

Abstract
Mutations in the RNA-binding protein FUS/TLS (FUS) have been linked to the neurodegenerative disease amyotrophic lateral sclerosis (ALS). Although predominantly nuclear, this heterogenous nuclear ribonuclear protein (hnRNP) has multiple functions in RNA processing including intracellular trafficking. In ALS, mutant or wild-type (WT) FUS can form neuronal cytoplasmic inclusions. Asymmetric arginine methylation of FUS by the class 1 arginine methyltransferase, protein arginine methyltransferase 1 (PRMT1), regulates nucleocytoplasmic shuttling of FUS. In motor neurons of primary spinal cord cultures, redistribution of endogenous mouse and that of ectopically expressed WT or mutant human FUS to the cytoplasm led to nuclear depletion of PRMT1, abrogating methylation of its nuclear substrates. Specifically, hypomethylation of arginine 3 of histone 4 resulted in decreased acetylation of lysine 9/14 of histone 3 and transcriptional repression. Distribution of neuronal PRMT1 coincident with FUS also was detected in vivo in the spinal cord of FUSR495X transgenic mice. However, nuclear PRMT1 was not stable postmortem obviating meaningful evaluation of ALS autopsy cases. This study provides evidence for loss of PRMT1 function as a consequence of cytoplasmic accumulation of FUS in the pathogenesis of ALS, including changes in the histone code regulating gene transcription.
INTRODUCTION
The neurodegenerative disease amyotrophic lateral sclerosis (ALS) is characterized by preferential loss of motor neurons, causing progressive paralysis leading to death from respiratory failure. Mutations in the FUS gene encoding fused in sarcoma/translated in liposarcoma (FUS/TLS) account for ∼5% of familial ALS cases [familial amyotrophic lateral sclerosis (fALS)], known as fALS6 (1–3). FUS functions as a heterogenous nuclear ribonuclear protein (hnRNP) with DNA/RNA-binding properties underlying roles in transcription (4), nuclear export and processing of mRNA (5) and transport of mRNA to dendritic spines (6). Although some of these functions require nucleocytoplasmic shuttling, FUS predominantly resides in the nucleus. Postmortem analysis of spinal cords from fALS6 patients revealed retention of FUS in the cytoplasm of some motor neurons and glia in the form of granular, vermiform and skein-like inclusions (1,3). Interestingly, FUS-positive cytoplasmic inclusions have been found in motor neurons in ALS cases without fALS6 mutations, i.e. with sporadic [sporadic amyotrophic lateral sclerosis (sALS)] or other types of fALS (7), suggesting FUS mislocalization could be associated more generally with pathogenesis of ALS.
Asymmetric dimethylation of arginine residues (ADMA) is a post-translational modification catalyzed by the class 1 family of protein arginine methyltransferases (PRMTs) and is characterized by the addition of two methyl groups to the same guanidino nitrogen atom (8). This post-translational modification regulates many cellular functions including nucleocytoplasmic shuttling of hnRNPs (8,9). We and others have reported that PRMT1, the most predominant class 1 arginine methyltransferase in mammalian cells (10), interacts with and methylates FUS and influences the nucleocytoplasmic distribution of wild-type (WT) and mutant FUS in a manner dependent on cell type and timing of PRMT1 inhibition (11–15). For our study (11), we established a primary culture model of fALS6 by expressing mutant or WT human FUS in motor neurons of murine spinal cord cultures. As in other models, the steady-state localization of mutant FUS, and to a lesser extent WT FUS, was shifted toward the cytoplasm. In those experiments, we observed a parallel change in the distribution of PRMT1 in motor neurons corresponding to FUS; PRMT1 was depleted from the nucleus when FUS was primarily cytoplasmic. We proposed that this redistribution of PRMT1 would result in hypomethylation of its nuclear substrates, including histones, which could have downstream effects on transcription. ADMA is known to regulate transcription via modification of histone proteins (16) as well as non-histone proteins including hnRNPs (17).
Histone proteins form nucleosome core particles that package DNA into a compact structure and can thereby regulate its accessibility. Each assembled nucleosome comprises an octamer containing two copies of each core histone (H2A, H2B, H3 and H4). The flexible N-terminal tails of core histones are susceptible to post-translational modifications that include methylation, acetylation, phosphorylation and ubiquitination (18,19). These modifications can alter interactions between core histone components and thereby influence DNA binding, the higher-order structure of chromatin, transcription factor binding, or access to the transcriptional machinery. Histone modifications can also act in a combinatorial manner, influencing additional post-translational modifications on the same or other histones (20). Such combinations of these modifications may serve important regulatory functions to coordinate changes in gene expression at specific loci across the genome in response to different cellular states. This regulation could be particularly relevant to motor neuron health, as transcriptional dysregulation has been reported in motor neurons of individuals with ALS (21–23).
PRMT1 catalyzes ADMA of arginine 3 of histone 4 (H4R3) (24), which can facilitate lysine acetylation of H4 at positions 5, 8, 12 and 16 (25) and H3 at positions 9 and 14 (26). These histone acetylation marks are associated with the formation of active chromatin. In contrast, loss of H4R3 ADMA is accompanied by the formation of repressive heterochromatin (26). In this study, we examined (i) the effect of cytoplasmic accumulation of FUS on the subcellular localization of PRMT1 in motor neurons and (ii) the consequence of PRMT1 redistribution to the cytoplasm on target histone modifications important for transcriptional regulation. Reduction in nuclear PRMT1 was accompanied by a dose-related decrease in the methylation of H4R3 and the acetylation of H3K9/K14 as well as chromatin condensation and reduction in RNA synthesis, suggesting that epigenetic mechanisms contribute to motor neuron dysfunction in ALS as downstream consequences of prolonged FUS mislocalization.
RESULTS
Cytoplasmic human FUS forms granular, vermiform and skein-like inclusions in cultured motor neurons
In our previous study, we established a primary culture model of fALS6 by expressing mutant or WT human FUS in motor neurons of murine spinal cord-dorsal root ganglion cultures (hereafter referred to as spinal cord cultures) by intranuclear microinjection of plasmid expression vectors (11). In that study, motor neurons overexpressing mutant human FUS (R521 mutations), and to a lesser extent FUSWT, presented with granular cytoplasmic inclusions within 3 days (also shown in Fig. 1A–C), the preponderance of which co-localized with the RNA-marker SYTO RNA Select (11). Redistribution of mutant FUS was examined over a longer time course in the present study (Fig. 1). Filamentous and skein-like inclusions developed in neurons expressing R521H for 6–7 days (Fig. 1D–G), reminiscent of the neuronal cytoplasmic inclusions observed in ALS spinal cord (1,3). Dendrites with skein-like inclusions also appeared atrophic (Fig. 1D and E).
Figure 1.

Mutant human FUS forms granular, linear and skein-like neuronal cytoplasmic inclusions and depletes nuclear endogenous FUS. (A–G) Immunolabeling of cultured motor neurons with anti-FLAG antibody on Day 3 (A–C) or Days 6–7 (D–G) after intranuclear microinjection of indicated plasmids. Arrows in A–C point to motor neuron nuclei. Mutant FUS was retained in the cytoplasm in the form of granular inclusions on Day 3 post-injection (A–C) and formed linear, donut and skein-like inclusions by 6 days, as illustrated in the neurons shown in (D–G). (E) 3D reconstruction of skein-like inclusions of eGFP-FUS from (D). (H –K) Double label of a motor neuron expressing FLAG-FUSR521H with anti-FLAG and anti-FUS to visualize both human and total FUS. Endogenous FUS is visible in the nuclei of background cells, but not in the motor neuron with only cytoplasmic FLAG labeling. (L) Fluorescence intensity profiles of anti-FLAG, anti-FUS and Hoechst labeling from profile line drawn on the motor neuron in K, demonstrating co-distribution of FLAG and FUS labeling, and thus human FUS with endogenous FUS. Scale bar = 20 µm.
