

Abstract
Congenital brain and craniofacial defects often occur together as a consequence of their developmental dependency on common progenitor tissue interactions and signaling pathways during embryogenesis. A classic example of this is perturbation of midline embryo development, and disruption of Hedgehog (Hh) signaling in the pathogenesis of holoprosencephaly. However, our understanding of how Hh signaling governs cell and tissue survival remains incomplete. Patched1 (Ptch1) is a well-known receptor for Hh ligands and Ptch1 overexpression is associated with cell and tissue-specific apoptosis. Here, we demonstrate that the X-linked inhibitory apoptosis protein (XIAP) associates with the C terminus of Ptch1 (Ptch1-C) in primary cilia to inhibit Ptch1-mediated cell death. Consistent with this observation, inhibition of XIAP suppresses cell proliferation, resulting in cell death and pathogenesis of an Hh loss-of-function phenotype. Thus, co-ordinated development of the brain and face is dependent in part upon XIAP mediation of Hh/Ptch1-regulated cell survival and apoptosis during embryogenesis.
INTRODUCTION
Craniofacial anomalies account for about one-third of all birth defects (1) and the well-known refrain ‘the face predicts the brain’ (2) reflects the high frequency of brain abnormalities that occur in association with craniofacial malformations. This is particularly true with respect to holoprosencephaly (HPE), which is one of the most common birth defects, occurring with an incidence of ∼1 in every 250 pregnancies (3). Individuals with HPE exhibit a variable failure in separation and growth of the left and right hemispheres of the brain, together with variable midline facial anomalies including hypotelorism, single nostrils and incisors, a narrow nose and closed mouth. HPE is therefore phenotypically a heterogeneous disorder and this is also true etiologically. Exposure to environmental teratogens such as alcohol (4,5) and retinoids (4) can result in HPE phenotypes. Gestational diabetes is also a factor as 1–2% of newborn infants of diabetic mothers exhibit HPE (6). HPE is also genetically heterogeneous and is currently associated with mutations in at least 12 different loci encompassing multiple signaling pathways such as BMP, NODAL, ZIC, SIX and Sonic Hedgehog (SHH) (7). What is common among many of the loci and signaling pathways is that they play important roles in the development of the ventral brain and midline structures of the embryo. This is particularly true for SHH signaling.
SHH binds to the 12-pass transmembrane receptor Patched1 (Ptch1), which leads to activation of a 7-pass transmembrane transducer, Smoothened (Smo), that in turn propagates SHH signaling through Glioma-associated oncogene homolog 1–3 (Gli1–3) transcription factors (8–10). Shh and Ptch1 are expressed in both the ventral forebrain (FB) and facial primordia of embryonic (E) 9.5–10.5 day mouse embryos and E2–3 day chick embryos where they regulate the patterning, proliferation and survival of the brain and craniofacial mesenchyme during embryogenesis (11–14). However, our understanding of how molecular signals co-regulate interdependent brain and facial development remains incomplete.
Here we explore the role of Ptch1 in cell proliferation and survival and its impact on the co-regulation of brain and facial development. We show that Ptch1 promotes cell and tissue-specific apoptosis via its regulation of Caspase9 (Casp9) activity and mitochondrial function. Furthermore, we discovered that the X-linked inhibitory apoptosis protein (XIAP) binds to the C terminus of Ptch1 and mediates the death-dependent function of Ptch1. Consistent with this observation, inhibition of XIAP induces cell death and suppresses cell proliferation. In addition, the association between Ptch1 and XIAP is observed in primary cilia in a Hedgehog (Hh) signaling-dependent manner. Thus, co-ordinated development of the brain and face is dependent in part upon XIAP mediation of Hh/Ptch1-regulated cell survival and apoptosis during embryogenesis.
RESULTS
Hh signaling regulation of cell survival in the ventral FB affects nasal process size and morphology
To explore how perturbed brain development affects facial development in the pathogenesis of HPE, we blocked Hh signaling in the brain of E9.5 mouse embryos via unilateral electroporation of short hairpin interfering RNAs (shRNAi) against mouse Shh (Shhi) (Supplementary Material, Fig. S1A and B) and via overexpression of the repressor form of mouse Gli3 (Gli3R) (15). After 24 h of culture, Shhi and Gli3R overexpression resulted in a decrease in the size of the brain vesicle on the electroporated (EP) side by18% compared with the non-EP control side (Fig. 1A and D, Shhi EP; brain vesicle, L/R rate = 0.818 ± 0.041, P = 0.0008; Fig. 1B and E, Gli3R EP; brain vesicle, L/R rate = 0.825 ± 0.096, P = 0.0014) as measured using surface anatomical landmarks (Supplementary Material, Fig. S2) (16). Furthermore, the reduction in tissue size in association with downregulated Hh signaling was associated with increased numbers of activated-Caspase3 (Casp3)-labeled neuroepithelial cells together with a considerable increase in cleaved-Casp9-labeled neuroepithelial cells (Fig. 1G and H). Casp9 is regarded as an intrinsic regulator of apoptosis. We also stained for the extrinsic apoptosis marker, cleaved-Caspase8 and the autophagy indicator, light chain isoform (LC3B) in Shhi EP embryos, but they were not significantly increased (Supplementary Material, Fig. S1B). This indicates that the Casp9-dependent intrinsic pathway is the primary mediator of apoptosis under Shh loss-of-function conditions. In association with increased cell death, Shhi and Gli3R overexpression resulted in a decrease in cell proliferation as evidenced by immunostaining with the M-phase cell proliferation marker, phospho-histone H3 (Ser10) (pHH3) (Fig. 1I and J). In fact, proliferation in the FB was reduced by up to 70% on the EP side compared with the control side.
Figure 1.

Hh signaling affects brain and facial development in mouse embryos. Frontal view of mouse embryos EP with Shh shRNAi (A), Gli3 repressor (Gli3R) (B), Gli3 activator (Gli3A) (C) and pEGFP as a reference. Light green area demarcates the brain vesicle (Brain ve.). The dark green lines represents the midline of the brain vesicle. Red colored areas demarcate the nasal processes (Nasal pr.). Inset panels show unilateral GFP EP. Comparisons of the relative size (mean ± s.d.) of brain vesicles (green bar) and nasal processes (red bar) from control non-EP sides (Ct) and EP sides for Shhi (D), Gli3R (E) and Gli3A (F). (G and H) Cleaved Casp3 and Casp9 labelling of frontal section of the front nasal process of EP embryos. Each half panels show unilateral GFP labeling in the brain. DAPI is a nuclear marker. (I) Double staining of frontal sections through the frontonasal process of Shhi EP embryos with neural crest marker, AP2α (green) and M-phase proliferation marker, phosphohiston H3 (pHH3, red). (J) Quantification of pHH3-positive cells in the forebrain (FB), medial nasal process (MNP) and nasal epithelium (NE). Md, mandibular process; Mx, maxillary process. Scale bars: 0.5 mm in (A)–(C); 0.2 mm in (G) and (H); 0.1 mm in (I).
Despite the blockade of Hh signaling specifically in the neuroepithelium, the nasal processes were also affected and were diminished in size by a similar proportion to the brain when compared with controls (Fig. 1A and D, Shhi EP; nasal process, EP/non-EP side rate = 0.810 ± 0.109, P = 0.0017; Fig. 1B and E, Gli3R EP; nasal process, EP/non-EP side rate = 0.740 ± 0.144, P = 0.00013). Similar to the EP neuroepithelium, the facial mesenchyme of the medial nasal process also exhibited increased numbers of Casp3- and Casp9-labeled cells (Fig. 1G and H) and a concomitant decrease in cell proliferation as measured by the number of pHH3-labeled mesenchymal cells compared with stage-matched controls (Fig. 1I and J). Interestingly, the pHH3-labeled mesenchymal cells were likely neural crest cells, as they expressed the transcription factor AP2 alpha (AP2α, activating enhancer-binding protein 2 alpha, also known as TFAP2A) rather than the endothelial cell marker PECAM-1 (platelet endothelial cell adhesion molecule, also known as cluster of differentiation 31 (CD31) (Fig. 1I, PECAM-1). These results suggest that Hh signaling-dependent regulation of cell survival in the brain affects the neighboring neural crest cell-derived nasal process mesenchyme, such that the size of the brain influences that of the facial mesenchyme.
In further support of this idea, upregulation of Hh signaling via Gli3A overexpression in the neuroepithelium, resulted in a significantly larger (28%) FB vesicle, together with a larger (13%) nasal process (Fig. 1C and F, brain vesicle, EP/non-EP side rate = 1.281 ± 0.238, P = 0.083; nasal process, EP/non-EP side rate = 1.128 ± 0.081, P = 0.014). Furthermore, in contrast to Shhi and Gli3R overexpression, there was no activation of Casp9. These results show that altering the size of the brain through modulation of Shh signaling-mediated cell survival, results in a corresponding change in the facial mesenchyme and size of the nasal process.
Midline defects in Shh mutant mice are associated with mitochondria-mediated cell death
Patients with mutations in SHH, and Shh mutant mice, exhibit similar growth defects in the brain and midline craniofacial tissues (17–19). Furthermore, elevated levels of apoptosis are thought to underlie the tissue hypoplasia and agenesis associated with Shh loss of function. However, the relationship between Hh signaling and cell death remains poorly understood. Ptch1 is known to function as a Hh ligand-dependent, cell death receptor (20), and recently the Casp9/DRAL complex was shown to regulate Ptch1 induction of cell death through the C-terminal domain of Ptch1 (Ptch1-C) (21).
To examine whether Shh loss of function induces caspase-dependent cell death, we performed immunohistochemistry on Shh mutant mouse embryos with specific markers of cell death. Numerous Casp3, Casp9 and transferase dUTP nick end labeling (TUNEL)-positive cells were detected in the anterior foregut and mesenchyme close to prospective FB in E8.5 mouse Shh−/− embryos compared with controls (Supplementary Material, Fig. S3A–D, white arrowhead). Subsequently, E10.5 Shh−/− embryos exhibit massively elevated levels of cell death in the craniofacial mesenchyme (Supplementary Material, Fig. S3E and F).
