A., Sawitzky M., Poirier R., Tuerck D., et al. Separation of fast from slow anabolism by site-specific PEGylation of insulin-like growth factor I (IGF-I) J. Biol. Chem. 2011;286:19501–19510. doi: 10.1074/jbc.M110.172189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schertzer J.D., Ryall J.G., Lynch G.S. Systemic administration of IGF-I enhances oxidative status and reduces contraction-induced injury in skeletal muscles of mdx dystrophic mice. Am. J. Physiol. Endocrinol. Metab. 2006;291:E499–E505. doi: 10.1152/ajpendo.00101.2006. [DOI] [PubMed] [Google Scholar]
- 27.Feeney E.J., Austin S., Chien Y.H., Mandel H., Schoser B., Prater S., Hwu W.L., Ralston E., Kishnani P.S., Raben N. The value of muscle biopsies in Pompe disease: identifying lipofuscin inclusions in juvenile- and adult-onset patients. Acta Neuropathol. Commun. 2014;2:2. doi: 10.1186/2051-5960-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prater S.N., Patel T.T., Buckley A.F., Mandel H., Vlodavski E., Banugaria S.G., Feeney E.J., Raben N., Kishnani P.S. Skeletal muscle pathology of infantile Pompe disease during long-term enzyme replacement therapy. Orphanet J. Rare Dis. 2013;8:90. doi: 10.1186/1750-1172-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klionsky D.J., Abdalla F.C., Abeliovich H., Abraham R.T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J.A., et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filosto M., Todeschini A., Cotelli M.S., Vielmi V., Rinaldi F., Rota S., Scarpelli M., Padovani A. Non-muscle involvement in late-onset glycogenosis II. Acta Myol. 2013;32:91–94. [PMC free article] [PubMed] [Google Scholar]
- 31.Katona I., Weis J., Hanisch F. Glycogenosome accumulation of the arrector pili muscle in Pompe disease. Orphanet J. Rare Dis. 2014;9:17. doi: 10.1186/1750-1172-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.ElMallah M.K., Falk D.J., Lane M.A., Conlon T.J., Lee K.Z., Shafi N.I., Reier P.J., Byrne B.J., Fuller D.D. Retrograde gene delivery to hypoglossal motoneurons using adeno-associated virus serotype 9. Hum. Gene Ther. Methods. 2012;23:148–156. doi: 10.1089/hgtb.2012.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elmallah M.K., Falk D.J., Nayak S., Federico R.A., Sandhu M.S., Poirier A., Byrne B.J., Fuller D.D. Sustained correction of motoneuron histopathology following intramuscular delivery of AAV in Pompe mice. Mol. Ther. 2013;22:702–712. doi: 10.1038/mt.2013.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thurberg B.L., Lynch Maloney C., Vaccaro C., Afonso K., Tsai A.C., Bossen E., Kishnani P.S., O'Callaghan M. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Lab. Invest. 2006;86:1208–1220. doi: 10.1038/labinvest.3700484. [DOI] [PubMed] [Google Scholar]
- 35.Sidman R.L., Taksir T., Fidler J., Zhao M., Dodge J.C., Passini M.A., Raben N., Thurberg B.L., Cheng S.H., Shihabuddin L.S. Temporal neuropathologic and behavioral phenotype of 6neo/6neo Pompe disease mice. J. Neuropathol. Exp. Neurol. 2008;67:803–818. doi: 10.1097/NEN.0b013e3181815994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raben N., Plotz P., Byrne B.J. Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease) Curr. Mol. Med. 2002;2:145–166. doi: 10.2174/1566524024605789. [DOI] [PubMed] [Google Scholar]
- 37.Byrne B.J., Kishnani P.S., Case L.E., Merlini L., Muller-Felber W., Prasad S., van der Ploeg A. Pompe disease: design, methodology, and early findings from the Pompe Registry. Mol. Genet. Metab. 2011;103:1–11. doi: 10.1016/j.ymgme.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Raben N., Wong A., Ralston E., Myerowitz R. Autophagy and mitochondria in Pompe disease: nothing is so new as what has long been forgotten. Am. J. Med. Genet. C Semin. Med. Genet. 2012;160:13–21. doi: 10.1002/ajmg.c.31317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pratt S.J., Shah S.B., Ward C.W., Inacio M.P., Stains J.P., Lovering R.M. Effects of in vivo injury on the neuromuscular junction in healthy and dystrophic muscles. J. Physiol. 2013;591:559–570. doi: 10.1113/jphysiol.2012.241679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valdez G., Tapia J.C., Lichtman J.W., Fox M.A., Sanes J.R. Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS One. 2012;7:e34640. doi: 10.1371/journal.pone.0034640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong L., Wang X., Choe D.W., Polley M., Burnett B.G., Bosch-Marce M., Griffin J.W., Rich M.M., Sumner C.J. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murray L.M., Comley L.H., Thomson D., Parkinson N., Talbot K., Gillingwater T.H. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008;17:949–962. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- 43.Fischer L.R., Glass J.D. Axonal degeneration in motor neuron disease. Neurodegener. Dis. 2007;4:431–442. doi: 10.1159/000107704. [DOI] [PubMed] [Google Scholar]
- 44.Hunter G., Aghamaleky Sarvestany A., Roche S.L., Symes R.C., Gillingwater T.H. SMN-dependent intrinsic defects in Schwann cells in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2013;23:2235–2250. doi: 10.1093/hmg/ddt612. [DOI] [PubMed] [Google Scholar]
- 45.Jessen K.R., Mirsky R. Control of Schwann cell myelination. F1000 Biol. Rep. 2010;2:pii: 19. doi: 10.3410/B2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trachtenberg J.T., Thompson W.J. Nerve terminal withdrawal from rat neuromuscular junctions induced by neuregulin and Schwann cells. J. Neurosci. 1997;17:6243–6255. doi: 10.1523/JNEUROSCI.17-16-06243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith I.W., Mikesh M., Lee Y., Thompson W.J. Terminal Schwann cells participate in the competition underlying neuromuscular synapse elimination. J. Neurosci. 2013;33:17724–17736. doi: 10.1523/JNEUROSCI.3339-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chou H.J., Lai D.M., Huang C.W., McLennan I.S., Wang H.D., Wang P.Y. BMP4 is a peripherally-derived factor for motor neurons and attenuates glutamate-induced excitotoxicity in vitro. PloS One. 2013;8:e58441. doi: 10.1371/journal.pone.0058441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murray L.M., Beauvais A., Bhanot K., Kothary R. Defects in neuromuscular junction remodelling in the Smn(2B/-) mouse model of spinal muscular atrophy. Neurobiol. Dis. 2012;49C:57–67. doi: 10.1016/j.nbd.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 50.Liu J.X., Brannstrom T., Andersen P.M., Pedrosa-Domellof F. Distinct changes in synaptic protein composition at neuromuscular junctions of extraocular muscles versus limb muscles of ALS donors. PLoS One. 2013;8:e57473. doi: 10.1371/journal.pone.0057473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Y., Thompson W.J. Nerve terminal growth remodels neuromuscular synapses in mice following regeneration of the postsynaptic muscle fiber. J. Neurosci. 2011;31:13191–13203. doi: 10.1523/JNEUROSCI.2953-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanes J.R., Lichtman J.W. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat. Rev. Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- 53.Raben N., Nagaraju K., Lee E., Kessler P., Byrne B., Lee L., LaMarca M., King C., Ward J., Sauer B., et al. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J. Biol. Chem. 1998;273:19086–19092. doi: 10.1074/jbc.273.30.19086. [DOI] [PubMed] [Google Scholar]
- 54.Fu H., DiRosario J., Kang L., Muenzer J., McCarty D.M. Restoration of central nervous system alpha-N-acetylglucosaminidase activity and therapeutic benefits in mucopolysaccharidosis IIIB mice by a single intracisternal recombinant adeno-associated viral type 2 vector delivery. J. Gene Med. 2010;12:624–633. doi: 10.1002/jgm.1480. [DOI] [PubMed] [Google Scholar]
- 55.Auld D.S., Robitaille R. Perisynaptic Schwann cells at the neuromuscular junction: nerve- and activity-dependent contributions to synaptic efficacy, plasticity, and reinnervation. Neuroscientist. 2003;9:144–157. doi: 10.1177/1073858403252229. [DOI] [PubMed] [Google Scholar]
- 56.Notterpek L., Shooter E.M., Snipes G.J. Upregulation of the endosomal-lysosomal pathway in the trembler-J neuropathy. J. Neurosci. 1997;17:4190–4200. doi: 10.1523/JNEUROSCI.17-11-04190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.