Live cell imaging of neurons expressing eGFP-tagged mutant FUS, using an Olympus VivaView incubator microscope, confirmed that large globular and filamentous inclusions formed as a result of growth and fusion of smaller aggregates. Time-lapse imaging of neurons expressing eGFP-FUSR521H, or eGFP-FUSWT, carried out between Days 4–7 post-injection showed FUS accumulating diffusely within the cytoplasm followed by a sudden coalescence into small granules throughout the neuron (Supplementary Material, Movie S1 showing imaging of a motor neuron expressing eGFP-FUSR521H as described in Materials and Methods). These inclusions moved, grew in size, fused and in some neurons lined up and fused into linear inclusions particularly in dendrites (formation of linear arrays in a motor neuron expressing eGFP-FUSR521H is shown in Supplementary Material, Movie S2). Ring-like FUS-positive inclusions were observed in the perikarya of some motor neurons (Fig. 1F). Consistent with previous studies, inclusions were apparent when either mutant or WT FUS accumulated in the cytoplasm, modeling inclusions that occur in fALS6 and sALS.
In contrast to human FUSWT, the FUSR521H and FUSP525L mutants, the truncation mutant FUS495X rarely formed inclusions when overexpressed in motor neurons but did accumulate in the cytoplasm (see Fig. 2A).
Figure 2.

Co-distribution of PRMT1 and FUS. (A) Double label of cultured motor neurons with antibodies to FUS and PRMT1. Distribution of PRMT1 mimicked that of endogenous, WT and mutant human FUS. Arrows point to motor neurons. Scale bar = 50 µm. (B and C) Quantitation of FUS and PRMT1 localization in neurons either uninjected (n = 6 cultures 17–62 neurons per culture) or injected with indicated FUS plasmids (n = 3 cultures per condition, 11–77 neurons per culture). Asterisks indicate significant shift in distribution from the nucleus to cytoplasm compared with endogenous proteins in uninjected neurons *P < 0.05, **P < 0.01, ***P < 0.001; t-test. (D and E) PRMT1 was not recruited to mutant FUS granular, linear and skein-like inclusions. Scale bars = 50 and 20 µm, respectively. (F and G) Co-labeling of either FUS or PRMT1 with synaptophysin. Neurons with cytoplasmic PRMT1/FUS were selected to determine whether cytoplasmic PRMT1 is recruited to synapses. Large arrows in (F) point to two dendrites emerging from the cell body to the left. Endogenous FUS, but not PRMT1, was recruited to synapses. Arrow heads point to some areas of FUS/synaptophysin co-localization. Scale bar = 10 µm. (H) Quantification of endogenous FUS and PRMT1 localization in cultured motor neurons at two stages of development of spinal cord cultures. Asterisks indicate significantly different distribution at 4 compared with 2 weeks, **P < 0.01, ***P < 0.001 (t-test, n = 3 per condition, 30–256 neurons per culture). More neurons have cytoplasmic FUS/PRMT1 as they develop and establish synaptic connections. (I) Quantification of endogenous FUS and PRMT1 localization in motor neurons in control cultures or cultures treated with 0.4 M sorbitol for 2 h showing significant redistribution of both proteins from the nucleus to the cytoplasm with osmotic stress. Asterisks indicate significant difference compared with control, *P < 0.05, **P < 0.01, ***P < 0.001; (t-test, n = 3 per condition, 28–58 neurons per culture).
To determine whether the endogenous mouse FUS co-distributed with ectopically expressed human FUS, FLAG-FUSR521H (Fig. 1) or FLAG-FUSR495X (not shown) was expressed in motor neurons, and cultures were co-labeled with anti-FLAG and anti-FUS (antibody 11570-1-AP) to visualize both human FUS and total FUS in the same neurons (Fig. 1H–L). When FLAG-FUS was completely cytoplasmic (by anti-FLAG labeling, Fig. 1H), there was no nuclear signal from the anti-FUS antibody (Fig. 1I), indicating that endogenous FUS was also depleted from the nucleus. Note endogenous FUS is visible in nuclei surrounding injected neurons indicating that the apparent disappearance of nuclear staining from the anti-FUS antibody was not due to imaging parameters too low to reveal endogenous FUS. A scan of fluorescence intensity through a plane of the cell including the nucleus (marked as a line in Fig. 1K) showed similar patterns of anti-FUS and anti-FLAG labeling (Fig. 1L).
Nuclear depletion of PRMT1 in neurons with cytoplasmic FUS
ADMA of FUS by PRMT1 regulates its subcellular localization and inhibition of PRMT1 activity retains WT or mutant FUS in the nucleus (11,12,14,15). Having noted redistribution of PRMT1 with FUS in motor neurons in those experiments, their co-distribution was evaluated quantitatively in the present study by double-label immunocytochemistry with anti-PRMT1 and anti-FLAG in neurons microinjected with constructs encoding FLAG-tagged WT or mutant (R521H, P525L and R495X) human FUS. The normal distribution of the endogenous murine proteins was evaluated in uninjected neurons. FUS and PRMT1 distribution was classified as either exclusively nuclear, exclusively cytoplasmic or distributed between both compartments, as illustrated in Supplementary Material, Figure S1A. The microinjection procedure had no effect on distribution of PRMT1 (Supplementary Material, Fig. S1B). The nuclear/cytoplasmic distribution of PRMT1 was congruent not only with endogenous FUS in control cultures (Fig. 2A and Supplementary Material, Fig. S1B) but also with ectopically expressed WT or mutant human FUS. Expression of human WT or ALS-linked mutants significantly shifted PRMT1 distribution to either mostly nuclear in neurons expressing FUSWT, or mostly cytoplasmic in neurons expressing mutant FUS (Fig. 2A–C). The greater redistribution of FUS and PRMT1 to the cytoplasm in neurons expressing FUSP525L was consistent with the juvenile onset and rapid disease progression in patients with this mutation relative to the milder phenotype of R521 mutations (27,28) (Fig. 2B and C). Although there was a significant shift in FUS/PRMT1 to the cytoplasm in neurons expressing FUSR495X, the concordance of their distribution was not as high as with other mutants. In no neurons was FUSR495X exclusively nuclear (Fig. 2B), yet 29% had exclusively nuclear PRMT1 (Fig. 2C). Retention of nuclear PRMT1 could have been a consequence of deletion of the PRMT1-interacting domain; however, despite missing the C-terminus containing a nuclear localization sequence, this mutant was capable of entering the nucleus, as 17% of neurons had both nuclear and cytoplasmic protein labeled by anti-FLAG, in agreement with Bosco et al. (39).
PRMT1 was not recruited to FUS inclusions. Even when PRMT1 was depleted from nuclei along with FUS, it was not detected in granular FUS inclusions of WT or mutant human FUS on Day 3 post-injection (Fig. 2D) nor in vermiform or skein-like inclusions on Day 6 post-injection (Fig. 2E), indicating that PRMT1 does not maintain association with aggregated FUS.