To further explore the relationship between Shh signaling and cell survival, with hypoplasia of the brain and craniofacial mesenchyme, we genetically ablated Shh producing ventral FB cells with diphtheria toxin (DT). E9.5 ShhcreGFP; iDTR (22,23) compound heterozygote embryos cultured in vitro for 24 h in the presence of 10 mg/ml DT, displayed a significant reduction in the size of the brain and nasal process (Fig. 2A, green and red region) as well as a considerable reduction in dorsal hemisphere separation (Fig. 2A, red arrow) and width between the nasal processes (Fig. 2A, blue bracket) compared with control embryos. The brain vesicle and nasal process were decreased by as much as 40% compared with the controls (Fig. 2B, midline length, Inducible diphtheria toxin receptor (iDTR)/control = 0.565 ± 0.089, P = 0.023; Fig. 2C, brain vesicle, iDTR/control = 0.623 ± 0.142, P = 0.035; nasal process, iDTR/C = 0.643 ± 0.0.059, P = 0.018). Interestingly, ShhcreGFP; iDTR embryos exhibited many TUNEL-positive apoptotic cells in the FB cavity and significantly elevated levels of cell death in the facial mesenchyme of the medial nasal processes as evidenced by elevated levels of Casp3- or Casp9-labeled cells (Fig. 2D–F, white arrows).
Figure 2.

Loss of Shh expressing cells is associated with mitochondrial-dependent cell death. (A) Frontal view of DT-treated control (DMSO) and ShhcreERT2; iDTR embryos. Green area demarcates the brain vesicle. Red area labels the nasal process. Blue brackets mark the length between left and right nasal processes. Red arrow indicate dorsal midline of forebrain hemispheres. (B and C) Quantification of midline length and relative size of (mean ± s.d.) of brain vesicles (Brain ve., green) and nasal processes (Nasal pro., red) from non-EP (Ct) and EP sides. (D–H) ShhcreERT2; iDTR embryos exhibit extensive cell death, altered dorso-ventral patterning and decreased proliferation. Shh expressing cells labelled with GFP in the ventral forebrain (D). Cell death labelling with cleaved casapse3 (Casp3), TUNEL (E) and cleaved Casp9 (F). Pax6 and Nkx2.1 (G) labelling of the dorsal-ventral forebrain, respectively, and phospho-histone H3 (pHH3) (H) labeling of mitotic cells. White arrows indicate cell death in the medial nasal process (E and F). White arrowheads indicate the boundary of Nkx2.1 and Pax6 expression (G).LNP, lateral nasal process; MNP, medial nasal process. Scale bars: 0.5 mm in (A); 0.2 mm in (D)–(G).
These results demonstrate the high sensitivity of the facial mesenchyme and medial nasal process to Shh loss of function, and is consistent with observations in Shh mutant embryos where the medial nasal process is missing while the lateral nasal process remains unaffected (18,19). Furthermore, ShhcreERT2; iDTR embryos exhibit FB dorsalization as evidenced by ventral expansion of the dorsal FB marker, Pax6 and diminished activity of the ventral FB marker, Nkx2.1 (Fig. 2G, white arrowheads). Taken together with decreased cell proliferation (Fig. 2H) this is similar to the Shh mutant embryo phenotype. Thus, irrespective of whether Shh signaling is ablated globally in Shh null mutants, or whether Shh expressing cells are specifically ablated spatiotemporally via DT, or whether Shh signaling is inhibited locally via electroporation, these results consistently and reproducibly indicate that the hypoplasia or agenesis of brain and craniofacial tissues that occurs due to perturbation of Shh signaling, is associated with caspase-mediated apoptosis.
Apoptosis is a form of programmed cell death that involves permeabilization of the outer membrane of mitochondria, and is initiated and executed by specific members of the Bcl2 and Caspase families of proteins, respectively (24). Activation of the Bcl2 family member, Bax, results in mitochondrial outer membrane permeabilzation and release of pro-apoptotic proteins such as cytochrome c, which in turn activates Casp9 (25). Once Casp9 is activated, it directly cleaves and activates Casp3 (26,27). Thus, Casp9 is regarded as an initiator caspase in mitochondria-related cell death, whereas Casp3 is considered an effector caspase (28). The elevated numbers of Casp9- and Casp3-labeled cells detected in the FB and facial mesenchyme in association with Shh loss of function suggest that Shh regulation of cell survival and proliferation may occur in part through the mitochondria.
Consistent with this idea, neuroepithelial overexpression of Bax, which regulates mitochondrial membrane permeability and functions as a pro-apoptotic factor, resulted in a 10–20% decrease in the size of the FB and nasal process on the EP side compared with the non-EP control side (Supplementary Material, Fig. S4A, FB, EP/non-EP side rate = 0.836 ± 0.0103, P = 0.012; nasal process, EP/non-EP side rate = 0.947 ± 0.033, P = 0.0036). Furthermore, Casp9 was widely activated in the Bax overexpressing cells in the neuroepithelium as well as in the facial mesenchyme in association with the decreased size of the brain and nasal process (Supplementary Material, Fig. S4C). This implies that mitochondrial dysfunction is associated with Casp9-mediated apoptosis in the regulation of tissue size.
Conversely, Bcl2, the founding member of the Bcl2 family of regulator proteins is considered an anti-apoptotic protein. Bcl2 encodes a mitochondrial membrane protein that functions as a pro-survival factor, and neuroepithelial overexpression of Bcl2 resulted in a 14–15% increase in the size of the brain and nasal process on the EP side compared with the non-EP control side (Supplementary Material, Fig. S4B), Brain vesicle, EP/non-EP side rate = 1.142 ± 0.110, P = 0.046; nasal process, EP/non-EP side rate = 1.154 ± 0.09, P = 0.0037). Furthermore in contrast to Bax overexpression, there was no ectopic activation of caspase activity in the brain or adjacent facial mesenchyme (Fig. 4D).
Figure 4.
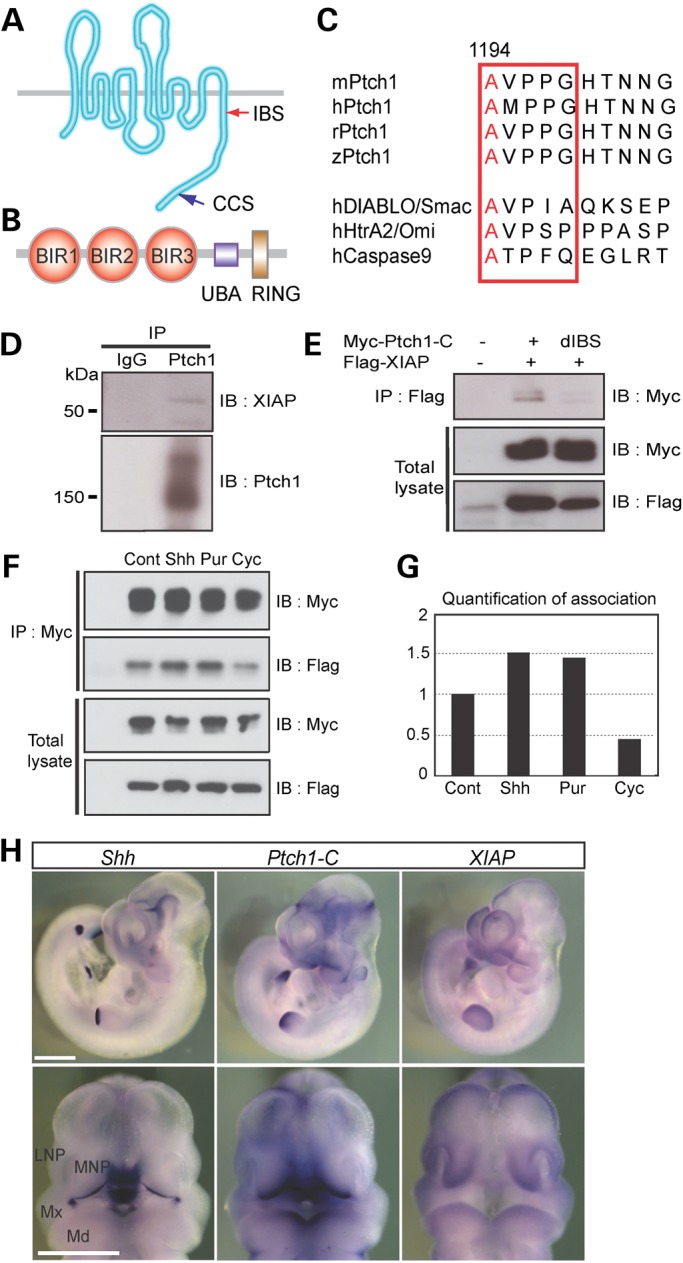
IAP-binding domain of Ptch1 associated with the BIR domain of XIAP. (A and B) Schematic representations of mouse Ptch1 (A) with IBS and caspase cleaved site (CCS) and XIAP (B) protein with Buculovirus IAP repeat domain (BIR, red circle), ubiquitin-associated domain (UBA, blue square) and RING zinc finger domain (RING, blown square). (C) Ptch1-C contains an IBS (red square) that is highly conserved and similar to known IAP-binding proteins. (D) Immunoprecipitation using Ptch1 antibody and western blot analysis of whole-cell lysates from E11.5 mouse embryos. Ptch1 interacts with XIAP. (E) Immunoprecipitation using flag antibody and western blot analysis of whole-cell lysates from myc-tagged Ptch1 C-terminal (Myc-Ptch1-C) or IBS deletion mutant (dIBS) and flag-tagged XIAP (Flag-XIAP) transfected human embryonic kidney cells (HEK293T). dIBS of Ptch1-C cannot associated with XIAP. (F) Association of Ptch1 and XIAP is affected by Shh ligand, purmorphamine (Pur, smoothened agonist) and cyclopamine (Cyc, smoothened antagonist). (G) Quantification of Ptch1 and XIAP association measured from (F). (H) Whole-mount in situ hybridization of Shh, Ptch1-C and XIAP mRNA expression in E10.5 mouse embryos. Upper panels show left side of whole embryos, whereas lower panels show frontal views. LNP, lateral nasal process; Md, mandibular process; MNP, medial nasal process; Mx, maxillary process. Scale bars: 1 mm in (H).
