

Abstract
4-(tert-Butyl)-2-((tert-butylamino)methyl)-6-(6-(trifluoromethyl)pyridin-3-yl)-phenol (JPC-2997) is a new aminomethylphenol compound that is highly active in vitro against the chloroquine-sensitive D6, the chloroquine-resistant W2, and the multidrug-resistant TM90-C2B Plasmodium falciparum lines, with 50% inhibitory concentrations (IC50s) ranging from 7 nM to 34 nM. JPC-2997 is >2,500 times less cytotoxic (IC50s > 35 μM) to human (HepG2 and HEK293) and rodent (BHK) cell lines than the D6 parasite line. In comparison to the chemically related WR-194,965, a drug that had advanced to clinical studies, JPC-2997 was 2-fold more active in vitro against P. falciparum lines and 3-fold less cytotoxic. The compound possesses potent in vivo suppression activity against Plasmodium berghei, with a 50% effective dose (ED50) of 0.5 mg/kg of body weight/day following oral dosing in the Peters 4-day test. The radical curative dose of JPC-2997 was remarkably low, at a total dose of 24 mg/kg, using the modified Thompson test. JPC-2997 was effective in curing three Aotus monkeys infected with a chloroquine- and pyrimethamine-resistant strain of Plasmodium vivax at a dose of 20 mg/kg daily for 3 days. At the doses administered, JPC-2997 appeared to be well tolerated in mice and monkeys. Preliminary studies of JPC-2997 in mice show linear pharmacokinetics over the range 2.5 to 40 mg/kg, a low clearance of 0.22 liters/h/kg, a volume of distribution of 15.6 liters/kg, and an elimination half-life of 49.8 h. The high in vivo potency data and lengthy elimination half-life of JPC-2997 suggest that it is worthy of further preclinical assessment as a partner drug.
INTRODUCTION
Malaria remains one of the deadliest diseases in tropical countries, with about 219 million cases in 2010 and an estimated 660,000 deaths, primarily in children from sub-Saharan Africa (1). Antimalarial drug resistance is a serious global health threat. To combat the development and spread of multiple drug resistance, artemisinin combination therapies (ACTs) such as artemether-lumefantrine, artesunate-amodiaquine, and dihydroartemisinin-piperaquine are now recommended worldwide for first-line treatment of uncomplicated Plasmodium falciparum malaria (2). Although ACTs remain highly effective, reports of P. falciparum resistance to artemisinins from four Southeast Asian countries (Cambodia, Myanmar, Thailand, and Vietnam) are alarming (3–6). This has led to a concerted effort by the WHO and national ministries of health to execute containment efforts in the affected countries. However, there is a genuine concern that artemisinin resistance will spread in a pattern similar to that of chloroquine and antifolate resistance, which spread from Southeast Asia to Africa in the 1970s and 1980s. In addition to the potential demise of the artemisinins, there is concern regarding the development and spread of resistance to the partner drugs. Thus, to avert a global health disaster, there is an urgent need to develop new and effective antimalarial drugs as new partner drugs or to supplement existing ACTs.
In the 1960s and 1970s, there was renewed interest in antimalarial chemotherapy research due to the development and spread of chloroquine resistance. During this period, the U.S. Army Research Program in Malaria tested over 250,000 compounds for antimalarial activity (7). By 1979, 43 different compounds from this program were selected for clinical trial evaluation. One of these, (4-(tert-butyl)-2-(tert-butylaminomethyl)-6-(4-chlorophenyl)phenol (WR-194,965), an aminomethylphenol, was highly potent against chloroquine-sensitive and chloroquine-resistant P. falciparum lines, with 50% inhibitory concentrations (IC50s) in the low-nanomolar range (8). In the rodent-P. berghei model, WR-194,965 had a 50% effective dose (ED50) of 2.2 mg/kg of body weight/day using the Peters 4-day test (9). WR-194,965 at an oral dose of 27 mg/kg produced a 90% cure in Aotus monkeys infected with the multidrug-resistant P. falciparum Vietnam Smith strain (10).
In a human challenge study, 750 mg WR-194,965 was orally administered every 12 h for 36 h to six human volunteers infected with the Vietnam Smith strain. Four of the six patients were cured. The two subjects who exhibited recrudescence (at 29 and 40 days posttreatment) were successfully cured with mefloquine, and subsequent in vitro tests showed that the parasites from these patients were sensitive to WR-194,965 (11). Attractive features of WR-194,965 are a nonquinoline structure and good tolerance, but a major disadvantage was limited potency, which led to a cessation in its clinical development by the U.S. Army. Recently, Powles et al. (12) from Merck Research Laboratories (Rahway, NJ) reported on the in vitro and in vivo efficacy of MK-4815 (2-aminomethyl-3,5-di-tert-butylphenol), confirming the utility of the same basic aminoethyl phenol structure and demonstrating an improvement in potency over WR-194,965.
We have conducted a comprehensive structure-activity relationship (SAR) study of this chemical series. This program led to the identification of 4-(tert-butyl)-2-((tert-butylamino)methyl)-6-(6-(trifluoromethyl) pyridin-3-yl)phenol (JPC-2997), a new aminomethylphenol which addresses the potency problems of WR-194,965. The SAR study will form the basis for a future publication. The structures of WR-194,965 and JPC-2997 are shown in Fig. 1.
FIG 1.

Chemical structures of (A) WR-194,965 and (B) JPC-2997.
In this study, we describe the in vitro antimalarial and cytotoxicity of JPC-2997 and its in vivo antimalarial efficacy against P. berghei in mice and P. vivax in monkeys. Preliminary pharmacokinetic properties of JPC-2997 in mice and monkeys are also presented.
(Part of this work was presented previously by David Jacobus at the annual meeting of the American Society of Tropical Medicine and Hygiene, November 11 to 17, 2013, Washington, DC, USA.)
MATERIALS AND METHODS
Drugs.