A major function of FUS is transport of specific mRNAs to dendritic spines (6). Presence of FUS at synapses has been demonstrated in cultured hippocampal neurons (6) and human and mouse brain (29). Similarly, double label of spinal cord cultures with antibodies to FUS (11570-1-AP) and synaptophysin revealed endogenous FUS at dendritic spines of motor neurons (Fig. 2F), whereas PRMT1 was not (Fig. 2G).
To further examine the co-distribution of PRMT1 with endogenous FUS, cultures at two stages of development were double-labeled with anti-FUS and anti-PRMT1: at 2 weeks in vitro when FUS is almost exclusively nuclear, and at 4 weeks when motor neurons have developed dendritic spines and a greater proportion of motor neurons contain cytoplasmic FUS at a point in time (Fig. 2H). Distribution of FUS and PRMT1 was categorized as in Supplementary Material, Figure S1B, i.e. exclusively nuclear, exclusively cytoplasmic or distributed between both compartments. As in Figure 2A, the distribution of PRMT1 paralleled FUS in both 2- and 4-week-old cultures, both being more cytoplasmic at 4 weeks in vitro compared with 2 weeks.
Endogenous FUS accumulates in the cytoplasm under conditions of cellular stress, including sorbitol-induced hyperosmotic stress (30). To determine whether PRMT1 would also redistribute with FUS to the cytoplasm with added stress, cultures were treated with 0.4 m sorbitol for 2 h and subsequently double-labeled with anti-FUS and anti-PRMT1. Treatment with sorbitol did indeed significantly shift both FUS and PRMT1 from the nucleus to the cytoplasm (Fig. 2I).
In summary, PRMT1 distributed with FUS in motor neurons under both physiological conditions and under stress conditions that result in cytoplasmic accumulation/nuclear depletion of FUS, including osmotic stress and expression of ALS-causing FUS mutants.
Nuclear depletion of PRMT1 reduces asymmetric arginine dimethylation of histone 4
Next we tested the hypothesis that depletion of PRMT1 from the nucleus in neurons with mislocalized FUS would result in hypomethylation of PRMT1’s nuclear substrates, using H4R3 as a representative substrate. PRMT1 is the major methyltransferase catalyzing asymmetric dimethylation of H4R3, a key regulator of transcription (24). The milder R521H mutant was used in these experiments in order to evaluate neurons with nuclear, cytoplasmic or combined nuclear/cytoplasmic distribution of FUS, the severe mutants being almost exclusively cytoplasmic (Fig. 2B). Methylation of H4R3 was evaluated by indirect immunocytochemistry with an antibody specifically recognizing asymmetrically methylated H4R3 [dimethylated arginine 3 of histone 4 (H4R3Me2)] (Fig. 3A) and quantified as pixel intensity of epifluorescence in motor neuron nuclei (Fig. 3B). This antibody recognizes asymmetrically, but not symmetrically methylated H4R3 (31). H4R3 ADMA was significantly decreased in motor neurons with predominantly cytoplasmic WT or R521H FLAG-FUS compared with neurons with nuclear FUS, 3 days after plasmid injection (Fig. 3B).
Figure 3.
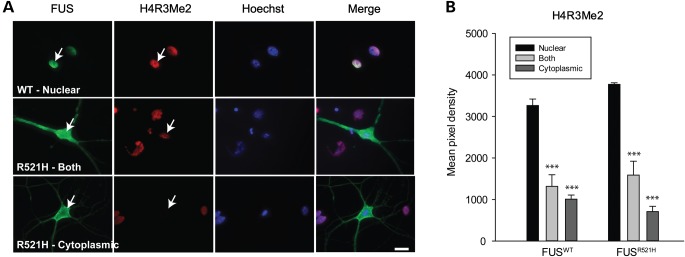
Decreased H4R3 ADMA in motor neurons with cytoplasmic FUS. (A) Double labeling of motor neurons expressing either WT or mutant FUS, with antibodies to FLAG and asymmetrically dimethylated H4R3 (H4R3Me2). Arrows point to motor neuron nuclei. (B) Mean fluorescence intensity of H4R3Me2 antibody labeling corresponding to localization of FUS. Presented are means ± SE of data collected in n = 38 neurons expressing FUSWT and n = 18 neurons expressing FUSR521H. *** significantly different from distribution when FUS was nuclear, P < 0.001; t-test. Scale bar = 20 µm.
Decrease in H4R3 methylation has been associated with the formation of repressive heterochromatin (26). In neurons overexpressing human FUS in which FUS was exclusively cytoplasmic, Hoechst staining was highly condensed (see Figs 1–4, Supplementary Material, Fig. S2), but this was not due to apoptosis as indicated by the absence of labeling by TUNEL-assay (Supplementary Material, Fig. S2). Interestingly, chromatin was not condensed in neurons of control cultures that exhibited both nuclear depletion of endogenous FUS and PRMT1 (Supplementary Material, Fig. S1B). Thus, severe chromatin condensation is not an obligate consequence, particularly under physiological conditions.
Figure 4.

H3K9/K14 acetylation is decreased in neurons with cytoplasmic FUS, downstream of H4R3 methylation. (A) Double labeling of motor neurons, expressing either WT or mutant FUS, with antibodies to FLAG and acetylated H3K9/K14 (H3K9/K14Ac) demonstrating decreased H3K9/K14 acetylation in neurons with cytoplasmic FUS. Arrows point to motor neuron nuclei. Presented are means ± SE of data collected in n = 43 neurons expressing WT FUS and n = 32 neurons expressing R521H FUS. (B) Mean fluorescence intensity of H3K9/K14Ac labeling corresponding to localization of FUS. Asterisks indicate significantly different compared with neurons with nuclear FUS, ***P < 0.001. (C) Western blot of purified histones from cultures treated with 20 µm AMI-1 (PRMT inhibitor), demonstrating a significant decrease in H3 acetylation, indicated by densitometric measurements of H3K9/K14Ac bands (n = 3 per condition), at 24 h of treatment. Coomassie-stained sister gel shows consistent levels of histone proteins among treatment groups. Asterisks indicate significantly different compared with untreated cultures, *P < 0.05. (D) Reduction in H4R3Me2 and H3K9/K14Ac in motor neurons after 24 h of exposure to 20 µm AMI-1 (**P = 0.01), presented as mean fluorescence intensity ± SE of antibody labeling in nuclei of motor neurons with nuclear FUS. (E and F) Mean fluorescence intensity of H3K9/K14Ac and H4R3Me2 labeling of neurons expressing R521H FUS in control cultures (vehicle treated) or cultures treated with the HDAC inhibitor, SAHA. SAHA significantly preserved H3 acetylation but did not prevent decreases in H4R3 methylation in neurons with cytoplasmic FUS. Asterisks indicate significantly different compared with vehicle treated ***P < 0.001; number of neurons evaluated is indicated on the graphs.
H3k9/K14 acetylation is decreased in motor neurons with cytoplasmic FUS
Acetylation of lysine residues of histones is a well-characterized post-translational modification, which plays a role in transcription by weakening the association between positively charged lysine residues and negatively charged DNA; this results in loosened organization of chromatin, allowing access to transcriptional machinery. Furthermore, histone modifications can act in a combinatorial manner (20), for example, H4R3 ADMA by PRMT1 results in H4 and H3 acetylation (25,26). We hypothesized that decreased H4R3 ADMA associated with redistribution of FUS and PRMT1 to the cytoplasm would result in decreased H3 acetylation at lysine residues 9 and 14 (H3K9/K14ac). Acetylation of these residues was detected using antibody recognizing only the acetylated form and quantified as pixel density of epifluorescence of antibody labeling 3 days following microinjection of motor neurons with constructs encoding WT or R521H FLAG-FUS (Fig. 4A). H3 acetylation was significantly decreased when either WT or R521H FUS was cytoplasmic compared with when FUS was nuclear (Fig. 4B).