These data strongly suggest that mitochondria-dependent cell survival in the neuroepithelium affects cell viability and proliferation in the nasal process such that the character of the brain influences that of the facial mesenchyme. Furthermore, the progenitor tissue hypoplasia and agenesis associated with Shh dysfunction may be mediated by Casp9 initiated, mitochondrial-dependent cell death.
Ptch1 C terminal governs mitochondria-mediated cell death
Recently, Ptch1-C was shown to regulate cell death through the Casp9 complex in vitro (21). This implies that Ptch1 may influence mitochondria-dependent cell survival. To determine whether Ptch1 and Ptch1-C can induce Casp9-associated mitochondrial cell death in vivo, we overexpressed Ptch1 full length and Ptch1-C in the brain of E9.5 mouse embryos by electroporation. The FB and nasal process were noticeably smaller on the EP side compared with the non-EP control side (Fig. 3A, brain vesicle, EP/non-EP side rate = 0.773 ± 0.040, P = 0.0003; nasal process, EP/non-EP side rate = 0.909 ± 0.059, P = 0.017, Fig. 3B, brain vesicle EP/non-EP side rate = 0.809 ± 0.108, P = 0.001; nasal process, EP/non-EP side rate = 0.923 ± 0.056, P = 0.044). Immunohistochemistry revealed that the brain and facial abnormalities observed after Ptch1 or Ptch1-C overexpression correlated with elevated numbers of Casp9-labeled cells in the neuroepithelium and facial mesenchyme (Fig. 3C–E). These results demonstrate that Ptch1 can regulate cell death during brain and facial development and may be associated with Casp9-dependent mitochondrial dysfunction.
Figure 3.

Caspase cleaved C-terminal Ptch1 translocates to mitochondria during cell death. (A and B) Frontal view of mouse embryos EP with myc-tagged Ptch1 full length (A) and Ptch1 C terminal (Ptch1-C) (B) with pEGFP as reference. Green area demarcates the brain vesicles. Red area indicates the nasal processes. Insets show unilateral electroporation of GFP in the forebrain. (C and D) Frontal sections of E9.5 mouse embryos labeled with cleaved Casp9 after unilateral brain electroporation with control, myc-tagged Ptch1 full-length (C) and Ptch1-C (D) vectors. Nuclei are stained with DAPI. (E) Ptch1 full-length overexpression (red) induces Casp9 activity (green) in mIMCD3 cells. (F) mIMCD3 cells transiently transfected with myc-tagged Ptc1-1/4C (Myc-Pct1-1/4C, green) co-localized with the mitochondria marker, MitoTracker Red. (G) Frontal views of control (DMSO) and 20 µm cyclopamine (Cyc, smoothened antagonist)-treated mouse embryos. (H) Frontal sections of cultured embryos, stained with Casp9 and AP2α. (I) Healthy mitochondria in mIMCD3 cells present with a long tubular shape. 5 µm cyclopamine-treated cells exhibit mitochondria with small bean-shaped morphology. (J) Flag-Ptch1-EGFP stable cells stained with MitoTracker Red under normal and cyclopamine-treated conditions. Mitochondrial localized Ptch1 is indicated by yellow color. (K) Quantification of cyclopamine induces Ptch1 localization in mitochondria. FB, forebrain; FE, foregut endoderm; E, eye; HB, hindbrain. Scale bars: 0.5 mm in (A), (B) and (G); 0.1 mm in (E); 0.2 mm in (C), (D) and (H); 50 µm in (I) and (J).
Ptch1 activity is reduced in Hh loss-of-function mutants, which is commensurate with Ptch1 being a regulatory target of Hh signaling (29). However, Ptch1's function as a dependence receptor depends on the presence or absence of Hh ligand. In the absence of Hh, Ptch1 induces apoptosis, whereas its pro-apoptotic activity is blocked in the presence of Hh. Interestingly, Ptch1 has many isoforms that are Gli dependent or independent (30) and we hypothesized that Ptch1 isoforms may persist in Shh−/− mutants to mediate the apoptosis observed in association with Shh loss of function. To determine whether Ptch1 isoforms are expressed in Shh mutants, we performed RT-PCR analysis using five distinct Ptch1 exon 1 isoform-specific forward primers in conjunction with exon 3 reverse primers. Five Ptch1 isoforms were expressed in wild-type embryos, and some of these including Ptch1 (E1C-E2-E3) and Ptch1-C (E21-E22-E23) were still expressed in Shh mutant embryos (Supplementary Material, Fig. S5A and B). This result suggests that specific isoforms of Ptch1 may regulate mitochondrial cell death in Shh mutant embryos (Supplementary Material, Fig. S5C).
Interestingly, substrates generated through cleavage by caspases have been shown to localize to specific organelles such as mitochondria in association with their induction of cell death (31,32). Since Ptch1-C is cleaved by Casp3 this raised the question as to whether the cleaved portion of Ptch1-C also localizes to specific organelles in association with its cell death function. To examine the localization of the post-cleavage terminal portion of Ptch1-C (Ptch1-1/4C), we overexpressed myc-tagged terminal Ptch1-C (Myc-Ptch1-1/4C) in mouse embryonic fibroblast, NIH3T3FRT cells. Myc-Ptc1-1/4C localization overlapped with the mitochondrial marker, MitoTracker Red (Fig. 3F), suggesting that Ptch1-1/4C, the cleaved product of Ptch1-C, translocates to mitochondria in association with the induction of cell death.
Shh mutant embryos exhibit Ptch1-C expression and mitochondria-associated cell death (Fig. 2; Supplementary Material, Fig. S3), suggesting that Shh/Ptch1 signaling dysfunction may correlate with mitochondria-related cell death. Therefore to explore this idea, we treated embryos with the Smo antagonist cyclopamine, and observed a reduction in the size of the forebrain and nasal process in association with elevated levels of Casp9 in the neuroepithelium and facial mesenchyme together with abnormal mitochondrial function (Fig. 3G and H). This association between Shh signaling dysfunction with altered mitochondrial function and cell death is further supported by recently published studies showing cyclopamine affects mitochondrial function by disrupting mitochondrial membrane potential, leading to cytochrome c release and apoptosis (33,34).
To further examine the effects of Hh signaling loss of function on mitochondrial morphology, we examined NIH3T3 cells cultured in the absence or presence of cyclopamine. Under normal culture conditions in the absence of cyclopamine, MitoTracker Red staining of mitochondria revealed these organelles to be long and tubular (Fig. 3I). In contrast, when NIH3T3cells were cultured in the presence of cyclopamine, labeled mitochondria were observed to be small and bean shaped, a distinctive morphology that is indicative of cells undergoing apoptosis (Fig. 3I). Thus, Hh signaling dysfunction is associated with altered mitochondrial morphology and function, and cell death.
Next, to ascertain whether Ptch1 may be involved in regulating mitochondrial function, we examined the localization of a dual N-terminal Flag/C-terminal EGFP-tagged Ptch1 in NIH3T3FRT cells. Ptch1 protein exhibited increased co-localization with small bean-shaped mitochondria in cyclopamine-treated cells compared with non-treated controls as evidenced by the overlap of GFP with the mitochondrial marker MitoTracker Red (Fig. 3J and K). This result suggests that Ptch1 or more specifically, the most C-terminal part of Ptch1 which is cleaved by Casp3, translocates to the mitochondria. Thus, Ptch1 induction of cell death appears to be mediated through translocation of the most C-terminal portion of Ptch1 to the mitochondria.
XIAP inhibits Ptch1 induction of cell death in an Hh-dependent manner
Ptch1-C induction of cell death appears to occur in association with mitochondrial dysfunction and Hh signaling loss of function (Fig. 3). Therefore, its activity needs to be tightly controlled. However, our understanding of how this occurs is poorly understood. Many mechanisms have been identified to play a role in the regulation of cell survival and this includes numerous inhibitors and inducers of cell death. Interestingly, the inhibitor of apoptosis (IAP) family can inhibit caspase activity (Supplementary Material, Fig. S6A), which led us to postulate that IAPs might be able to limit the cell death function of Ptch1.
Consistent with this hypothesis, we found that the mouse Ptch1-C tail contains one predicted IAP-binding motif (IBM), AVPP (1194–1197), which is similar to the consensus sequence A-(V/T/I)-(P/A)-(F/Y/I/V) of IAP-binding proteins (Fig. 4A and C) (35). These motifs are conserved among several species of vertebrates but not invertebrates (Fig. 4C). To determine whether IAP family proteins could interact with Ptch1-C, we carried out binding assays via immunoprecipitaion and western blotting. Among several IAP family members, mouse X-linked IAP (mXIAP) was strongly associated with Ptch1-C (Fig. 4D, Supplementary Material, Fig. S6B). Interestingly, XIAP is the strongest inhibitor among the Birc family and can inhibit Casp9 activity through binding of the Baculovirus IAP repeat 3 (BIR3) domain (Fig. 4B) (28). Furthermore, deletion analysis of mXIAP and Ptch1-C confirmed binding between the AVPP site of Ptch1-C and XIAP (Fig. 4E). This led us to postulate that XIAP might inhibit induction of Casp9-mediated cell death by binding to the IBM of Ptch1-C.