4-(tert-Butyl)-2-((tert-butylamino)methyl)-6-(6-(trifluoromethyl)-pyridin-3-yl)phenol (JPC-2997 hydrochloride) and 4-(tert-butyl)-2-(tert-butylaminomethyl)-6-(4-chlorophenyl)phenol (WR-194,965 hydrochloride) were provided by Jacobus Pharmaceutical Company, and their synthesis will be described in detail elsewhere. Dihydroartemisinin was obtained from Vital Health Care (Mumbai, India) and Central Pharmaceutical Factory No. 1 (Hanoi, Vietnam), chloroquine diphosphate and mefloquine hydrochloride were obtained from Sigma (St. Louis, MO), atovaquone from GlaxoSmithKline (Middlesex, United Kingdom), pyronaridine tetraphosphate from Shin Poong Pharm (Seoul, South Korea), and piperaquine tetraphosphate from Waterstone Technology (St. Carmel, IN) and Central Pharmaceutical Factory No. 1 (Hanoi, Vietnam).
Parasites.
Three laboratory-adapted P. falciparum lines were used in this study: D6 from Sierra Leone is chloroquine and pyrimethamine sensitive but mefloquine resistant, W2 from Indochina is chloroquine and pyrimethamine resistant, and TM90-C2B from Thailand is atovaquone, chloroquine, and mefloquine resistant. Isolates were cultured in RPMI 1640-LPLF medium (Gibco, Invitrogen Corporation, CA) containing 5.97 g/liter HEPES-free acid (Sigma St. Louis, MO), 2.0 g/liter d-glucose, 0.05 g/liter hypoxanthine, and 40 mg/liter gentamicin with the pH adjusted to 6.9 and freshly supplemented with 0.21% sodium bicarbonate and 10% human plasma. Cultures were maintained at 1 to 8% parasitemia and 4% hematocrit (O+ red blood cells) in an atmosphere of 90% N2, 5% O2, 5% CO2, at 37°C (13).
In vitro antimalarial activity.
For in vitro antimalarial activity assessment, WR-194,965 and JPC-2997 compounds were dissolved in dimethyl sulfoxide (DMSO) and dihydroartemisinin was dissolved in 100% methanol, whereas piperaquine and chloroquine were dissolved in 50% methanol–50% water. All drugs were subsequently diluted in culture medium (without hypoxanthine) containing 10% human plasma. Residual methanol and DMSO concentrations were found to be too low to inhibit parasite growth (data not shown). Testing of parasite susceptibility to JPC-2997 and chloroquine was carried out by measuring the inhibition of [3H]hypoxanthine uptake (14). Briefly, highly synchronous ring-stage parasite cultures produced by several rounds of d-sorbitol selection (15) were exposed to 10 2-fold dilutions of the compounds (in triplicate) in 96-well flat-bottom microtiter plates at 37°C for a total of 48 h. Each well contained 100 μl of parasite culture at 2% hematocrit and 1% parasitemia in culture medium prepared as described above but lacking hypoxanthine. A 20-μl (0.2-μCi) aliquot of [3H]hypoxanthine was added to each well ∼24 h after commencement of the assay. Parasite DNA was harvested onto glass fiber filters, and radioactivity was counted to generate [3H]hypoxanthine uptake (counts per minute) versus drug concentration (log values) curves. IC50s were determined using a nonlinear regression analysis (GraphPad Prism V5.0; GraphPad Software, Inc., CA) and were defined as the concentrations producing 50% inhibition of uptake of [3H]hypoxanthine by parasites compared with compound-free samples (controls).
In vitro cytotoxicity assay.
Compounds were tested for in vitro cytotoxicity against two human cell lines, HEK293 (human embryonic kidney) and HepG2 (human hepatocellular carcinoma), and BHK (baby hamster kidney) cells by the alamarBlue (Invitrogen Corporation, CA) fluorescent cell viability assay (16). Cell cultures were maintained in RPMI 1640 medium (Sigma, MO) containing 10% fetal bovine serum and 0.03% l-glutamine (termed complete medium) in 75-cm2 flasks at 37°C, with the medium changed twice weekly. Cells from 60 to 80% confluent cultures were trypsinized, washed in complete medium, and plated at 5 × 103 cells per well in 135 μl of complete medium in 96-well flat-bottom plates for 24 h at 37°C prior to the addition of the compounds. Triplicate 15-μl aliquots of JPC-2997 and WR-194,965 compounds and the reference drugs chloroquine, dihydroartemisinin, and piperaquine covering 11 2-fold dilutions were added to the wells and mixed gently, and plates were incubated for 72 h more. Controls included compound-free wells with DMSO (vehicle) as positive controls and cell-free wells with medium only, which were used for background subtraction.
Following incubation for 72 h, the culture medium was removed, and 50 μl of 10% alamarBlue in complete medium was added to each well for 2 h more. The fluorescence from the wells was measured using a GENios Plus microplate reader and XFluor4 software, using a 530-nm excitation filter and a 595-nm emission filter. Data were obtained from at least two independent experiments for each cell line and analyzed using GraphPad Prism V5.0. The selectivity index (SI) was calculated using the IC50s derived from mammalian cells divided by the IC50s obtained against the P. falciparum D6 line.
In vivo antimalarial studies of P. berghei in mice.
The in vivo efficacy of JPC-2997 and WR-194,965 was determined in three murine malaria models: the Peters 4-day test (17), the modified Thompson test (18) and the test for onset of action and time to recrudescence (19, 20). The Peters 4-day test measures the suppressive activity of blood schizontocides over 4 days at a tolerated dose that does not cause physical stress in healthy mice. Swiss outbred ARC (Animal Resource Centre, Murdoch, Western Australia, Australia) female mice 5 to 6 weeks of age (weight 24 to 32 g), in groups of six, were inoculated intraperitoneally with 20 × 106 P. berghei-infected erythrocytes of the chloroquine-sensitive ANKA strain. Untreated control mice typically die between days 6 and 7 postinfection. Drugs were dissolved in Tween 80-ethanol (7:3, vol/vol), diluted 10-fold in distilled water, and administered subcutaneously (s.c.) or orally (p.o.) at about 2 h after parasite inoculation (D0) and daily for 3 consecutive days. Thin blood smears were made on D4 and stained with Giemsa. The degree of infection (parasitemia, expressed as a percentage of infected erythrocytes) was determined microscopically. The ED50 and ED90 (50% and 90% effective doses) were calculated by nonlinear regression analysis using GraphPad Prism V5.0, and mean ED50 and ED90 were based on at least two independent experiments.