Similar changes in histone marks were observed in spinal cord cultures treated with the PRMT inhibitor AMI-1, which preferentially, although not exclusively, inhibits PRMT1 (32). Histones were extracted via acid solubilization from control cultures and cultures treated with 20 µm AMI-1 for 24, 48 and 72 h and western blots were probed with antibodies to asymmetrically dimethylated H4R3 and acetylated H3K9/K14. H3K9/K14 acetylation was significantly reduced in cultures treated with AMI-1 for 24 h compared with untreated cultures (Fig. 4C). This decreased H3K9/K14 acetylation was not maintained in cultures treated longer than 24 h, suggesting desensitization to the compound's effect. By western analysis, H4R3me did not appear as affected by AMI-1 as H3K9/K14, even though both were significantly reduced in motor neurons, measured as pixel density of epifluorescence of H4R3Me2 and H3K9/14Ac labeling in nuclei of motor neurons with nuclear FUS (Fig. 4D). In terms of the western analysis, it is possible that the methylation status of H4R3 changes during histone purification or that the effect of AMI-1 is more prominent in motor neurons than other cell types in the mixed culture.
Inhibiting histone deacetylase (HDAC) preserved H3K9/K14 acetylation in neurons expressing mutant FUS (Fig. 4). Cultures were treated with 7.5 µm Vorinostat [suberanilohydroxamic acid (SAHA)] or vehicle for 48 h, beginning 24 hµ post-injection of motor neurons with FLAG-FUSR521H construct. H3K9/K14 acetylation was maintained and even increased with SAHA treatment (Fig. 4E) without significantly affecting H4R3 ADMA (Fig. 4F).
Transcriptional activity is decreased in neurons with cytoplasmic FUS
Reduction in these histone modifications would be expected to have consequences on the transcriptional activity of the cell (33). Because of the wide-ranging effects of FUS on gene transcription and RNA splicing (34–36), overall transcriptional activity was assessed in neurons with nuclear or cytoplasmic FUS/PRMT1 by employing a 5-bromouridine (BrU) incorporation assay to detect newly synthesized RNA. Indeed, BrU incorporation within the nucleus, measured as intensity of anti-BrdU immunolabeling, was significantly less in neurons with cytoplasmic endogenous FUS, compared with neurons with predominantly nuclear FUS (Fig. 5A). Inhibiting PRMT activity with AMI-1 also reduced BrU incorporation, even in neurons with nuclear FUS, indicating that loss of PRMT1 function did indeed contribute to the decrease in transcription with loss of nuclear FUS (Fig. 5B).
Figure 5.

RNA synthesis is decreased in neurons with cytoplasmic FUS and following inhibition of PRMT activity by AMI-1. (A) Mean fluorescence intensity of anti-BrdU antibody labeling of neurons incubated with 5 mm BrU for 2 h to assess BrU incorporation in newly synthesized RNA. Transcriptional activity was reduced in neurons with cytoplasmic FUS (n = 14) compared with those with nuclear FUS (n = 12). (B) Transcriptional activity was also reduced in neurons with nuclear FUS treated with AMI-1 (n = 9) compared with untreated (n = 13). *P < 0.05, ***P < 0.001.
Unfortunately, the BrU assay was not meaningful in neurons overexpressing human FUS from plasmid DNA because of high background resulting from transcription of FUS from the plasmid CMV promoter. However, these data do demonstrate the importance of FUS localization in transcriptional regulation under normal conditions through its effect on PRMT1 function.
Coincident distribution of PRMT1 with WT or mutant FUS in murine spinal motor neurons in situ
PRMT1 distribution also was evaluated in a transgenic mouse model produced by the Hayward lab (University of Massachusetts Medical School) (Fig. 6). Mice harboring WT or R495X human FUS transgenes were established by cloning cDNAs into MoPrP.Xho (ATCC #JHU-2) containing the promoter, 5′-intronic and 3′-untranslated sequences of the murine prion protein (PrP) gene (Lawrence Hayward, personal communication). R495X lines, including the line PX78 used in this study, had 11–15 transgene copies and 3- to 5-fold FUS overexpression. While mice from the mutant FUS transgenic lines display no obvious weakness, abnormalities were detected by electromyography (EMG), including fibrillation potentials, muscle denervation and reduction in motor unit number estimation at 8–12 months. These abnormalities did not occur in mice from lines expressing nuclear human FUSWT (lines PWT17 or PWT52, limited to three transgene copies, <2-fold FUS overexpression). Non-transgenic littermates served as controls.
Figure 6.

Co-distribution of PRMT1 with FUS in murine spinal motor neurons in situ in non-transgenic, and WT and mutant FUS transgenic mice. Double immunolabeling of cross-sections of spinal cord from non-transgenic mice (A–C: 154 days old, line PX78 littermate), FUSWT transgenic mice (D–E: 163 days old, line PWT17) and FUS495X transgenic mice (F–I: 154 days old, line PX78; J–L: 378 days old, line PX78) with mouse anti-FUS (sc-47711) and rabbit anti-PRMT1. PRMT1 distribution paralleled FUS, being nuclear in motor neurons of non-transgenic mice and FUSWT transgenics, but to varying degrees cytoplasmic as well as nuclear in neurons of FUSR495X mice. Scale bar = 20 µm.
The distribution of FUS and PRMT1 assessed by double-label immunocytochemistry is shown in Figure 6. Human FUSWT (Fig. 6D) was mostly nuclear, similar to endogenous FUS in non-transgenic littermates (Fig. 6A). A higher burden of cytoplasmic FUSR495X was observed in the perikarya of motor neurons in the anterior horn of the spinal cord (Fig. 6G and J), indicating a shift in the equilibrium of the mutant toward the cytoplasm in addition to its presence in the nucleus. Most neurons had a mixture of nuclear and cytoplasmic labeling, but the amount of cytoplasmic mutant FUS was variable as can be seen by comparing Figure 6G and J. No inclusions were identified by FUS immunohistochemistry (or by ubiquitin or p62 inclusion markers; data not shown). Like FUS, PRMT1 was predominantly nuclear in cross-sections of spinal cord from non-transgenic (Fig. 6B) and FUSWT transgenic (Fig. 6E) mice, but cytoplasmic labeling was detected in neurons in sections from FUSR495X mice (Fig. 6H and K) in amounts relative to FUS.
Postmortem interval affects subcellular distribution of PRMT1
Cytoplasmic FUS inclusions have also been observed in spinal motor neurons in sporadic ALS (7). However, in control experiments using archived autopsied spinal cord tissue from three individuals dying of non-neurological causes, we noticed that rather than the predominantly nuclear anti-PRMT1 labeling expected in motor neurons, labeling was mostly cytoplasmic (Fig. 7A). Thus, we investigated the possibility that PRMT1 is lost from nuclei postmortem. This hypothesis was investigated in mice by fixing lumbar spinal cord immediately following euthanasia or after a postmortem interval of 6 h, considered to be a short postmortem interval for processing of human autopsy tissue. Cross-sections of spinal cord immunolabeled with antibody to PRMT1 or FUS are illustrated in Figure 7B. PRMT1 was depleted from the nucleus of motor neurons as early as 6 h of postmortem, despite persistence of nuclear FUS. Even following immediate immersion in formalin fixative, preservation of PRMT1 distribution was variable compared with perfusion fixation (as in Fig. 6) or cryopreservation (data not shown). Thus, PRMT1 is labile postmortem, obviating reliable studies with archived human autopsy specimens.