To address this hypothesis, we explored the association between Ptch1 and XIAP in the presence of Shh ligand, a Smo agonist (purmorphamine) and a Smo antagonist (cyclopamine). Association between Ptch1 and XIAP increased in the presence of Shh- and purmorphamine-treated cells, but decreased in cyclopamine-treated cells (Fig. 4F and G). Since cyclopamine induces abnormal mitochondrial morphology and Ptch1-C induces changes in mitochondrial morphology, the dissociation of Ptch1-C and XIAP in cyclopamine-treated cells could underlie the induction of cell death. Consistent with this phenomenon, XIAP expression analyses in Shh−/− mutant embryos via RT-PCR and chromatin immunoprecipitation (ChIP) analysis (Supplementary Material, Fig. S7) showed that mouse XIAP is an Hh signaling target (36). In addition, Ptch1-C and XIAP mRNA are strongly expressed in vivo in neuroepithelial cells and facial mesenchyme adjacent to Shh producing ventral forebrain and oral epithelium in E10.5 mouse embryos (Fig. 4H). These results indicate that XIAP may inhibit Ptch1 induction of cell death in an Hh-dependent manner.
XIAP regulates cell survival in the brain and face of mouse embryos
Our data suggests that XIAP may regulate Ptch1-associated cell death during brain and facial development. Therefore, we explored a spatiotemporally specific role for XIAP in survival during brain development. XIAP shRNAi (XIAPi) overexpression (Supplementary Material, Fig. S1C and D) via unilateral electroporation resulted in a significantly smaller (24%) forebrain, and smaller (28%) nasal processes on the EP side compared with the control non-EP side (Fig. 5A, brain vesicle, EP/non-EP side rate = 0.857 ± 0.025, P = 0.0052; nasal process, EP/non-EP side rate = 0.824 ± 0.058, P = 0.017). These morphological changes were associated with elevated Casp9-induced cell death (Fig. 5B). In addition, overexpression of the XIAP inhibitor, Diablo (direct IAP-binding protein with low pl, also known as Smac), by electroporation into the neuroepithelium also resulted in Casp9 induction of cell death in the ventral forebrain (Fig. 5C and D, brain vesicle, EP/non-EP side rate = 0.85 ± 0.087, P = 0.0158). Consistent with our other observations of co-dependent brain and midline facial development, we also observed a corresponding increase in Casp9-related cell death in the adjacent facial mesenchyme resulting in a smaller nasal process (nasal process, EP/non-EP side rate = 0.789 ± 0.118, P = 0.0047). These results show that XIAP functions to promote cell survival and may help co-ordinate brain and facial development.
Figure 5.

XIAP inhibitor treated embryos develop HPE like phenotypes. (A and C) Frontal view of mouse embryos EP with XIAP shRNAi, and XIAP inhibitor, Diablo and pEGFP as a reference. Green area demarcates the brain vesicle. Red area indicates the nasal processes. Insets show unilateral electroporation of GFP in the forebrain. Quantification of relative size of (mean ± s.d.) of brain vesicles (Brain ve., green) and nasal processes (Nasal pro., red) from non-EP (Ct) and EP sides. (D) Diablo expression induces cell death in the ventral forebrain, but not in dorsal forebrain. (B, D) Cleaved Casp9 and AP2α staining in frontal sections of the front nasal region of EP embryos. Nuclei are stained with DAPI. (E) Frontal views of control (DMSO) and Embelin (100, 200 μm)-treated embryos. Blue brackets indicate the distance between the nasal processes. Red arrows illustrate the dorsal midline of the forebrain. (F and G) Quantification of midline length and relative size of (mean ± s.d.) of brain vesicles (Brain ve., green) and nasal processes (Nasal pro., red) from non-EP (Ct) and EP sides. (H–J) Embelin-treated embryos stimulate Casp9-positive cell death in ventral forebrain and neural crest (AP2α)-derived mesenchyme of medial nasal process, in associated with a decrease in Shh, Islet1/2 (Isl1/2) and Nkx2.1 activity in the ventral forebrain (I). Conversely, the domain of Pax6 expanded ventrally consistent with altered dorso-ventral patterning. White arrowheads demarcate the dorso-ventral border between Nkx2.1 and Pax6 expression. LNP, lateral nasal process; MNP, medial nasal process. Scale bars: 0.5 mm in (A)–(H) and (J); 0.2 mm in (I).
Embelin, a natural benzoquinone-derivative originally isolated from Embelia ribes plant, is a chemical inhibitor of XIAP that can associate with the BIR3 domain of XIAP and induce cell death in cultured cells (37). To determine whether Embelin also affects XIAP function in organisms, we treated whole mouse embryos in culture in the presence of Embelin. E8.5 embryos treated with 100 μm Embelin exhibited developmental arrest including cessation of heart beating and circulation at E9.5 (Supplementary Material, Fig. S8A). Embelin-treated embryos exhibited small body size, together with heart and tail looping defects (Supplementary Material, Fig. S8A) in association with extensive AP2α-labeled neural crest cell Casp9/Casp3-positive apoptosis and forebrain dorsalization through decreased Nkx2.1 and expanded Pax6 (Supplementary Material, Fig. S8B–D). More importantly, Embelin cultured embryos exhibit dose-dependent phenotypic severity including hyperplasia or agenesis of midline tissue (Fig. 5E–G, blue square brackets, brain vesicle, Embelin 100 µm/ dimethyl sulfoxide (DMSO) control = 0.697 ± 0.125, P = 0.04; Embelin 200 µm/DMSO control = 0.622 ± 0.040, P = 0.008, nasal process, Embelin 100 µm/DMSO control = 0.713 ± 0.087, P = 0.001, Embelin 200 µm/DMSO control = 0.0.569 ± 0.048, P < 0.001, midline length, Embelin 100 µm/DMSO control = 0.811 ± 0.050, P = 0.009, Embelin 200 µm/DMSO control = 0.591 ± 0.054, P = 0.001). These phenotypes are similar to Shh signaling mutants (11,38), cyclopamine-treated embryos (Fig. 3G) and humans with HPE. It is also reflective of the effects of a conditional deletion of Shh expressing cells (Fig. 2A). Embelin-treated embryos showed increased Casp9-positive cells in the ventral forebrain and in association with AP2α-positive facial mesenchyme in the medial nasal process (Fig. 5H). This effect correlates with a decrease in Shh signaling and in further support of this phenomenon, well-known downstream targets of Shh signaling including neuronal markers such as Islet1/2 (Fig. 5I), and the ventral forebrain marker, Nkx2.1 were decreased, while the domain of the dorsal marker, Pax6 was expanded ventrally (Fig. 5J). These changes are indicative of forebrain dorsalization which is a well-known outcome of Shh loss of function. Consistent with these results, RT-PCR analysis of Shh signaling molecules and IAP markers in Embelin-treated embryos revealed downregulation of the Shh signaling pathway (Supplementary Material, Fig. S9). These results suggested that XIAP plays an important role in Hh signaling during brain and facial development.
XIAP inhibits Ptch1 induction of cell death
To test whether XIAP can inhibit Ptch1 induction of cell death (Fig. 6A), we overexpressed XIAP and Ptch1 in human embryonic kidney (HEK293T) cells and in the neuroepithelium of avian embryos. XIAP overexpression did indeed block Ptch1 full-length and C-terminal (Ptch1-C) induction of cell death (Fig. 6B–D). Ptch1 full-length (39.17 ± 0.027%, P = 0.018) and Ptch1-C (45.21 ± 0.0036%, P = 0.021) overexpressing HEK293T cells displayed a 21–27% increase in Trypan blue-positive cell death compared with control cells (17.79% ± 0.010%) (Fig. 6B). Similarly, overexpression via electroporation in the neuroepithelium of HH10 stage avian embryos showed that Ptch1 induction of cell death (TUNEL positive, 16.8 ± 1.12 cells) can be reduced by co-electroporation of XIAP in a dose-dependent manner (0.5 µg DNA, 5.8 ± 2.35; 1 µg, 5.6 ± 3.57; 2 µg, 2.9 ± 1.79, P < 0.001) (Fig. 6C and D). These results indicate that XIAP can inhibit Ptch1 induction of cell death.
Figure 6.

XIAP inhibits Ptch1 induction of cell death. (A) Schematic illustration of inhibition of mouse Ptch1 induction of cell death by XIAP. (B) Quantification of XIAP inhibition of Ptch1 full-length and C-terminal induction of Trypan blue-positive cell death in HEK293T cells. (C and D) XIAP inhibits Ptch1 induction of TUNEL-positive cell death in chick neural tube. (E) XIAP rescued nasal process morphology in Diphtheria toxin treated ShhcreGFP; iDTR embryos. (F and G) Quantification of relative area of brain vesicles (Brain ve., green) and nasal processes (Nasal pro., red) from non-EP (Ct) and XIAP EP sides of Control and ShhcreGFP; iDTR embryos. (H) XIAP inhibits Casp9-dependent cell death in the ventral forebrain and mesenchyme of the medial nasal processes (white arrows). Scale bars: 0.1 mm in (C) and (J); 0.5 mm in (E) and (H).
Furthermore to examine whether XIAP can also inhibit cell death in the developing mouse brain, we electroporated XIAP into wild-type and ShhcreGFP; iDTR embryos. The XIAP EP side displayed a bigger (21%, 27%) forebrain vesicle and (28%, 24%) nasal process in both wild-type (brain vesicle, EP/non-EP side rate = 1.24 ± 0.033, P = 0.035; nasal process, EP/non-EP side rate = 1.28 ± 0.045, P = 0.037) and ShhcreGFP; iDTR (brain vesicle, EP/non-EP side rate = 1.27 ± 0.041, P = 0.034; nasal process, EP/non-EP side rate = 1.24 ± 0.046, P = 0.044) embryos (Fig. 6E–G). Interestingly, the medial nasal process mesenchyme in ShhcreGFP; iDTR embryos exhibited decreased Casp9-positive cell death on the EP side compared with the non-EP control side (Fig. 6H, arrows). These results demonstrate that XIAP can inhibit cell death in the forebrain and medial nasal process, preventing the effects of Shh loss of function.