The modified Thompson test measures the survival of mice and parasite clearance in an established infection following administration of the drug at D3 to D5 postinfection. CD-1 male or female mice 5 weeks of age (Charles River Laboratories, Wilmington, MA; weight, 24 to 30 g), in groups of seven, were infected with 5 × 105 parasitized erythrocytes of chloroquine-sensitive P. berghei KBG-173 strain. By D3 postinfection, parasitemia was about 1%. This strain produces reliable parasitemia and death in CD-1 mice. The strain is maintained in vivo in Swiss mice and has been used for more than 20 years to test susceptibility to a wide range of antimalarial drugs. Drugs were mixed in 0.5% hydroxyethylcellulose and 0.1% Tween 80 and administered via oral gavage either once daily at 24-h intervals or twice daily 6 h apart on days 3, 4, and 5 postinfection. Blood smears were taken on D6 and twice a week thereafter, from day 10 until the end of the test on day 31. Mortality data were tabulated for 31 days. Mice that survived for 31 days and were blood film negative were considered cured.
The test for onset of drug action and time of recrudescence measures the speed of parasite clearance by a candidate compound and the time at which recrudescence occurs after a single dose. This was done by administering a single oral dose of 100 mg/kg at day 4 after infection of groups of ARC female mice (n = 6) intraperitoneally with 10 × 106 P. berghei-infected erythrocytes (ANKA strain). Untreated control mice typically achieve a parasitemia of about 20% by day 4 postinfection. Drugs were mixed in ethanol-Tween 80-water (10:10:80 [vol/vol/vol]). To monitor the reduction in parasitemia blood smears were collected at 12 h intervals for the first 96 h after dosing, and then daily from day 5 to 17, followed by collections every 2 to 3 days until day 30.
Preliminary pharmacokinetics of JPC-2997 in mice.
Single-dose escalating studies of JPC-2997 were carried out in healthy ARC female mice (aged 6 to 9 weeks, weighing between 23 and 40 g). Groups of five mice were treated with 2.5, 5, 10, 20, and 40 mg/kg of JPC-2997. The mice, anesthetized with carbon dioxide, were sacrificed by cardiac puncture at 0 h (before dosing), at 1, 3, 6, 12, 24, 48, 72, 96, and 120 h, and then at days 7 and 14 following oral administration. Blood samples (∼0.7 ml) were collected using lithium heparin as the anticoagulant. After 0.2 ml of blood had been separated, the remaining blood was centrifuged at 16,000 × g at 4°C for 5 min, and plasma was separated. Blood and plasma samples were stored at −80°C until analysis, and concentrations of JPC-2997 were measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (see the supplemental material). The lower limit of quantification (LLOQ) of JPC-2997 in blood and plasma was 0.5 ng/ml, with an inaccuracy of <4.1%, using 50 μl of sample. The interassay precision of analysis (percent coefficient of variation [CV]) for JPC-2997 in blood over the concentration range of 0.5 ng/ml to 2,000 ng/ml was <4.6% and in plasma over the concentration range of 0.5 ng/ml to 4,000 ng/ml was <9.4%.
Pharmacokinetic parameters were maximum drug concentration (Cmax), time to reach maximum drug concentration (Tmax), area under the concentration-time curve from 0 h to the last data point (AUC0→last) and from the last data point to infinity (AUClast→∞), terminal half-life (t1/2), apparent oral clearance (CL/F), and apparent volume of distribution (V/F). These parameters were determined from the blood and plasma concentration-time data by noncompartmental methods (21). The blood-to-plasma concentration ratio was calculated using the ratio of AUCblood to AUCplasma.
In vivo efficacy of JPC-2997 in the Aotus monkey-P. vivax model.
Naive splenectomized Aotus monkeys (n = 3), weighing between 960 and 1,126 g, were inoculated intravenously with 1 × 106 parasites of the P. vivax AMRU1 strain, which is resistant to chloroquine (22). Three to four days after the onset of patency, monkeys were treated with 20 mg/kg JPC-2997 daily at 24-h intervals for 3 days by nasogastric tubulation. The drug was prepared and administered in 10% ethanol–1% Tween 80–89% distilled water. Heparinized blood samples (20 μl) were collected predose on day 0 (i.e., immediately before JPC-2997 administration), on days 1 and 2 immediately before the next dose, and then on days, 3, 4, 7, 11, 14, 21, and 28 at the same time of day after the start of treatment. Blood JPC-2997 concentrations were measured by LC-MS/MS. After treatment of monkeys, thick and thin blood smears were examined daily by counting parasitized erythrocytes against 500 leukocytes or 10,000 erythrocytes. When no parasites were detected, follow-up thick blood smears were examined twice a week for 62 days after the start of treatment.
Animal ethics.
The animal studies for the Peters 4-day test, onset and duration assessment of antimalarial activity, monkey studies, and studies of the pharmacokinetics of JPC-2997 were approved by the Army Malaria Institute Animal Ethics Committee in accord with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. The animal studies for the modified Thompson test were approved by the University of Miami Institutional Animal Care and Use Committee.
RESULTS
Aminomethylphenols are active against asexual blood stages of P. falciparum and have low cytotoxicity.
The mean IC50s of JPC-2997 against the chloroquine-sensitive D6 and chloroquine-resistant W2 lines were 13 nM and 6.6 nM, respectively (Table 1). In comparison to WR-194,965, JPC-2997 was 2.3-fold and 4.2-fold more active against the D6 and W2 lines, respectively. JPC-2997 was less active against the multidrug-resistant TM90-C2B line, with an IC50 of 34 nM, but was 1.6-fold more active than WR-194,965. JPC-2997 was markedly more active than chloroquine but less active than dihydroartemisinin and piperaquine against the TM90-C2B P. falciparum line.