Figure 7.

PRMT1 is depleted from the nucleus in archived autopsied human spinal cord and in mouse spinal cord fixed after postmortem interval. (A) Cross-section of postmortem spinal cord from a non-neurological control case (immersion-fixed and paraffin-embedded after excision) immunolabeled with antibody to PRMT1 and counterstained with hematoxylin. Note that PRMT1 is cytoplasmic rather than the expected nuclear distribution. Image to the right is a higher magnification of the boxed area in the left image. (B) Mouse spinal cord was excised and fixed by immersion in buffered neutral formalin immediately either after euthanasia or 6 h of postmortem. Cross-sections from paraffin embedded tissue were labeled with antibody to PRMT1 or FUS. Note the loss of nuclear PRMT1 with postmortem interval, despite persistence of nuclear FUS. Scale bar = 20 µm.
DISCUSSION
The majority of fALS6 patients present with mutations at the C-terminal region of FUS containing the nuclear localization sequence (37). Cytoplasmic inclusions containing FUS have been observed in motor neurons of fALS and sALS patients (7), implicating altered trafficking and distribution of this hnRNP more generally in ALS pathogenesis. Furthermore, the degree to which ALS-linked FUS mutants accumulate in the cytoplasm of cultured cells generally correlates with severity of disease in patients (38,39).
We previously reported that asymmetric arginine dimethylation (ADMA) of FUS by PRMT1 is a strong regulator of FUS nucleocytoplasmic shuttling and that both WT and FUS mutants interact with PRMT1 (11), findings also reported by other labs (12–15). In the present study, we further developed the primary culture model in which WT or mutant human FUS is expressed in motor neurons of dissociated spinal cord cultures (11) and investigated how the interaction of PRMT1 and FUS might lead to loss of nuclear function of PRMT1 and contribute to toxicity.
In cultured motor neurons expressing R521H or P525L mutants, small cytoplasmic inclusions containing FUS developed by Day 3 post-microinjection of expression plasmid, resembling those in cell lines (11,38–40). However, by Day 7 of expression, large circular, vermiform and skein-like inclusions had developed, modeling the inclusions observed in human autopsy material. Time-lapse imaging showed that these large inclusions formed by either growth or fusion of small inclusions, or both. Motor neurons with cytoplasmic FUS also presented with atrophic dendrites, a common finding in ALS (41). The truncation mutant R495X (39,42) was investigated in both cultured motor neurons and transgenic mice. Cytoplasmic FUS was increased in motor neurons expressing FUSR495X, both in the culture model and in transgenic mice, yet significant levels were retained in the nucleus (this mutant was both nuclear and cytoplasmic in >40% of neurons in the culture model and almost all spinal motor neurons in the transgenic mice). Thus, despite missing the nuclear localization sequence in the deleted C-terminus, FUSR495X can be imported into the nucleus, as previously reported by Bosco et al. (39).
Using primary spinal cord cultures, we demonstrated that nuclear depletion of FUS resulted in loss of PRMT1 function in the nucleus, leading to downregulation of transcription through loss of major histone modifications. PRMT1 was depleted from the nucleus of cultured motor neurons with cytoplasmic endogenous FUS or ectopically expressed mutant or WT human FUS. Although there was a definite and statistically significant shift in PRMT1 localization to the cytoplasm in motor neurons expressing FUSR495X, nuclear depletion of PRMT1 was not as complete as in neurons expressing FUSR521H or FUSP525L. This could be influenced by the continued presence of nuclear FUS in neurons expressing FUSR495X.
The mechanism underlying loss of nuclear PRMT1 is unclear. PRMT1 does not necessarily maintain association with FUS even though ADMA by PRMT1 is important for FUS trafficking, and the two proteins can be co-immunoprecipitated (11,13–15). PRMT1 was clearly not recruited to synapses with endogenous FUS, nor was it associated with cytoplasmic inclusions of WT or mutant human FUS in motor neurons. In contrast, others have reported that some PRMT1 was associated with inclusions of truncated FUS, as well as being diffusely cytoplasmic, and in stress granules in oxidatively challenged SH-SY5Y cells (15). In addition, PRMT1 and PRMT8 accumulated in inclusion bodies in COS cells expressing FUS mutants (14). There are 10 known alternatively spliced PRMT1 isoforms, only one of which, PRMT1v2, contains a nuclear export signal (43). Shifting the proportion of alternatively spliced variants to PRMT1v2 cannot be excluded as an alternative explanation for the observed loss of nuclear PRMT1.
Redistribution of PRMT1 observed in the culture model was validated in vivo in mice. PRMT1 was predominantly nuclear in spinal motor neurons of non-transgenic mice and transgenic mice carrying the human FUSWT transgene, but both nuclear and cytoplasmic in FUSR495X transgenic mice, similar to the distribution of FUS. This pattern was consistent with observations in cultured motor neurons (this study) and HEK-293 cells [Bosco et al. (2010)] (i.e. increased cytoplasmic FUS/PRMT1, but considerable retention in the nucleus) and with the very mild phenotype of these mice, only detected by EMG (39).
Unfortunately, we could not reliably assess the distribution of PRMT1 in human tissue or whether it is mislocalized to the cytoplasm in neurons of ALS patients. By immunohistochemistry, PRMT1 was cytoplasmic in spinal motor neurons in the archived control tissue tested (spinal cord from individuals dying of non-neurological causes). A study in mice comparing PRMT1 distribution with time of processing demonstrated that postmortem interval before tissue fixation is the most likely reason for its cytoplasmic distribution in control autopsy tissue. Best results were obtained using perfusion fixation with paraformaldehyde (PFA) or cryopreservation; distribution of PRMT1 was compromised even with immediate immersion fixation in formalin or PFA. Similarly, endogenous PRMT1 was not retained in the nuclear fraction upon subcellular fractionation of cultured cells (Tibshirani, unpublished data) suggesting that interaction with its substrates is transient or labile. Loss of nuclear PRMT1 under these circumstances could be explained by compromised integrity of the nuclear membrane leading to leakage of small, soluble proteins such as PRMT1 into the cytoplasm and/or loss of energy-dependent protein interactions (44). Although isoform switching to PRMT1v2 is a possibility in living cells, it seems unlikely postmortem. These experiments stress the importance of determining postmortem effects on cellular processes being investigated in such specimens.
Decreased H4R3Me2 and H3K9/K14Ac resulted from nuclear depletion of FUS and PRMT1. The normal function of FUS includes recruiting HDAC1 to sites of induced DNA damage, and this process is perturbed in cells expressing mutant FUS (45). However, we observed decreased H3K9/K14Ac when endogenous FUS or either WT or R521H FUS were depleted from the nucleus, indicating this decrease was inherent upon the localization of FUS rather than brought on by a functional consequence of the mutation. Decreased H4R3Me2 and H3K9/K14Ac had consequences on transcriptional activity. BrU incorporation into RNA was reduced in motor neurons with cytoplasmic endogenous FUS/PRMT1 compared with those with nuclear FUS/PRMT1, indicating this was a normal physiological process with FUS trafficking. The data are more than correlative. Reduction of PRMT1 expression has physiological consequences to motor neurons that relate to the ALS phenotype. These include reduction in mitochondrial size (11) and dendritic branching (Supplementary Material, Fig. S3).