Hh signaling-dependent Ptch1 and XIAP association and separation in primary cilia
Endogenous XIAP localizes in primary cilia on the apical surface of the ventral forebrain cells close to the medial nasal process (Fig. 7A). In addition, XIAP localizes in the cytoplasm during interphase, but later during mitotic pro-, prometa-, meta- and anaphase, XIAP becomes predominantly localized at the centrosome (Supplementary Material, Fig. S10A). Interestingly, Ptch1 shows a similar subcellular localization during the entire cell cycle (Supplementary Material, Fig. S10B). These similar patterns of localization led us to postulate that Ptch1 induction of cell death and its regulation by XIAP occurs in primary cilia. To examine any changes in localization of Ptch1 and XIAP, we established NIH3T3 cell lines which stably expressed EGFP-tagged Ptch1 and mCherry-tagged XIAP. Under serum starvation and purmorphamine (Smo antagonist)-treated conditions, Ptch1 (EGFP) and XIAP (mCherry) were observed to be co-localized in cilia (Fig. 7B). In contrast, in the presence of Shh, Ptch1 protein was no longer observed in primary cilia, but XIAP remained localized in cilia. Conversely, in cyclopamine-treated cells, Ptch1 protein localized to primary cilia, whereas XIAP did not (Supplementary Material, Fig. S10). These results are consistent with a separation of Ptch1 and XIAP during the regulation of cell death in response to Hh signaling loss of function (Figs 4F, G and 7).
Figure 7.

Ptch1 and XIAP are associated in primary cilia. (A) XIAP and acetylated-tubulin (Ace-tub) expression in frontal sections of E10.5 mouse embryos. Lower panels are high magnification of white dashed square of upper panels. XIAP protein localized to primary cilia (white arrowheads) and cytoplasm in ventral forebrain neuroepithelium. Ace-tub and DAPI label primary cilia and nuclei, respectively. (B) N-terminal Flag/C-terminal EGFP-tagged Ptch1 and mCherry-tagged XIAP are stably expressed in NIH3T3FRT cells under serum starvation (SS), Sonic Hh condition medium (Shh), purmorphamine (Smo agonist, Pur) and cyclopamine (Smo antagonist, Cyc)-treated conditions. (C) Model of Ptch1 (blue) and XIAP (green circle) association with/without Hh (pink circle) ligand in primary cilia. Without Hh, Ptch1 separates from XIAP (1). Ptch1 C-terminal domain is cleaved (2) and then C-terminal product induces mitochondrial dysfunction leading to cell death (3). When Hh binds to Ptch1 (4), Hh and Ptch1 are degraded leading to cell survival (5, 6). Scale bars: 50 µm in (A); 5 µm in (B).
DISCUSSION
Our data present new insights into the co-ordination of brain and facial development during mouse embryogenesis. We observed that Hh signaling, co-regulates cell proliferation and apoptosis in the forebrain and adjacent facial mesenchyme and thus subsequently influences the size of both the brain and face (Fig. 1). Our observations that Shh signaling regulates cell survival in the brain, which subsequently affects the adjacent nasal process and thus facial development, is supported by several mouse mutant and chick embryo analyses (39–41). For example, Shh and Six3 double heterozygote mice exhibit HPE. Six3 is a positive transcriptional regulator of Shh in the forebrain neuroepithelium and hence the compound mutant phenotype is thought to be caused by a tissue-specific decrease of Shh activity (12,13,39,40,42–44). Furthermore, a Six3-cre deletion of Shh specifically in the ventral forebrain elicits an HPE phenotype similar to Shh−/− mutant embryos. Thus, midline tissues such as the ventral forebrain and the signals that emanate from them such as Shh are essential for co-ordinately regulating brain and facial growth and development.
In this study, we focused on the functions of Ptch1 as an Hh receptor and cell death inducer via mitochondria in integrating brain and facial development. A role for the mitochondria in regulating forebrain and adjacent facial mesenchyme proliferation and apoptosis was supported by Bcl2, Bax, Ptch1 and XIAP overexpression. Bcl2 encodes a mitochondrial membrane protein that functions as a pro-survival factor and overexpression of Bcl2 led to an increase in forebrain and facial prominence size. In contrast, overexpression of Bax, which regulates mitochondrial membrane permeability and functions as a pro-apoptotic factor, resulted in a corresponding decrease in forebrain and facial prominence size and was associated with elevated levels of Casp9-positive cells in the neuroepithelium and adjacent facial mesenchyme. Thus, perturbation of mitochondrial-specific proteins specifically in the neuroepithelium, affected neuroepithelial and facial mesenchyme proliferation and apoptosis, which had a direct subsequent effect on the size of the brain and nasal process. Thus the function of the mitochondria itself appears to be a strong regulator of brain and facial development and growth.
Similar to the outcome of enhanced Bax activity, overexpression of Ptch1, which is a receptor for Shh and can localize to mitochondria, resulted in elevated Casp9-mediated cell death in the neuroepithelium and facial mesenchyme and a correspondingly smaller forebrain and adjacent nasal process. Furthermore, blocking the function of XIAP which is an IAP, also resulted in a reduction in the size of the forebrain and adjacent nasal process similar to the effects of Bax or Ptch1 overexpression.
Recently, the co-receptor CDON was also shown to function as a ‘dependence receptor’, in that it actively triggers apoptosis in the absence (45).The pro-apoptotic activity of unbound CDON (cell adhesion molecule-related/downregulated by oncogenes) protein in activating Casp9 is similar to the results we obtained with respect to Ptch1 function. This raises some interesting questions as to whether CDON and Ptch1 may function via a similar mechanism. Interestingly, the intracellular domain of mouse CDON contains a candidate IAP-binding site (AVP; 1090–1092 aa), which suggests that both Ptch1 and CDON may function as receptors in the Hh signaling pathway and regulate cell survival via XIAP or another IAP family protein member. Furthermore, it is interesting to note that other cell death dependence receptors have candidate IAP-binding sites in their intracellular C terminus, suggesting that the regulation of survival by IAP family members may be a common part of their machinery.
Our observation that the Ptch1-C can translocate to mitochondria in association with inhibition of Hh signaling (Fig. 3) suggests that Ptch1 localization may be associated with mitochondrial dysfunction. Interestingly, it has been reported that C-terminal cleavage of ErbB2 by caspases inhibits Bcl2 in mitochondria and induces cell death (46). Ptch1 C-terminus translocation may also lead to inactivation of mitochondrial cell survival proteins during the induction of apoptosis.
Interestingly, we found that XIAP can bind to the C terminus of Ptch1 through an evolutionary conserved motif but more importantly, XIAP can suppress the cell death inducing function of Ptch1. Thus, XIAP promotes the survival of forebrain neuroepithelium and neural crest cell-derived facial mesenchyme by inhibiting the Hh receptor, Ptch1, and thereby blocking mitochondria-associated cell death.
It is well established that Ptch1 localizes to primary cilia in the regulation of Hh signaling (10) and databases of cilia-associated proteins such as Cildb (http://cildb.cgm.cnrs-gif.fr/) (47) and CepDB (Centrosome and cilia protein database, http://www.cebi.sdu.dk/CepDB/) further support this idea. As a mechanistic part of our studies exploring the interactions between Ptch1 and XIAP, we discovered that both the C-terminal potion of Ptch1 and XIAP were capable of co-localization in primary cilia, and that XIAP binds Ptch1 (Figs 4F, G and 7A, B; Supplementary Material, Fig. S10). Furthermore, the interaction between Ptch1 and XIAP and their co-localization in cilia are Hh signaling dependent. These observations suggest that XIAP may associate with Ptch1 in primary cilia and protect Ptch1 from translocation into mitochondria providing an important survival mechanism in cilia. Thus, XIAP plays an important role in coordinating brain and facial development by regulating the effects of Hh signaling through Ptch1 in mitochondria and cilia-mediated cell survival and apoptosis.
This represents an important advance in our understanding of the pathogenesis of HPE. Mitochondria regulate many cell functions including energy conversion (ATP production, aerobic respiration) and apoptosis, or programmed cell death (48,49). Furthermore, mitochondrial dysfunction is known to contribute to hypoxia resulting in reduced oxygen concentration in aerobic tissues (49). Previous reports indicate that hypoxia may be an environmental factor that influences the variable spectrum of brain and craniofacial anomalies characteristic of HPE (50,51). Thus, our observations support the notion that mitochondrial dysfunction may be a developmental risk factor associated with the pathogenesis of HPE.
Mutations in Shh signaling are associated not only with developmental defects such as HPE but also with specific brain and skin cancers such as meduloblastoma and basal cell carcinoma. Furthermore, Hh signaling is constitutively active in ∼25% of human tumors and in Nevoid basal cell carcinoma syndrome (also known as Gorlin syndrome) in which patients with mutations in PATCHED1 exhibit tumors of the brain, skin and muscle. It is also interesting to note that XIAP is upregulated in many human tumors (52,53). Our evidence for roles of Ptch1 and XIAP in cell survival demonstrate that not only is XIAP a key regulator of brain and facial development but it also implies that XIAP could be a novel potential target for chemical inhibition in the treatment of Hh signaling-related tumors.
MATERIALS AND METHODS
Constructs
Mouse Bcl2 (NM_009741), Bax (NM_007527), Birc2 (NM_007465.2), Birc3 (NM_007464.3), Birc5 (Survivin40, NM_009689.2) and Diablo (Smac, NM_023232) were cloned into pGEM-T Easy vector (Promega) by standard PCR cloning using specific primer sets. mPtch1-pRK5 (gift from Dr Beachy), and Flag-mGli1, Flag-mGli2 (gift from Dr Hui) and Flag-XIAP (gift from Dr Silke) were used as templates for PCR. Constructs were subcloned into N-terminal Flag and Myc-tagged pcDNA5/FRT vector (Invitrogen, gift from Dr Sato). Mouse Ptch1-C and XIAP shRNAs were cloned into pSuper (OligoEngine) using the following DNA oligos. All pSuper-based shRNAi constructs were checked via western blot (Supplementary Material, Fig. S1A and B).