TABLE 1.
In vitro antimalarial activities of JPC-2997 against Plasmodium falciparum and and cytotoxicities for mammalian cell lines, with selectivity indexesa
| Compound | Activity (IC50, nM) against: |
HEK293 |
HepG2 |
BHK |
|||||
|---|---|---|---|---|---|---|---|---|---|
| D6 | W2 | TM90-C2B | IC50 (μM) | SI | IC50 (μM) | SI | IC50 (μM) | SI | |
| JPC-2997 | 13 ± 2 | 6.6 ± 1.7 | 34 ± 1 | 40 ± 5 | 3077 | 42 ± 10 | 3,230 | 36 ± 9 | 2,769 |
| WR-194,965 | 30 ± 5 | 28 ± 4 | 53 ± 5 | 31 ± 3 | 1033 | 28 ± 1 | 933 | 35 ± 1 | 1,167 |
| Piperaquine | 23 ± 2 | 36 ± 5 | 22 ± 2 | 87 ± 27 | 3783 | >120 | >5217 | >120 | >5217 |
| Chloroquine | 13 ± 1 | 143 ± 28 | 144 ± 28 | 51 ± 27 | 3923 | 27 ± 13 | 2,077 | 33 ± 16 | 2,538 |
| Dihydroartemisinin | 1.9 ± 0.1 | 1.9 ± 0.3 | 1.7 ± 0.7 | 4 ± 1 | 2105 | 30 ± 9 | 15,789 | 107 ± 13 | 5,632 |
Values are means ± SD from ≥2 observations. The SI is the IC50 for the mammalian cell line divided by that for the Plasmodium falciparum D6 line.
The mean IC50s for JPC-2997 determined in the cytotoxicity assay in three mammalian cell lines were relatively high (≥36 μM), resulting in SI values of ≥2,769, which are similar to those of chloroquine. Compared with WR-194,965, the SI of JPC-2997 was about 3-fold higher. Overall, JPC-2997 has a high selectivity index when antimalarial activity is compared to mammalian cytotoxicity.
JPC-2997 is highly active in vivo against rodent malaria.
In the Peters 4-day test, JPC-2997 suppressed the P. berghei ANKA strain in mice with an ED50 of 0.5 mg/kg/day following either subcutaneous or oral dosing (Table 2). JPC-2997 was 1.8-fold more active than either chloroquine (0.9 mg/kg/day) or piperaquine (0.9 mg/kg/day) but 2.5-fold less potent than dihydroartemisinin (0.2 mg/kg/day) after subcutaneous administration. When JPC-2997 was administered orally, it was more active than the reference standards chloroquine (1.1 mg/kg/day) by 2.2-fold, dihydroartemisinin (1.3 mg/kg/day) by 2.6-fold, and piperaquine (2.4 mg/kg/day) by 4.8-fold, based on their respective ED50s.
TABLE 2.
In vivo antimalarial activity of JPC-2997 in mice infected with the Plasmodium berghei ANKA strain in the Peters 4-day testa
| Route of administration | Compound | ED50 (mg/kg/day) | ED90 (mg/kg/day) |
|---|---|---|---|
| Subcutaneous | JPC-2997 | 0.5 ± 0.0 | 0.8 ± 0.1 |
| Chloroquine | 0.9 ± 0.3 | 1.7 ± 0.5 | |
| Dihydroartemisinin | 0.2 ± 0.1 | 0.4 ± 0.1 | |
| Piperaquine | 0.9 ± 0.4 | 4.3 ± 0.8 | |
| Oral | JPC-2997 | 0.5 ± 0.1 | 1.0 ± 0.0 |
| Chloroquine | 1.1 ± 0.6 | 4.7 ± 2.5 | |
| Dihydroartemisinin | 1.3 ± 0.2 | 4.2 ± 0.8 | |
| Piperaquine | 2.4 ± 0.9 | 6.7 ± 3.2 |
Values are means ± SD for ≥2 observations.
In the modified Thompson test, JPC-2997 produced 100% cure in mice infected with the P. berghei KBG-173 strain after single oral daily doses of 8 mg/kg for 3 days (total dose, 24 mg/kg) (Table 3). In comparison, piperaquine was only 71% effective in curing mice when administered the same treatment regimen. Both JPC-2997 and piperaquine at a dose of 8 mg/kg given twice daily for 3 days were equally curative. WR-194,965 was 2-fold less active than JPC-2997, requiring a total dose of 96 mg/kg (i.e., 16 mg/kg twice daily for 3 days) to produce 100% cure (data not shown). In contrast to WR-194,965 and piperaquine, chloroquine administered at a total dose of 192 mg/kg cured no mice, and dihydroartemisinin at 192 mg/kg cured only 14% of mice.
TABLE 3.
In vivo efficacy of JPC-2997 and reference antimalarials administered orally either daily or twice daily over 3 days in mice infected with the Plasmodium berghei KBG-173 strain in the modified Thompson test
| Administration and compound | Dose (mg/kg) | Total dose (mg/kg) | Median survival time (days) | No. of mice alive at day 31 (n = 7) | % cured on day 31a |
|---|---|---|---|---|---|
| Once dailyb | |||||
| JPC-2997 | 8 | 24 | >31 | 7 | 100 |
| JPC-2997 | 4 | 12 | 19 | 6 | 86 |
| Piperaquine | 8 | 24 | 24 | 5 | 71 |
| Piperaquine | 4 | 12 | 21 | 4 | 57 |
| Dihydroartemisinin | 64 | 192 | 18 | 0 | 0 |
| Twice daily | |||||
| JPC-2997 | 8 | 48 | >31 | 7 | 100 |
| JPC-2997 | 2 | 12 | 25 | 6 | 71 |
| WR-194,965 | 8 | 48 | 23 | 4 | 57 |
| WR-194,965 | 2 | 12 | 17 | 0 | 0 |
| Piperaquine | 8 | 48 | >31 | 7 | 100 |
| Piperaquine | 2 | 12 | 22.5 | 5 | 71 |
| Chloroquine | 32 | 192 | 16 | 0 | 0 |
| Dihydroartemisinin | 32 | 192 | 18 | 1 | 14 |
Percentage of mice that were blood film negative on day 31 after the start of drug treatment.