Overall, the data indicate that PRMT1 redistribution is a normal consequence of FUS localization. This conclusion is supported by the coincidence of FUS and PRMT1 localization in motor neurons of developing spinal cord cultures and those subjected to osmotic shock by treatment with sorbitol, both conditions associated with increased cytoplasmic distribution of endogenous FUS. It is possible that reducing the neuron's transcriptional activity while FUS delivers its cargo mRNAs to synapses could be an adaptation to prevent the production of excessive RNAs until the return of FUS to the nucleus. Regardless, should alterations in transcription be sustained, as in pathological retention of FUS brought on by mutation or long-term stress, consequences for neuronal function would be expected, particularly synaptic and dendritic maintenance. A difference between redistribution of endogenous FUS and ectopically expressed human FUS was the condensation of chromatin in the latter. This could reflect a difference in trafficking kinetics, a hypothesis being investigated, or other consequences of disrupted nuclear function of FUS. Regardless, chromatin condensation was not associated with apoptosis (neurons were not TUNEL-positive).
In summary, our results point to a sustained loss of nuclear PRMT1 function as a contributing mechanism of mutant FUS toxicity in ALS. Loss of nuclear PRMT1 correlated with cytoplasmic mislocalization of FUS and was associated with changes in histone marks linked to transcriptional inhibition. The extent to which this repression is general or affects particular gene families remains to be determined. Given that FUS itself can affect transcription of a large number of genes by binding to RNA polymerase (46), its redistribution to the cytoplasm could, through a physiological process, influence transcription both directly and indirectly through PRMT1. Although cycling of PRMT1 between the nucleus and the cytoplasm is a normal physiological process, sustained loss of nuclear PRMT1 would be expected to have significant consequences on neuronal function in the context of pathological cytoplasmic FUS accumulation in ALS.
MATERIALS AND METHODS
Dissociated spinal cord cultures
Preparation of cultures of spinal cords (with attached dorsal root ganglia) from dissociated E13 mouse embryos, culture in hormone- and growth factor-supplemented medium, and identification of motor neurons in mixed cultures was as previously described (47,48). Cultures were used in experiments 3–7 weeks after plating.
Gene transfer by intranuclear microinjection
Gene transfer to motor neurons was accomplished by intranuclear microinjection of plasmid vectors as described previously (48), motor neurons in these long-term cultures not being amenable to transfection. Human FUS-encoding plasmids (WT and the mutants R521H, P525L and R495X) with an N-terminal FLAG-tag and in some cases an eGFP-tag (11), were injected at a concentration of 20 ng/µl. 70-kDa dextran–fluorescein isothiocyanate was included in the injectate as an inert marker.
Immunocytochemistry and immunohistochemistry
Cultured cells were fixed in 3% PFA in phosphate-buffered saline (PBS) for 10 min, permeabilized in 0.5% NP-40 for 1 min and fixed once more for 2 min. Cells were then blocked in 5% horse serum in PBS for 30 min. Incubations in primary and secondary antibodies were for 30 min, followed by 3 × 5 min washes in PBS. Coverslips were mounted on microscope slides using DAKO mounting medium (Carpinteria, CA, USA).
FUSWT and FUSR495X transgenic mice and non-transgenic littermates were perfused with 4% PFA in PBS for 30 min. Samples of brain and spinal cord were stored in 4% PFA at 4°C. Immunolabeling was carried out on 4-µm sections from paraffin blocks. Antigen retrieval was performed by boiling tissues for 10 min with BD Retrievagen A pH 6.0 (BD Biosciences, ON, Canada). Tissues were incubated with primary antibody overnight at 4°C followed by incubation in AlexaFluor-conjugated secondary antibody, then mounted in Prolong Gold Antifade Mountant with DAPI (Life Technologies, Inc., Burlington, ON, Canada).
For human spinal cord and mice used for the postmortem interval study, tissues were fixed in 10% neutral buffered formalin, embedded in paraffin and cut in 4 -µm sections. Two cases of sporadic ALS and two non-neurological control cases were examined, with postmortem intervals ranging from 6 to 26 h. Antigen retrieval was performed by boiling in sodium citrate buffer. Tissues were blocked in 5% BSA + 0.03% Triton X-100 in PBS for 1 h at RT. Samples were then incubated with primary antibody overnight at 4°C followed by biotinylated secondary antibody. Labeling was visualized using Vectastain Elite ABC kit (Vector Labs, Burlington, ON, Canada) using diaminobenzidine as substrate and ImmPACT DAB peroxidase substrate kit (Vector Labs). The same protocol was used to mimic postmortem interval in mice. Adult C57Bl6 (∼12 months old) were euthanized by ketamine/xylazine intraperitoneal injection. Tissues were either perfuse-fixed with 4% PFA or immersion-fixed in 10% neutral buffered saline immediately or with postmortem interval.
Imaging
Images were viewed using a Zeiss Observer Z1 microscope (Carl Zeiss Canada Ltd, Toronto, ON, Canada) equipped with a Hamamatsu ORCA-ER cooled CD camera (Hamamatsu, Japan). Filter sets 02, 13 and 00 were used for epifluorescence microscopy (Carl Zeiss Canada Ltd). Images were acquired and fluorescence intensity profiles were analyzed with Axiovision software (Carl Zeiss Canada Ltd).
Long-term time-lapse imaging of FUS inclusion formation was carried out in an Olympus VivaView incubator microscope (Olympus Canada, Inc., Richmond Hill, ON, Canada). Neurons were injected with eGFP-FUSR521H and imaged every 15 min from Days 4–6 post-injection. The scale bar on Supplementary Materials, Movies S1 and S2 in Supplementary Materials represents 20 µm.
Analysis of histone post-translational modifications
Histones were extracted from spinal cord cultures according to a modified protocol (49). All reagents were pre-chilled on ice. Cells were washed in PBS, harvested into microfuge tubes and pelleted by centrifugation at 2800 g. To obtain nuclear and cytoplasmic fractions, the pellets were pipetted vigorously in 0.1% NP40 in PBS, then nuclei were pelleted by centrifugation at 2800 g. Nuclear pellets were dissolved in 0.4 N HCl and incubated on ice for 1.5 h. Acid-insoluble proteins were precipitated by centrifugation at 15 000 g. The supernatant was collected, and histones were precipitated by addition of trichloroacetic acid to a final concentration of 30% and incubated on ice for 1.5 h. Proteins were pelleted at 16 000 g for 10 min, and pellets were washed twice with ice-cold acetone. After air drying, pellets were dissolved in Laemmli buffer. Samples were separated by 15% SDS–PAGE and transferred to nitrocellulose membrane. Consistent loading of histone proteins was verified by Coomassie-staining of sister gels. Nitrocellulose membranes were blocked in 5% skim milk in TBS and probed with primary and HRP-conjugated secondary antibodies in 5% skim milk in TBS with 3 × 15 min washes with TBST. Optical density of bands was performed using NIH Image J software. Levels of H3 acetylation in samples from treated cultures were normalized to untreated control indicated by western blot and normalized once more to amount loaded compared with control indicated by Coomassie-staining.
Bromouridine incorporation assay
Cultures were incubated in 5 mm BrU in culture medium for 2 h at 37°C. Cells were incubated in 3% PFA in PBS for 15 min at 37°C, and incorporation was detected by immunocytochemistry with anti-BrdU antibody.