Cloning primers sets and DNA oligos
Mouse Bcl2-F; 5′-TAACTCGAGATGGCGCAAGCCGGGA-3′
Mouse Bcl2-R; 5′-AGGGTACCTCACTTGTGGCCCAGGT-3′
Mouse Bax-F; 5′-TTAACTCGAGATGGACGGGTCCGGGG-3′
Mouse Bax-R; 5′-TTGCGGCCGCTCAGCCCATCTTCTTC-3′
Mouse Birc2-F; 5′-CCGCTCGAGATGGACAAAACTGTCT-3′
Mouse Birc2-R; 5′-AAGCGGCCGCTCATGAGAGAAATGT-3′
Mouse Birc3-F; 5′-AAGTCGACATGGTTCAAGACAGCGC-3′
Mouse Birc3-R; 5′-AAGGATCCTCAGGAGAGAAATGTGC-3′
Mouse Birc5-F; 5′-TTAACTCGAGATGGGAGCTCCGGCG-3′
Mouse Birc5-R; 5′-CGGGATCCTCAGTCCTTATTCTC-3′
Mouse Diablo-F; 5′-CTCGAGATGGCGGCTCTGAGAAGTT-3′
Mouse Diablo-R; 5′-TCAATCTTCACGCAGGTAGGCCTCC-3′
Mouse Ptch1-C-F; 5′-CTCGAGATGGGACCGTGTCCTGA-3′
Mouse Ptch1-C-R; 5′-TCATTGGGAGCTGCTCCCCCACGGC-3′
Mouse XIAP-F; 5′-CGCTCGAGATGACTTTTAACAGT-3′
Mouse XIAP-R; 5′-GCGGCCGCCTAAGACATAAAAATTT-3′
mShhi-1-F; 5′-GATCCCCGCAGTTTATTCCCAACGTATTCAAGAGATACGTGGG GAATAAACTGCTTTTTA-3′
mShhi-1-R; 5′-AGCTTAAAAAGCAGTTTATTCCCAACGTATCTCTTGAATACGTTGGGAATAAACTGCGGG-3′
mShhi-2-F; 5′-GATCCCCGGGAAGATCACAAGAAACTTTCAAGAGAAGTTTCTTGTGATCTTCCCTTTTTTA-3′
mShhi-2-R; 5′-AGCTTAAAAAGGGAAGATCACAAGAAACTTCTCTTGAAAGTTTCTTGTGATCTTCCCGGG-3′
hmXIAPi-1-F; 5′-GATCCCCGGAGCAGCTTGCAAGAGCTTTCAAGAGAAGCTCTTGCAAGCTGCTCCTTTTTA-3′
hmXIAPi-1-R; 5′-AGCTTAAAAAGGAGCAGCTTGCAAGAGCTTCTCTTGAAAGCTCTTGCAAGCTGCTCCGGG-3′
hmXIAPi-2-F; 5′-GATCCCCGTAGTAGTCCTGTTTCAGCTTCAAGAGAGCTGAAACAGGACTACTACTTTTTA-3′
hmXIAPi-2-R; 5′-AGCTTAAAAAGTAGTAGTCCTGTTTCAGCTCTCTTGAAGCTGAAACAGGACTACTACGGG-3′
hmXIAPi-3-F; 5′-GATCCCCGTCATTTCAGAACTGGCCGTTCAAGAGACGGCCAGTTCTGAAATGACTTTTTA-3′
hmXIAPi-3-R; 5′-AGCTTAAAAAGTCATTTCAGAACTGGCCGTCTCTTGAACGGCCAGTTCTGAAATGACGGG-3′
Chromatin immunoprecipitation
Flag-mGli1-EGFP and Flag-mGli2-EGFP stable NIH3T3FRT cell lines (Invitrogen) were made via transfection and hygromycin selection. Three Gli transcriptional binding sites were identified in the 5′ upstream locus of XIAP using the UCSC genome browser (http://genome.ucsc.edu/). ChIP experiments were performed essentially as previously described (54) using FlagM2 antibody (Sigma). PCR was performed using the following primer sets.
Chip primers
Mouse XIAP-GBS1-Chip-F; 5′-CCTGTCTCGTCATTCCTTC-3′
Mouse XIAP-GBS1-Chip-R; 5′-AACCATGGAAGGCAGAACAC-3′
Mouse XIAP-GBS2-Chip-F; 5′-CCAGCCCTCGCTTGGAATTA-3′
Mouse XIAP-GBS2-Chip-R; 5′-AACTTGAGTCTGTTCTCTCC-3′
Mouse XIAP-GBS3-Chip-F; 5′-GCTCATCTCAGCACTCAGCA-3′
Mouse XIAP-GBS3-Chip-R; 5′-CTGGCTGCAAACTTGCTATG-3′
Quantitative RT-PCR
The heads of cultured embryos treated with DMSO and Embelin were collected and processed with TRIzol regent (Invitrogen) for total mRNA isolation (Supplementary Material, Fig. S7, red line). First-strand cDNA was synthesized using the SuperScript III first-strand synthesis system (Invitrogen) followed by PCR with specific primer sets to measure cDNA concentration.
RT-PCR primer sets
GAPDH-140-F; 5′-GACCCCTTCATTGACCTCAACTAC-3′
GAPDH-140-R; 5′-TCTCGCTCCTGGAAGATGGTGATG-3′
Ptch1-E1-RT-F; 5′-ATGGGAGAGGGAGGAAAGATCCTT-3′
Ptch1-E1-RT-R; 5′-AGATGGACCACGGTTGCTCTAGAT-3′
Ptch1-E1B-RT-F; 5′-AACATGGCCTCGGCTGGTAAC-3′
Ptch1-E1C-RT-F; 5′-TTGATGTGAACCTCACGGTCAGCG-3′
Ptch1-E13-RT-R; 5′-TAATTCTCGACTCACTCGTCCACC-3′
Ptch1-Exon21-F; 5′-TCCTTCTTTGGACCGTGTCCTGAG-3′
Ptch1-Exon22-F; 5′-TCTCCAGCCAATGGCCTAAACCGA-3′
Ptch1-Exon22-R; 5′-GACCGGGCAAAGACAGGGTTTTCT-3′
Ptch1-Exon22-R; 5′-CTAGGGCCAGAATGCCCTTCAGTA-3′
Shh-RT-F; 5′-GCCTACAAGCAGTTTATTCCCAAC-3′
Shh-RT-R; 5′-ACTCCAGGCCACTGGTTCATCACA-3′
XIAP-F; 5′-CTCAGTTAACAAGGAGCAGCTTGC-3′
XIAP-R; 5′-TGTTCCCAAGGGTCTTCACTTGGC-3′
Mouse strains and embryos
iDTR (22) and Ptch1+/− mice were obtained from the Jackson Laboratory (Bar Harbor, ME) (55). Shh+/− mice were obtained from Dr Phil Beachy (Stanford, CA). Shhflox and ShhcreGFP mice were obtained from Dr Brain Harfe (Gainesville, Florida) (23,56). The generation and genotyping of mutant embryos were performed as described (55,57).
Study approval
Mice were housed in the Laboratory Animal Services Facility at the Stowers Institute for Medical Research according to IACUC animal welfare guidelines.
In situ hybridization
Whole-mount digoxigenin in situ hybridization was performed according to standard procedures (58) using the following probes: Ptch1, Shh and XIAP. Whole-mount embryos were photographed under a dissecting microscope with a digital camera (Leica and Nikon).
Immunofluorescence for cell line and embryonic tissue
Cultured cells were fixed in 1.6% paraformaldehyde (PFA)/0.1% phosphate-buffered saline (PBS) for 10 min at RT and then permeabilized with 0.2% Triton X-100/PBS for 10 min. Dissected mouse and chick embryos were fixed in 4% PFA/PBS for 1.5–2 h or 1% formamide overnight at 4°C and then were embedded in OCT and frozen. Alternatively, embryos were fixed in Bouin's fixative (saturated picric acid : formaldehyde : glacial acetic acid = 15 : 5 : 1) overnight at 4°C and then were embedded in paraffin (Paraplast, Sigma). For antigen retrieval, tissue sections were treated twice with 0.01% sodium citrate, pH 6, solution for 15 min in a microwave oven. Three percent of bovine serum albumin/0.1% PBS/0.1% Tween 20 was used for blocking and antibody incubation. First antibodies; mouse anti-acetylated tubulin (1/2000, Sigma, T6793), mouse anti-AP2α (1/50, 3B5, Iowa hybridoma), rabbit polyclonal anti-cleaved Casp3 (1/300, Cell Signaling Technologies, #9661S), anti-cleaved Casp9 (1/100, Cell Signaling Technologies, #9509), anti-GFP (1/1000, Invitrogen, A6455), chicken anti-GFP (1/1000, Aves labs Inc., GFP-1020), anti-Pax6 (1/100, Millipore, AB2237), rat monoclonal anti-PECAM (CD31) (1/100, BD Bioscience, #553370), anti-phosho-histone H3 (Ser10) (1/300, Millipore, 06-570) and anti-Shh (1/50, Iowa hybridoma, 5E1) were incubated overnight at 4°C. Second antibodies were donkey anti-mouse, anti-rabbit and anti-goat Alexa Fluor488, 546 and 647 (1/200–1/500, Invitrogen). MitoTraker Red CMXRos (100 nm, Invitrogen, M7512) was used for mitochondrial staining in cell lines. DAPI (1/1000, Sigma, D9564) was used for nuclear staining. Fluorescent confocal images were acquired using a LSM-510-VIS and Upright Pascal (Carl Zeiss). Statistical analyses were performed using a two-tailed Student's t-test. Quantification of Ptch1/XIAP localization together with acetylated tubulin was performed using ImageJ software (Supplementary Material, Fig. S10; Fig. 4F and G).