Single daily doses of WR-194,965 and chloroquine were not assessed.
The onset of action of JPC-2997 in the treatment of an established infection of P. berghei of about 20% parasitemia was compared with that of seven standard antimalarial drugs (Fig. 2). The order of parasitemia clearance and the time of first blood smear-negative slides were as follows: dihydroartemisinin (36 h), piperaquine (36 h), pyronaridine (36 h), amodiaquine (60 h), chloroquine (60 h), atovaquone (96 h), JPC-2997 (120 h), and mefloquine (120 h) (Fig. 2A). After clearance of parasitemia, recrudescences commenced at the following times: 2.5 days for dihydroartemisinin, 6 days for chloroquine, 8 days for both amodiaquine and atovaquone, 12 days for mefloquine, and 20 days for JPC-2997 (Fig. 2B). Of note, only two of six mice treated with JPC-2997 exhibited recrudescence, with the four mice remaining blood smear negative up to day 30 of follow-up. No mice treated with either piperaquine or pyronaridine exhibited recrudescence up to day 30 of observation.
FIG 2.
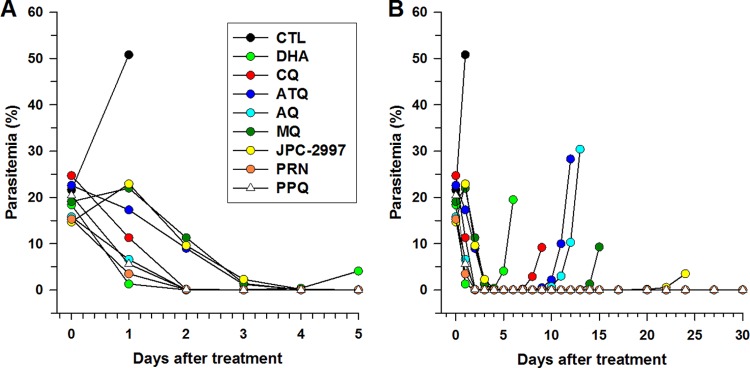
(A) Onset of action and (B) recrudescence after a single oral dose of 100 mg/kg of amodiaquine (AQ), atovaquone (ATQ), chloroquine (CQ), dihydroartemisinin (DHA), JPC-2997, mefloquine (MQ), piperaquine (PPQ), and pyronaridine (PRN) to groups of six mice on day 4 after infection with Plasmodium berghei ANKA. The control (CTL) mice were administered the vehicle.
Preliminary pharmacokinetics of JPC-2997 in mice.
The mean blood and plasma concentration-versus-time profiles of JPC-2997 following single oral doses of 5 mg/kg and 20 mg/kg JPC-2997 are shown in Fig. 3. The pharmacokinetics of JPC-2997, following escalating single oral doses from 2.5 to 40 mg/kg of JPC-2997 are summarized in Table 4, and plasma JPC-2997 concentration-versus-time profiles are shown in Fig. S1 in the supplemental material. Although the blood sampling collection times did not provide an opportunity to determine the maximum plasma concentration and the time it was reached, it appears that JPC-2997 is rapidly absorbed, with the highest plasma concentrations being measured at 1 h postdose. Plasma concentrations of JPC-2997 in mice declined biphasically, with an elimination half-life of 49.8 h. The oral pharmacokinetics of JPC-2997 in mice over the dose range of 2.5 to 40 mg/kg were in general linear (Table 4). JPC-2997 has a CL/F of 0.22 liters/h/kg with a V/F of 15.6 liters/kg. In mice, blood concentrations of JPC-2997 paralleled those of plasma (Fig. 3), and the blood-to-plasma concentration ratio at various time points after the administration of 5 mg/kg or 20 mg/kg JPC-2997 was estimated at 0.7. The administered doses of JPC-2997 were well tolerated in the mice, with no observed adverse events such as vomiting, behavioral changes, or body weight changes.
FIG 3.
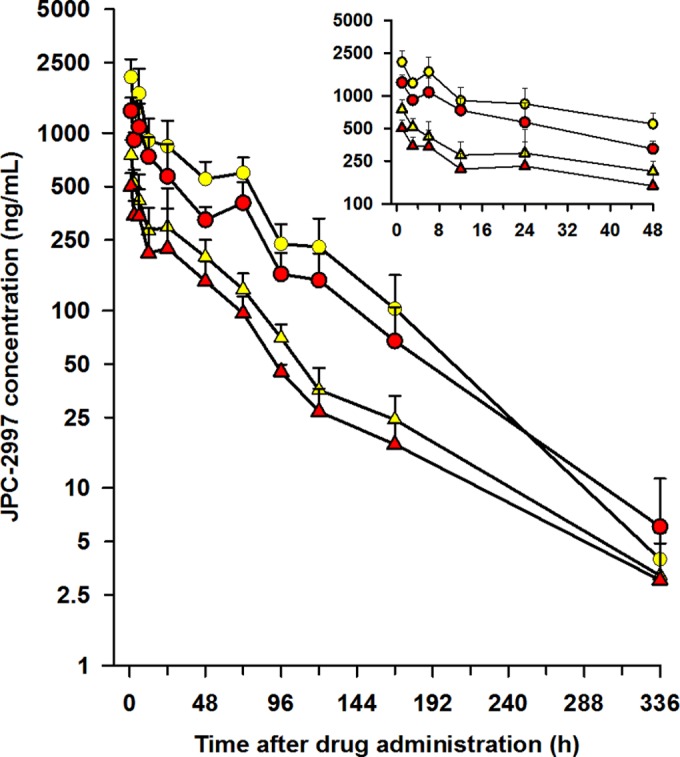
Mean (SD) plasma and blood concentrations versus time for JPC-2997 in healthy mice administered a single oral dose of 5 mg/kg (red triangles, blood; yellow triangles, plasma) and 20 mg/kg (red circles, blood; yellow circles, plasma) of JPC-2997. (Inset) Mean (SD) plasma and blood concentration-time profiles of JPC-2997 over the first 48 h after dosing with 5 mg/kg and 20 mg/kg of JPC-2997.