Antibodies and reagents
Antibodies used for this study were: mouse anti-FLAG M2 (Sigma–Aldrich, St. Louis, MO, USA, F1804, 1 : 400), rabbit anti-FLAG (Sigma–Aldrich F2555, 1 : 50), mouse anti-FUS (Santa Cruz Biotechnology, Inc., Dallas, TX, USA, sc-47711, 1 : 200), rabbit anti-FUS (Proteintech, Chicago, IL, USA, 11570-1-AP, 1 : 400), rabbit anti-PRMT1 (Abcam, Cambridge MA, USA, ab73246, 1 : 400), mouse anti-synaptophysin (Santa Cruz Biotechnology, Inc., sc-12737, 1 : 400), rabbit anti-methylated H4R3 (Active Motif 39705 from Cedarlane Laboratories, 1 : 400 ICC, 1 : 500 WB), rabbit anti-acetylated H3K9/K14 (Cell Signaling Technology, Beverly, MA, USA, 9677, 1 : 400 ICC, 1 : 1000 WB), mouse anti-MAP2 (Novus Biologicals Canada, Oakville, ON, Canada, NBP1-92711, 1 : 400), mouse anti-BrdU (Sigma–Aldrich SAB4700630, 1 : 250), anti-mouse Cy2-IgG (Rockland, Gilbertsville, PA, USA, 610-711-124,1 : 300), anti-rabbit Cy3-IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA, 715-165-150, 1 : 300), anti-rabbit Cy2-IgG; Cy3-IgG (Jackson ImmunoResearch Laboratories, Inc., 711-225-152; 711-165-152, 1 : 300), anti-rabbit HRP-IgG (Jackson ImmunoResearch Laboratories, Inc., 111-035-045, 1 : 2500), anti-rabbit Alexa Fluor 188-IgG and anti-mouse Alexa Fluor 594-IgG (Life Technologies), AMI-1 (Santa Cruz Biotechnology, Inc., sc-205928), 5-Bromouridine (Sigma–Aldrich 851087), DeadEnd™ Fluorometric TUNEL System (Promega G3250).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by the ALS Association (69F361 to H.D.D.); the Muscular Dystrophy Association (255651 to H.D.D.); the National Institutes of Health (NINDS 1RC1NS068391 to L.H.) and ALS Therapy Alliance (to L.H.).
Supplementary Material
REFERENCES
- 1.Hewitt C., Kirby J., Highley J.R., Hartley J.A., Hibberd R., Hollinger H.C., Williams T.L., Ince P.G., McDermott C.J., Shaw P.J. Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2010;67:455–461. doi: 10.1001/archneurol.2010.52. [DOI] [PubMed] [Google Scholar]
- 2.Kwiatkowski T.J., Jr, Bosco D.A., Leclerc A.L., Tamrazian E., Vanderburg C.R., Russ C., Davis A., Gilchrist J., Kasarskis E.J., Munsat T., et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 3.Vance C., Rogelj B., Hortobagyi T., De Vos K.J., Nishimura A.L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P., et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Blitterswijk M., Landers J.E. RNA processing pathways in amyotrophic lateral sclerosis. Neurogenetics. 2010;11:275–290. doi: 10.1007/s10048-010-0239-4. [DOI] [PubMed] [Google Scholar]
- 5.Kovar H. Dr. Jekyll and Mr. Hyde: the two faces of the FUS/EWS/TAF15 protein family. Sarcoma. 2011;2011:837474. doi: 10.1155/2011/837474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujii R., Takumi T. TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J. Cell Sci. 2005;118:5755–5765. doi: 10.1242/jcs.02692. [DOI] [PubMed] [Google Scholar]
- 7.Deng H.X., Zhai H., Bigio E.H., Yan J., Fecto F., Ajroud K., Mishra M., Ajroud-Driss S., Heller S., Sufit R., et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 2010;67:739–748. doi: 10.1002/ana.22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bedford M.T., Clarke S.G. Protein arginine methylation in mammals: who, what, and why. Mol. Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu M.C. The role of protein arginine methylation in mRNP dynamics. Mol. Biol. Int. 2011;2011:163827. doi: 10.4061/2011/163827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang J., Frankel A., Cook R.J., Kim S., Paik W.K., Williams K.R., Clarke S., Herschman H.R. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 2000;275:7723–7730. doi: 10.1074/jbc.275.11.7723. [DOI] [PubMed] [Google Scholar]
- 11.Tradewell M.L., Yu Z., Tibshirani M., Boulanger M.C., Durham H.D., Richard S. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum. Mol. Genet. 2012;21:136–149. doi: 10.1093/hmg/ddr448. [DOI] [PubMed] [Google Scholar]
- 12.Dormann D., Madl T., Valori C.F., Bentmann E., Tahirovic S., Abou-Ajram C., Kremmer E., Ansorge O., Mackenzie I.R., Neumann M., et al. Arginine methylation next to the PY-NLS modulates transportin binding and nuclear import of FUS. EMBO J. 2012;31:4258–4275. doi: 10.1038/emboj.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du K., Arai S., Kawamura T., Matsushita A., Kurokawa R. TLS and PRMT1 synergistically coactivate transcription at the survivin promoter through TLS arginine methylation. Biochem. Biophys. Res. Commun. 2011;404:991–996. doi: 10.1016/j.bbrc.2010.12.097. [DOI] [PubMed] [Google Scholar]
- 14.Scaramuzzino C., Monaghan J., Milioto C., Lanson N.A., Jr, Maltare A., Aggarwal T., Casci I., Fackelmayer F.O., Pennuto M., Pandey U.B. Protein arginine methyltransferase 1 and 8 interact with FUS to modify its sub-cellular distribution and toxicity in vitro and in vivo. PLoS One. 2013;8:e61576. doi: 10.1371/journal.pone.0061576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamaguchi A., Kitajo K. The effect of PRMT1-mediated arginine methylation on the subcellular localization, stress granules, and detergent-insoluble aggregates of FUS/TLS. PLoS One. 2012;7:e49267. doi: 10.1371/journal.pone.0049267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Lorenzo A., Bedford M.T. Histone arginine methylation. FEBS Lett. 2011;585:2024–2031. doi: 10.1016/j.febslet.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee Y.-H., Stallcup M.R. Minireview: protein arginine methylation of nonhistone proteins in transcriptional regulation. Mol. Endocrinol. 2009;23:425–433. doi: 10.1210/me.2008-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan A.U., Krishnamurthy S. Histone modifications as key regulators of transcription. Front. Biosci. 2005;10:866–872. doi: 10.2741/1580. [DOI] [PubMed] [Google Scholar]
- 19.Cheung P., Allis C.D., Sassone-Corsi P. Signaling to chromatin through histone modifications. Cell. 2000;103:263–271. doi: 10.1016/s0092-8674(00)00118-5. [DOI] [PubMed] [Google Scholar]
- 20.Lee J.S., Smith E., Shilatifard A. The language of histone crosstalk. Cell. 2010;142:682–685. doi: 10.1016/j.cell.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergeron C., Beric-Maskarel K., Muntasser S., Weyer L., Somerville M.J., Percy M.E. Neurofilament light and polyadenylated mRNA levels are decreased in amyotrophic lateral sclerosis motor neurons. J. Neuropathol. Exp. Neurol. 1994;53:221–230. doi: 10.1097/00005072-199405000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Jiang Y.M., Yamamoto M., Kobayashi Y., Yoshihara T., Liang Y., Terao S., Takeuchi H., Ishigaki S., Katsuno M., Adachi H., et al. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 2005;57:236–251. doi: 10.1002/ana.20379. [DOI] [PubMed] [Google Scholar]