Cell death assay
Cell death was determined by immunohistochemistry using anti-cleaved Casp3 (1/300, Cell signaling), anti-cleaved Casp9 (1/100, Cell signaling) and by Terminal deoxynucleotidyl TUNEL analysis. TUNEL assay was performed with in situ cell death detection kit (Roche Diagnostics Corporation) according to the manufacturer's instructions.
In vivo chick and in vitro mouse brain electroporation
Plasmid DNA was prepared in PBS at 1–5 μg/μl with 0.01% Fast Green (OmniPur). All constructs were EP into the mouse brain at E9.5 (59) and into the chick neural tube of Hamburger–Hamilton Stages 10–12 (60). A 70 V (mouse embryos) or 25 V (chick embryos), 50 ms square pulse was applied five times via chamber-type electrodes (CUY520P20, NEPA GENE) or platinum electrodes using the electroporator CUY 21 (BEX CO. LTD). After culture, the mouse head, and the neural tube at the forelimb level of chick embryos were sectioned frontally or transversely and processed for fluorescence immunohistochemistry.
Whole embryo culture and chemical treatment
Pregnant mice were euthanized, and E8.0–8.5 and E9.5 embryos were EP and incubated for 24 h in culture bottles containing 1 ml of 100% rat serum (Harlan Laboratory) supplemented with 2 mg/ml glucose and penicillin–streptomycin under N2 balanced 5% O2 for E8.0–8.5 or 20% O2 for E9.5. Cyclopamine (5 mm in DMSO stock, Toronto Research Chemicals, C988400), DT (10 mg/ml in rat serum, MBL, RK-01-517), Embelin (50 mm in DMSO stock, Sigma-Aldrich, E1406-10MG) and Purmorphamine (10 mm in DMSO stock, EMD Biosciences. #540220) were added directly to the culture medium.
Quantification of brain vesicle and nasal process area
After culture, frontal and lateral images of embryos were collected with a Leica dissection microscope and camera. The outline of the brain vesicle and nasal process in cultured embryos were demarcated by standard landmarks (Supplementary Material, Fig. S2) (16). For embryos treated with DT, cyclopamine and embelin, the normalization of individual embryonic size was based on the distance between the nose and isthmus (Supplementary Material, Fig. S2B). For EP embryos, the area of control non-EP side and control-treated embryos was established as a grid of 1.0 area. The area of EP and chemically treated embryos was compared with controls. Quantification of brain vesicle and nasal process area was calculated using a minimum of three embryos using ImageJ software. Results were evaluated using the Student's t-test.
Immunoprecipitation and western blot
For the production of whole-cell lysates, cells were lysed in RIPA buffer (20 mm Tris–HCl, pH 8.0, 150 mm NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and protease inhibitor) or buffer B (20 mm HEPES, 0.5% Nonidet P-40, 250 mm NaCl, 10% glycerol, 2 mm EDTA and protease inhibitor). For immunoprecipitation, antibodies were coupled to Protein A/G beads (Santa Cruz). Myc- and Flag-tagged proteins were immunopurified from clarified supernatant with the use of anti-Myc and anti-Flag affinity gel, respectively (Sigma). The primary antibodies used were mouse anti-Flag (Sigma, 1/10 000), anti-Myc (Sigma, 1/1000), anti-α-tubulin (Sigma, 1/5 000), rabbit anti-Ptch1 (Santa Cruz, 1/500) and goat anti-Ptch1 (Santa Cruz, 1/500). Secondary antibodies and conjugates included donkey anti-mouse, anti-rabbit horseradish peroxidase (HRP) antibodies (GE HealthCare, 1/10 000) and anti-goat HRP antibodies (Jackson Lab, 1/10 000) and mouse and rabbit TrueBlot™ ULTRA HRP antibodies (eBioscience, 1/1000). After overnight binding at 4°C, the beads were washed with buffer B and eluted with 2× SDS buffer or Flag peptide (Sigma). Quantification of band intensity in immunoprecipitation and western blot experiments was performed with ImageJ software (Fig. 4F and G).
Cell culture and chemicals treatment
Human embryonic kidney 293 cells (HEK293T, ATCC), NIH3T3FRT (Invitrogen) and Mouse Inner Medullary Collecting Duct Cells (mIMCD3, ATCC) were maintained at 37°C in 5% CO2 in air in high glucose DMEM (GIBCO) media containing 10% fetal bovine serum (Sigma-Aldrich) and penicillin–streptomycin (GIBCO). Cells grown to 60–80% confluency were transfected with constructs using standard transfection regents, Fugne6 (Roche Molecular Biochemicals) or Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Shh-conditioned media was generated by transfection of an Shh expression vector into HEK293T cells. The Smoothened agonist (purmorphamine, 10 µm) and antagonist (cyclopamine, 10 µm) were added directly to the culture medium.
SUPPLEMENTARY MATERIAL
FUNDING
This work supported by the Stowers Institute for Medical Research and the National Institute of Dental and Craniofacial Research (R01 DE 016082).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs P.A. Beachy and B. Harfe for the Shh+/−, and Shhfx/+ and ShhcreGFP+/− mice, respectively, and Drs P.A. Beachy, R. Krumlauf, A.P. McMahon, S. Sato, C. Sato, M.P. Scott and J. Silke for providing probes for in situ hybridization, expression vectors, antibodies and cell line.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Trainor P.A. Craniofacial birth defects: the role of neural crest cells in the etiology and pathogenesis of Treacher Collins syndrome and the potential for prevention. Am. J. Med. Genet. A. 2010;152A:2984–2994. doi: 10.1002/ajmg.a.33454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Demyer W., Zeman W., Palmer C.G. The face predicts the brain: diagnostic significance of median facial anomalies for holoprosencephaly (arhinencephaly) Pediatrics. 1964;34:256–263. [PubMed] [Google Scholar]
- 3.Cohen M.M., Jr Perspectives on holoprosencephaly: part III. Spectra, distinctions, continuities, and discontinuities. Am. J. Med. Genet. 1989;34:271–288. doi: 10.1002/ajmg.1320340232. [DOI] [PubMed] [Google Scholar]
- 4.Cohen M.M., Jr, Shiota K. Teratogenesis of holoprosencephaly. Am. J. Med. Genet. 2002;109:1–15. doi: 10.1002/ajmg.10258. [DOI] [PubMed] [Google Scholar]
- 5.Aoto K., Yayoi S., Higashiyama D., Shiota K., Motoyama J. Fetal ethanol exposure activates protein kinase A and impairs Shh expression in prechordal mesendoderm cells in the pathogenesis of holoprosencephaly. Birth Defects Res. A Clin. Mol. Teratol. 2008;82:224–231. doi: 10.1002/bdra.20447. [DOI] [PubMed] [Google Scholar]
- 6.Barr M., Jr, Hanson J.W., Currey K., Sharp S., Toriello H., Schmickel R.D., Wilson G.N. Holoprosencephaly in infants of diabetic mothers. J. Pediatr. 1983;102:565–568. doi: 10.1016/s0022-3476(83)80185-1. [DOI] [PubMed] [Google Scholar]
- 7.Wallis D., Muenke M. Mutations in holoprosencephaly. Hum. Mutat. 2000;16:99–108. doi: 10.1002/1098-1004(200008)16:2<99::AID-HUMU2>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 8.Varjosalo M., Li S.P., Taipale J. Divergence of hedgehog signal transduction mechanism between Drosophila and mammals. Dev. Cell. 2006;10:177–186. doi: 10.1016/j.devcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 9.Briscoe J., Therond P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013;14:416–429. doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- 10.Rohatgi R., Milenkovic L., Scott M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 11.Jeong J., Mao J., Tenzen T., Kottmann A.H., McMahon A.P. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 2004;18:937–951. doi: 10.1101/gad.1190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu D., Marcucio R.S. A SHH-responsive signaling center in the forebrain regulates craniofacial morphogenesis via the facial ectoderm. Development. 2009;136:107–116. doi: 10.1242/dev.026583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu D., Helms J.A. The role of sonic hedgehog in normal and abnormal craniofacial morphogenesis. Development. 1999;126:4873–4884. doi: 10.1242/dev.126.21.4873. [DOI] [PubMed] [Google Scholar]
- 14.Echelard Y., Epstein D.J., St-Jacques B., Shen L., Mohler J., McMahon J.A., McMahon A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- 15.Persson M., Stamataki D., te Welscher P., Andersson E., Bose J., Ruther U., Ericson J., Briscoe J. Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. 2002;16:2865–2878. doi: 10.1101/gad.243402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parsons T.E., Schmidt E.J., Boughner J.C., Jamniczky H.A., Marcucio R.S., Hallgrimsson B. Epigenetic integration of the developing brain and face. Dev. Dyn. 2011;240:2233–2244. doi: 10.1002/dvdy.22729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roessler E., Belloni E., Gaudenz K., Jay P., Berta P., Scherer S.W., Tsui L.C., Muenke M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat. Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- 18.Chiang C., Litingtung Y., Lee E., Young K.E., Corden J.L., Westphal H., Beachy P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- 19.Aoto K., Shikata Y., Imai H., Matsumaru D., Tokunaga T., Shioda S., Yamada G., Motoyama J. Mouse Shh is required for prechordal plate maintenance during brain and craniofacial morphogenesis. Dev. Biol. 2009;327:106–120. doi: 10.1016/j.ydbio.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 20.Thibert C., Teillet M.A., Lapointe F., Mazelin L., Le Douarin N.M., Mehlen P. Inhibition of neuroepithelial patched-induced apoptosis by sonic hedgehog. Science. 2003;301:843–846. doi: 10.1126/science.1085405. [DOI] [PubMed] [Google Scholar]