TABLE 4.
Pharmacokinetics of JPC-2997 in healthy mice administered single oral doses of JPC-2997 between 2.5 and 40 mg/kg
| Dose (mg/kg) | Concn at 1 ha (μg/liter) | Concn/dose (%) | AUC/dose (%) | AUC (μg · h/liter) | t1/2 (h) | V/F (liters/kg) | CL/F (liters/h/kg) |
|---|---|---|---|---|---|---|---|
| 2.5 | 288 | 0.38 | 16.7 | 12,555 | 60.1 | 17.3 | 0.20 |
| 5 | 753 | 0.5 | 17.5 | 26,213 | 48 | 13.2 | 0.19 |
| 10 | 1,013 | 0.34 | 14 | 41,847 | 53.5 | 18.5 | 0.24 |
| 20 | 2,067 | 0.34 | 15.1 | 90,370 | 40.1 | 12.8 | 0.22 |
| 40 | 3,235 | 0.27 | 13.9 | 166,370 | 47.4 | 16.4 | 0.24 |
| Mean ± SD | 0.37 ± 0.08 | 15.4 ± 1.6 | 49.8 ± 7.5 | 15.6 ± 2.5 | 0.22 ± 0.02 |
Highest plasma JPC-2997 concentration measured with the bleed schedule used.
JPC-2997 is highly potent in the treatment of malaria in the Aotus monkey-P. vivax model.
Monkeys were patent by day 5 after intravenous inoculation of the AMRU1 strain. When parasitemia reached between 31,691 and 51,620 parasites/μl of blood (geometric mean, 38,002 parasites/μl; n = 3), the monkeys were treated with 20 mg/kg JPC-2997 per day for 3 days. The geometric mean parasitemia declined to 29,944 parasites/μl (range, 22,231 to 38,340/μl) at 24 h and 11,371 parasites/μl (range, 3,920 to 26,460/μl) at 48 h after starting JPC-2997 treatment, a decrease in parasitemia of 21.2% and 70.1%, respectively, from the predose parasitemia value. The majority of parasites showed an irregular microscopic appearance, such as compact dark staining of cytoplasm and nonpigmented parasites, consistent with parasites being drug affected by day 2 posttreatment. By day 5, parasitemia had been cleared in all monkeys. Over a 62-day follow-up period there was no recurrence of parasites. The maximum blood concentration of JPC-2997 was 744 ± 82 ng/ml (mean ± standard deviation [SD]) at day 4 after the start of treatment, and by day 28 the concentration had declined to 171 ± 82 ng/ml, with an estimated elimination half-life of about 10.8 days (see Fig. S2 in the supplemental material). The monkeys appeared to tolerate the treatment regimen well, with no observed adverse events such as vomiting, behavioral changes, or body weight changes.
DISCUSSION
In vitro antimalarial and cytotoxicity analyses.
In this study, we showed JPC-2997 to be about 2- to 4-fold more active in vitro than WR-194,965 against both drug-sensitive and multidrug-resistant P. falciparum lines. There was a lack of cross-resistance between JPC-2997 and chloroquine with increased activity of JPC-2997 against the chloroquine-resistant W2 line but similar activity against the chloroquine-sensitive D6 line. In addition to its high antimalarial activity, JPC-2997 was several thousand-fold less cytotoxic in murine and human cell lines. The SI of JPC-2997 showed that it was about 3-fold less toxic than WR-194,965. Based on JPC-2997's superior in vitro antimalarial activity and favorable cytotoxicity profile compared to those of WR-194,965, further studies were performed to assess the in vivo antimalarial efficacy of JPC-2997, with comparisons to standard antimalarials.
In vivo efficacy in murine P. berghei models.
The efficacy of JPC-2997 against patent infection in mice was demonstrated using two murine models. Studies against the chloroquine-sensitive P. berghei ANKA strain showed JPC-2997 to be highly active in suppressing infection in the Peters 4-day test, with a mean ED50 of 0.5 mg/kg/day following either s.c. or p.o. dosing. The efficacy of JPC-2997 was about 2-fold higher than that of chloroquine and piperaquine. For comparison, Peters and Robinson observed similar activity between WR-194,965 (ED50 of 2.2 mg/kg/day) (9), and Peters observed similar activity between WR-194,965 and chloroquine (ED50 of 1.9 mg/kg/day) (17) following s.c. administration in mice infected with the chloroquine-sensitive P. berghei N strain.
In the modified Thompson test, JPC-2997 showed remarkably potent activity against the P. berghei KBG-173 strain, with a total dose of 24 mg/kg curing all infected mice, and was better than piperaquine when given once daily for 3 days. JPC-2997 was more effective than WR-194,965 and considerably superior to either chloroquine or dihydroartemisinin when given twice daily for 3 days. In addition to JPC-2997's impressive suppression and radical cure of blood-stage murine malaria, the drug was effective in preventing recrudescence in 66% (four of six) of infected mice treated with a single oral dose of 100 mg/kg, whereas recrudescence occurred in all mice treated with amodiaquine, atovaquone, chloroquine, dihydroartemisinin, and mefloquine. Overall, the oral potency of JPC-2997 is a major improvement over that of WR-194,965.
In vivo efficacy in the Aotus monkey-P. vivax model.