- 23.Campos-Melo D., Droppelmann C., He Z., Volkening K., Strong M. Altered microRNA expression profile in amyotrophic lateral sclerosis: a role in the regulation of NFL mRNA levels. Mol. Brain. 2013;6:26. doi: 10.1186/1756-6606-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strahl B.D., Briggs S.D., Brame C.J., Caldwell J.A., Koh S.S., Ma H., Cook R.G., Shabanowitz J., Hunt D.F., Stallcup M.R., et al. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr. Biol. 2001;11:996–1000. doi: 10.1016/s0960-9822(01)00294-9. [DOI] [PubMed] [Google Scholar]
- 25.Wang H., Huang Z.Q., Xia L., Feng Q., Erdjument-Bromage H., Strahl B.D., Briggs S.D., Allis C.D., Wong J., Tempst P., et al. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001;293:853–857. doi: 10.1126/science.1060781. [DOI] [PubMed] [Google Scholar]
- 26.Huang S., Litt M., Felsenfeld G. Methylation of histone H4 by arginine methyltransferase PRMT1 is essential in vivo for many subsequent histone modifications. Genes. Dev. 2005;19:1885–1893. doi: 10.1101/gad.1333905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conte A., Lattante S., Zollino M., Marangi G., Luigetti M., Del Grande A., Servidei S., Trombetta F., Sabatelli M. P525L FUS mutation is consistently associated with a severe form of juvenile amyotrophic lateral sclerosis. Neuromuscul. Disord. 2012;22:73–75. doi: 10.1016/j.nmd.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Baumer D., Hilton D., Paine S.M., Turner M.R., Lowe J., Talbot K., Ansorge O. Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology. 2010;75:611–618. doi: 10.1212/WNL.0b013e3181ed9cde. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aoki N., Higashi S., Kawakami I., Kobayashi Z., Hosokawa M., Katsuse O., Togo T., Hirayasu Y., Akiyama H. Localization of fused in sarcoma (FUS) protein to the post-synaptic density in the brain. Acta. Neuropathol. 2012;124:383–394. doi: 10.1007/s00401-012-0984-6. [DOI] [PubMed] [Google Scholar]
- 30.Sama R.R., Ward C.L., Kaushansky L.J., Lemay N., Ishigaki S., Urano F., Bosco D.A. FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J. Cell. Physiol. 2013;228:2222–2231. doi: 10.1002/jcp.24395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun L., Wang M., Lv Z., Yang N., Liu Y., Bao S., Gong W., Xu R.-M. Structural insights into protein arginine symmetric dimethylation by PRMT5. Proc. Natl. Acad. Sci. 2011;108:20538–20543. doi: 10.1073/pnas.1106946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonham K., Hemmers S., Lim Y.H., Hill D.M., Finn M.G., Mowen K.A. Effects of a novel arginine methyltransferase inhibitor on T-helper cell cytokine production. FEBS J. 2010;277:2096–2108. doi: 10.1111/j.1742-4658.2010.07623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X., Hu X., Patel B., Zhou Z., Liang S., Ybarra R., Qiu Y., Felsenfeld G., Bungert J., Huang S. H4R3 methylation facilitates beta-globin transcription by regulating histone acetyltransferase binding and H3 acetylation. Blood. 2010;115:2028–2037. doi: 10.1182/blood-2009-07-236059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiu H., Lee S., Shang Y., Wang W.Y., Au K.F., Kamiya S., Barmada S.J., Finkbeiner S., Lui H., Carlton C.E., et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Invest. 2014;124:981–999. doi: 10.1172/JCI72723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoell J.I., Larsson E., Runge S., Nusbaum J.D., Duggimpudi S., Farazi T.A., Hafner M., Borkhardt A., Sander C., Tuschl T. RNA targets of wild-type and mutant FET family proteins. Nat. Struct. Mol. Biol. 2011;18:1428–1431. doi: 10.1038/nsmb.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y., Liu S., Liu G., Ozturk A., Hicks G.G. ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. 2013;9:e1003895. doi: 10.1371/journal.pgen.1003895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lagier-Tourenne C., Cleveland D.W. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136:1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dormann D., Rodde R., Edbauer D., Bentmann E., Fischer I., Hruscha A., Than M.E., Mackenzie I.R., Capell A., Schmid B., et al. ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. EMBO J. 2010;29:2841–2857. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bosco D.A., Lemay N., Ko H.K., Zhou H., Burke C., Kwiatkowski T.J., Jr, Sapp P., McKenna-Yasek D., Brown R.H., Jr, Hayward L.J. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 2010;19:4160–4175. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ito D., Seki M., Tsunoda Y., Uchiyama H., Suzuki N. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann. Neurol. 2011;69:152–162. doi: 10.1002/ana.22246. [DOI] [PubMed] [Google Scholar]
- 41.Nakano I., Hirano A. Atrophic cell processes of large motor neurons in the anterior horn in amyotrophic lateral sclerosis: observation with silver impregnation method. J. Neuropathol. Exp. Neurol. 1987;46:40–49. doi: 10.1097/00005072-198701000-00004. [DOI] [PubMed] [Google Scholar]
- 42.Waibel S., Neumann M., Rosenbohm A., Birve A., Volk A.E., Weishaupt J.H., Meyer T., Muller U., Andersen P.M., Ludolph A.C. Truncating mutations in FUS/TLS give rise to a more aggressive ALS-phenotype than missense mutations: a clinico-genetic study in Germany. Eur. J. Neurol. 2013;20:540–546. doi: 10.1111/ene.12031. [DOI] [PubMed] [Google Scholar]
- 43.Goulet I., Gauvin G., Boisvenue S., Côté J. Alternative splicing yields protein arginine methyltransferase 1 isoforms with distinct activity, substrate specificity, and subcellular localization. J. Biol. Chem. 2007;282:33009–33021. doi: 10.1074/jbc.M704349200. [DOI] [PubMed] [Google Scholar]
- 44.Boisvert F.-M., Lam Y.W., Lamont D., Lamond A.I. A quantitative proteomics analysis of subcellular proteome localization and changes induced by DNA damage. Mol. Cell. Prot. 2010;9:457–470. doi: 10.1074/mcp.M900429-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang W.Y., Pan L., Su S.C., Quinn E.J., Sasaki M., Jimenez J.C., Mackenzie I.R., Huang E.J., Tsai L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013;16:1383–1391. doi: 10.1038/nn.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwartz J.C., Ebmeier C.C., Podell E.R., Heimiller J., Taatjes D.J., Cech T.R. FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes. Dev. 2012;26:2690–2695. doi: 10.1101/gad.204602.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roy J., Minotti S., Dong L., Figlewicz D.A., Durham H.D. Glutamate potentiates the toxicity of mutant Cu/Zn-superoxide dismutase in motor neurons by postsynaptic calcium-dependent mechanisms. J. Neurosci. 1998;18:9673–9684. doi: 10.1523/JNEUROSCI.18-23-09673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Durham H.D., Roy J., Dong L., Figlewicz D.A. Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J. Neuropathol. Exp. Neurol. 1997;56:523–530. doi: 10.1097/00005072-199705000-00008. [DOI] [PubMed] [Google Scholar]
- 49.Shechter D., Dormann H.L., Allis C.D., Hake S.B. Extraction, purification and analysis of histones. Nat. Prot. 2007;2:1445–1457. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.