- 21.Mille F., Thibert C., Fombonne J., Rama N., Guix C., Hayashi H., Corset V., Reed J.C., Mehlen P. The Patched dependence receptor triggers apoptosis through a DRAL-caspase-9 complex. Nat. Cell Biol. 2009;11:739–746. doi: 10.1038/ncb1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buch T., Heppner F.L., Tertilt C., Heinen T.J., Kremer M., Wunderlich F.T., Jung S., Waisman A. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat. Methods. 2005;2:419–426. doi: 10.1038/nmeth762. [DOI] [PubMed] [Google Scholar]
- 23.Harfe B.D., Scherz P.J., Nissim S., Tian H., McMahon A.P., Tabin C.J. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell. 2004;118:517–528. doi: 10.1016/j.cell.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 24.Cory S., Adams J.M. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 25.Wei M.C., Zong W.X., Cheng E.H., Lindsten T., Panoutsakopoulou V., Ross A.J., Roth K.A., MacGregor G.R., Thompson C.B., Korsmeyer S.J. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li P., Nijhawan D., Budihardjo I., Srinivasula S.M., Ahmad M., Alnemri E.S., Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 27.Srinivasula S.M., Ahmad M., Fernandes-Alnemri T., Alnemri E.S. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol. Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- 28.Riedl S.J., Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- 29.Wong S.Y., Reiter J.F. The primary cilium at the crossroads of mammalian hedgehog signaling. Curr. Top Dev. Biol. 2008;85:225–260. doi: 10.1016/S0070-2153(08)00809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimokawa T., Svard J., Heby-Henricson K., Teglund S., Toftgard R., Zaphiropoulos P.G. Distinct roles of first exon variants of the tumor-suppressor Patched1 in Hedgehog signaling. Oncogene. 2007;26:4889–4896. doi: 10.1038/sj.onc.1210301. [DOI] [PubMed] [Google Scholar]
- 31.Kurokawa M., Kornbluth S. Caspases and kinases in a death grip. Cell. 2009;138:838–854. doi: 10.1016/j.cell.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naresh A., Long W., Vidal G.A., Wimley W.C., Marrero L., Sartor C.I., Tovey S., Cooke T.G., Bartlett J.M., Jones F.E. The ERBB4/HER4 intracellular domain 4ICD is a BH3-only protein promoting apoptosis of breast cancer cells. Cancer Res. 2006;66:6412–6420. doi: 10.1158/0008-5472.CAN-05-2368. [DOI] [PubMed] [Google Scholar]
- 33.Han M.E., Lee Y.S., Baek S.Y., Kim B.S., Kim J.B., Oh S.O. Hedgehog signaling regulates the survival of gastric cancer cells by regulating the expression of Bcl-2. Int. J. Mol. Sci. 2009;10:3033–3043. doi: 10.3390/ijms10073033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Autret A., Martin S.J. Emerging role for members of the Bcl-2 family in mitochondrial morphogenesis. Mol. Cell. 2009;36:355–363. doi: 10.1016/j.molcel.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 35.Shi Y. A conserved tetrapeptide motif: potentiating apoptosis through IAP-binding. Cell Death Differ. 2002;9:93–95. doi: 10.1038/sj.cdd.4400957. [DOI] [PubMed] [Google Scholar]
- 36.Kurita S., Mott J.L., Cazanave S.C., Fingas C.D., Guicciardi M.E., Bronk S.F., Roberts L.R., Fernandez-Zapico M.E., Gores G.J. Hedgehog inhibition promotes a switch from Type II to Type I cell death receptor signaling in cancer cells. PLoS ONE. 2011;6:e18330. doi: 10.1371/journal.pone.0018330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nikolovska-Coleska Z., Xu L., Hu Z., Tomita Y., Li P., Roller P.P., Wang R., Fang X., Guo R., Zhang M., et al. Discovery of embelin as a cell-permeable, small-molecular weight inhibitor of XIAP through structure-based computational screening of a traditional herbal medicine three-dimensional structure database. J. Med. Chem. 2004;47:2430–2440. doi: 10.1021/jm030420+. [DOI] [PubMed] [Google Scholar]
- 38.Byrd N., Becker S., Maye P., Narasimhaiah R., St-Jacques B., Zhang X., McMahon J., McMahon A., Grabel L. Hedgehog is required for murine yolk sac angiogenesis. Development. 2002;129:361–372. doi: 10.1242/dev.129.2.361. [DOI] [PubMed] [Google Scholar]
- 39.Geng X., Speirs C., Lagutin O., Inbal A., Liu W., Solnica-Krezel L., Jeong Y., Epstein D.J., Oliver G. Haploinsufficiency of Six3 fails to activate Sonic hedgehog expression in the ventral forebrain and causes holoprosencephaly. Dev. Cell. 2008;15:236–247. doi: 10.1016/j.devcel.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeong Y., Leskow F.C., El-Jaick K., Roessler E., Muenke M., Yocum A., Dubourg C., Li X., Geng X., Oliver G., et al. Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat. Genet. 2008;40:1348–1353. doi: 10.1038/ng.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willnow T.E., Hilpert J., Armstrong S.A., Rohlmann A., Hammer R.E., Burns D.K., Herz J. Defective forebrain development in mice lacking gp330/megalin. Proc. Natl. Acad. Sci. USA. 1996;93:8460–8464. doi: 10.1073/pnas.93.16.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chong H.J., Young N.M., Hu D., Jeong J., McMahon A.P., Hallgrimsson B., Marcucio R.S. Signaling by SHH rescues facial defects following blockade in the brain. Dev. Dyn. 2012;241:247–256. doi: 10.1002/dvdy.23726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu D., Marcucio R.S. Neural crest cells pattern the surface cephalic ectoderm during FEZ formation. Developmental Dynamics. 2012;241:732–740. doi: 10.1002/dvdy.23764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marcucio R.S., Cordero D.R., Hu D., Helms J.A. Molecular interactions coordinating the development of the forebrain and face. Developmental Biology. 2005;284:48–61. doi: 10.1016/j.ydbio.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 45.Delloye-Bourgeois C., Gibert B., Rama N., Delcros J.G., Gadot N., Scoazec J.Y., Krauss R., Bernet A., Mehlen P. Sonic Hedgehog promotes tumor cell survival by inhibiting CDON pro-apoptotic activity. PLoS Biol. 2013;11:e1001623. doi: 10.1371/journal.pbio.1001623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strohecker A.M., Yehiely F., Chen F., Cryns V.L. Caspase cleavage of HER-2 releases a Bad-like cell death effector. J. Biol. Chem. 2008;283:18269–18282. doi: 10.1074/jbc.M802156200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arnaiz O., Malinowska A., Klotz C., Sperling L., Dadlez M., Koll F., Cohen J. Cildb: a knowledgebase for centrosomes and cilia. Database (Oxford) 2009;2009:bap022. doi: 10.1093/database/bap022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galluzzi L., Kepp O., Trojel-Hansen C., Kroemer G. Mitochondrial control of cellular life, stress, and death. Circ. Res. 2012;111:1198–1207. doi: 10.1161/CIRCRESAHA.112.268946. [DOI] [PubMed] [Google Scholar]
- 49.Galluzzi L., Kepp O., Kroemer G. Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012;13:780–788. doi: 10.1038/nrm3479. [DOI] [PubMed] [Google Scholar]
- 50.Siebert J.R. Cyclopia, aprosencephaly, and acardiac twinning: is hypoxia-ischemia a unifying mechanism? Am. J. Med. Genet. A. 2007;143A:3100–3106. doi: 10.1002/ajmg.a.32027. [DOI] [PubMed] [Google Scholar]
- 51.Smith F., Hu D., Young N.M., Lainoff A.J., Jamniczky H.A., Maltepe E., Hallgrimsson B., Marcucio R.S. The effect of hypoxia on facial shape variation and disease phenotypes in chicken embryos. Dis. Model Mech. 2013;6:915–924. doi: 10.1242/dmm.011064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Almagro M.C., Vucic D. The inhibitor of apoptosis (IAP) proteins are critical regulators of signaling pathways and targets for anti-cancer therapy. Exp. Oncol. 2012;34:200–211. [PubMed] [Google Scholar]
- 53.Lum L., Beachy P.A. The Hedgehog response network: sensors, switches, and routers. Science. 2004;304:1755–1759. doi: 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- 54.Raha D., Hong M., Snyder M. ChIP-Seq: a method for global identification of regulatory elements in the genome. Curr. Protoc. Mol. Biol. 2010 doi: 10.1002/0471142727.mb2119s91. Chapter 21:Unit 21 19 21-14. [DOI] [PubMed] [Google Scholar]
- 55.Goodrich L.V., Milenkovic L., Higgins K.M., Scott M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 56.Dassule H.R., Lewis P., Bei M., Maas R., McMahon A.P. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development. 2000;127:4775–4785. doi: 10.1242/dev.127.22.4775. [DOI] [PubMed] [Google Scholar]
- 57.St-Jacques B., Dassule H.R., Karavanova I., Botchkarev V.A., Li J., Danielian P.S., McMahon J.A., Lewis P.M., Paus R., McMahon A.P. Sonic hedgehog signaling is essential for hair development. Curr. Biol. 1998;8:1058–1068. doi: 10.1016/s0960-9822(98)70443-9. [DOI] [PubMed] [Google Scholar]
- 58.Wilkinson D.G., Nieto M.A. Detection of messenger RNA by in situ hybridization to tissue sections and whole mounts. Methods Enzymol. 1993;225:361–373. doi: 10.1016/0076-6879(93)25025-w. [DOI] [PubMed] [Google Scholar]
- 59.Takahashi M., Nomura T., Osumi N. Transferring genes into cultured mammalian embryos by electroporation. Dev. Growth Differ. 2008;50:485–497. doi: 10.1111/j.1440-169X.2008.01046.x. [DOI] [PubMed] [Google Scholar]
- 60.Hamburger V., Hamilton H.L. A series of normal stages in the development of the chick embryo. 1951. Dev. Dyn. 1992;195:231–272. doi: 10.1002/aja.1001950404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.