JPC-2997 at a dose of 20 mg/kg daily for 3 days was effective in curing Aotus monkeys infected with the P. vivax AMRU1 strain. The same regimen of chloroquine does not cure Aotus monkeys infected with AMRU1 (22). Clearance of P. vivax malaria by JPC-2997 was comparable to that of P. berghei, taking about 4 to 5 days. Although parasitemia clearance was not rapid in the splenectomized monkeys, most parasites appeared to be drug affected 24 h to 48 h after the start of treatment, and in nonsplenectomized monkeys, parasite clearance might have been faster. We are planning to assess the in vivo efficacy of JPC-2997 against the chloroquine-resistant P. falciparum FVO strain in Aotus monkeys in the near future.
Pharmacokinetic analysis.
Based on these favorable in vivo efficacy data, the pharmacokinetics of JPC-2997 in healthy mice showed linear kinetics in the oral-dose range of 2.5 to 40 mg/kg. It appears to be well absorbed and exhibits biphasic decline in both blood and plasma concentrations, with a relatively long elimination half-life of 49.8 h. JPC-2997's elimination half-life is longer than those of dihydroartemisinin (25 min) (23), amodiaquine (∼7 h) (24), atovaquone (12 h) (25), and mefloquine (17 h) (26), and comparable to that of chloroquine (47 h) (27) but shorter than that of piperaquine (15 days) (28). Unlike the quinoline antimalarials amodiaquine (29), chloroquine (30), and pyronaridine (31), which concentrate in erythrocytes, JPC-2997 does not appear to accumulate in erythrocytes, with a blood-to-plasma concentration ratio of 0.7. However, accurately measuring JPC-2997's uptake into erythrocytes requires not only measurement of the blood-to-plasma concentration ratio but also knowledge of the unbound fraction of JPC-2997 in plasma, which is presently unknown, and the hematocrit of the blood sample. JPC-2997 has a low CL/F of 0.22 liters/h/kg, and its V/F of 15.6 liters/kg indicates that the drug is distributed to body tissues. From a pharmacokinetic perspective, the relatively long elimination half-life justifies JPC-2997 being a potential partner drug for antimalarial treatment or being used alone for prophylaxis.
Comparison with MK-4815.
JPC-2997 and MK-4815 have comparable in vitro antimalarial activities in the low-nanomolar range against a broad spectrum of P. falciparum lines, with various levels of susceptibility to standard antimalarials. However, JPC-2997 is at least 2-fold more potent than MK-4815 against P. berghei KBG-173 strain, with no mice cured after a total dose of 48 mg/kg MK-4815 (16 mg on 3 days) using the modified Thompson test (data not shown). In contrast to JPC-2997, MK-4815 (12) has a higher clearance (1.26 liters/h/kg versus 0.22 liters/h/kg) and a much shorter elimination half-life in mice (4.5 h versus 49.8 h) and monkeys (14 h versus 259 h), suggesting that MK-4815 would have less posttreatment prophylaxis activity than JPC-2997.
Future application.
The quinoline-derived Mannich bases amodiaquine and pyronaridine are in current use for the treatment of uncomplicated P. falciparum malaria as partner drugs with artesunate (2). Artesunate-amodiaquine (ASAQ Winthrop; Sanofi-Aventis) is a widely available ACT, with high efficacy in Africa (32–34) but variable potency in Southeast Asian countries because of tolerant amodiaquine strains (35). In the past, amodiaquine administered alone for treatment or long-term prophylaxis of malaria has been associated with the severe adverse events agranulocytosis, hepatotoxicity, and neurotoxicities (involuntary movements/dystonia) (36, 37). These adverse events are believed to be associated with the oxidative metabolism of amodiaquine forming toxic reactive iminoquinone metabolites (38, 39). Since, JPC-2997 is not a quinoline derivative, it does not form toxic iminoquinone metabolites.
Compared with JPC-2997, amodiaquine is not as potent in suppressing murine malaria in the Peters 4-day test, with an ED50 of 2.1 mg/kg/day (40), and in the present study it was less effective in preventing recrudescence of the P. berghei ANKA strain. These favorable in vivo efficacy findings for JPC-2997 suggest that it may be a suitable partner drug with artesunate or as an addition to an existing ACT, such as dihydroartemisinin-piperaquine, to inhibit potential development of drug resistance. The latter concept is worthy of further consideration, as the elimination half-life of JPC-2997 lies between those of dihydroartemisinin and piperaquine.
In designing the next generation of drugs for malaria control and eradication, the Medicines for Malaria Venture (MMV) has developed target candidate profiles (TCP) that provide a “job description” for a new molecule to enter clinical development (41). JPC-2997 fulfills the TCP-2 definition of a long-duration partner drug, capable of killing residual parasites that are not eliminated by a rapid-asexual-stage-clearance TCP-1 compound. Furthermore, because of JPC-2997's long half-life, it may be a suitable TCP-4 candidate compound for chemoprotection.
In summary, JPC-2997 is a highly potent compound against chloroquine-resistant P. falciparum lines with low cytotoxicity in mammalian cell lines. It possesses potent in vivo efficacy in the murine P. berghei and Aotus monkey-P. vivax models, with long-lasting action in preventing recrudescences. After oral dosing, JPC-2997 is rapidly absorbed with a relatively long elimination half-life in mice and monkeys. Overall, the in vitro and in vivo antimalarial potency of JPC-2997 and its favorable pharmacokinetic properties suggest that the compound is worthy of further studies, including toxicological and metabolic evaluation and preclinical assessment.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the technical excellence of Kerryn Rowcliffe for in vitro drug testing and thank Donna MacKenzie, Stephen McLeod-Robertson, and Thomas Travers for rodent studies using the P. berghei ANKA strain and efficacy studies in Aotus monkeys. We thank Ivor Harris and John Hunter for veterinarian support for the Aotus monkey study. We are grateful to the Australian Red Cross Blood Service for the provision of human blood and sera for in vitro cultivation of P. falciparum lines.
This research was funded by the Australian Defence Organization and Jacobus Pharmaceutical Company Inc.
H.-M.S, G.D.H., W.Z., P.E.K., K.W.S., J.T., G.A.S., L.R.J., and D.P.J. work for Jacobus Pharmaceutical Company. We have no other conflicts of interest to declare.
The opinions expressed are those of the authors and do not necessarily reflect those of the Defense Health Service or any extant Australian Defense Force policy.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03762-14.
REFERENCES
- 1.World Health Organization. 2012. World malaria report 2012. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.World Health Organization. 2010. Guidelines for the treatment of malaria, 2nd ed. World Health Organization. Geneva, Switzerland. [Google Scholar]
- 3.Carrara VI, Zwang J, Ashley EA, Price RN, Stepniewska K, Barends M, Brockman A, Anderson T, McGready R, Phaiphun L, Proux S, van Vugt M, Hutagalung R, Lwin KM, Phyo AP, Preechapornkul P, Imwong M, Pukrittayakamee S, Singhasivanon P, White NJ, Nosten F. 2009. Changes in the treatment responses to artesunate-mefloquine on the northwestern border of Thailand during 13 years of continuous deployment. PLoS One 4:e4551. doi: 10.1371/journal.pone.0004551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dondorp AM, Nosten F, Das PYD, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hien TT, Thuy-Nhien NT, Phu NH, Boni MF, Thanh NV, Nha-Ca NT, Thai LH, Thai CQ, Toi PV, Thuan PD, Long LT, Dong LT, Merson L, Dolecek C, Stepniewska K, Ringwald P, White NJ, Farrar J, Wolbers M. 2012. In vivo susceptibility of Plasmodium falciparum to artesunate in Binh Phuoc Province, Vietnam. Malar J 11:355. doi: 10.1186/1475-2875-11-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saunders D, Vanachayangkul P, Lon C, U.S. Army Military Malaria Research Program, National Center for Parasitology, Entomology, and Malaria Control (CNM), Royal Cambodian Armed Forces . 2014. Dihydroartemisinin-piperaquine failure in Cambodia. N Engl J Med 371:484–485. doi: 10.1056/NEJMc1403007. [DOI] [PubMed] [Google Scholar]
- 7.World Health Organization. 1981. New antimalarial drugs under development, p 92–101. http://whqlibdoc.who.int/monograph/WHO_MONO_27_(2ed)_(chp4).pdf World Health Organization, Geneva, Switzerland. [Google Scholar]
- 8.Peters W, Irare SG, Ellis DS, Warhurst DC, Robinson BL. 1984. The chemotherapy of rodent malaria. XXXVII. Studies on the activity of three new antimalarials (WR 194,965, WR 228,258 and WR 225,448) against rodent and human malaria parasites (Plasmodium berghei and P. falciparum)I. Ann Trop Med Parasitol 78:567–579. [PubMed] [Google Scholar]
- 9.Peters W, Robinson BL. 1984. The chemotherapy of rodent malaria. XXXVII. The in vivo action of two Mannich bases, WR 194,965 and WR 228,258 and an 8-aminoquinoline WR 225,448. Ann Trop Med Parasitol 78:561–565. [PubMed] [Google Scholar]
- 10.Schmidt LH, Crosby R. 1978. Antimalarial activities of WR-194,965, an alpha-amino-o-cresol derivative. Antimicrob Agents Chemother 14:672–679. doi: 10.1128/AAC.14.5.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cosgriff TM. 1984. Evaluation of WR 194,965 in the treatment of multi-drug resistant P. falciparum, p B2-B14. In WR 194,965 H3P04. 4-(t-butyl)-2-t:-butylaminomethyl)-6-(4-chloro-phenyl)phenol phosphate. Investigational new drug, supplement no. 6. Walter Reed Army Institute of Research, Washington, DC. [Google Scholar]
- 12.Powles MA, Allocco J, Yeung L, Nare B, Liberator P, Schmatz D. 2012. MK-4815, a potential new oral agent for treatment of malaria. Antimicrob Agents Chemother 56:2414–2419. doi: 10.1128/AAC.05326-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 14.Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. 1979. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother 16:710–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lambros C, Vanderberg JP. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65:418–420. [PubMed] [Google Scholar]
- 16.O'Brien J, Wilson I, Orton T, Pognan F. 2000. Investigation of the Alamar blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem 267:5421–5426. [DOI] [PubMed] [Google Scholar]
- 17.Peters W. 1975. The chemotherapy of rodent malaria, XXII. The value of drug-resistant strains of P. berghei in screening for blood schizontocidal activity. Ann Trop Med Parasitol 69:155–171. [PubMed] [Google Scholar]
- 18.Ager AL. 1984. Experimental models: rodent malaria models (in vivo), p 225–254. In Peters W, Richards WHG (ed), Handbook of experimental pharmacology: antimalarial drugs, vol 68 Springer Verlag, New York, NY. [Google Scholar]
- 19.Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, Chiu FC, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H, McIntosh K, Padmanilayam M, Santo Tomas J, Scheurer C, Scorneaux B, Tang Y, Urwyler H, Wittlin S, Charman WN. 2004. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 430:900–904. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- 20.Rottmann M, McNamara C, Yeung BK, Lee MC, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, González-Páez GE, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck HP, Brun R, Nosten F, Renia L, Dartois V, Keller TH, Fidock DA, Winzeler EA, Diagana TT. 2010. Spiroindolones, a potent compound class for the treatment of malaria. Science 329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibaldi M, Perrier D. 1982. Pharmacokinetics, 2nd ed. Marcel Dekker Inc., New York, NY. [Google Scholar]
- 22.Obaldia N, Rossan RN, Cooper RD, Kyle DE, Nuzum EO, Rieckmann KH, Shanks GD. 1997. WR 238605, chloroquine, and their combinations as blood schizonticides against a chloroquine-resistant strain of Plasmodium vivax in Aotus monkeys. Am J Trop Med Hyg 56:508–510. [DOI] [PubMed] [Google Scholar]
- 23.Batty KT, Gibbons PL, Davis TM, Ilett KF. 2008. Pharmacokinetics of dihydroartemisinin in a murine malaria model. Am J Trop Med Hyg 78:641–642. [PubMed] [Google Scholar]
- 24.O'Neill PM, Shone AE, Stanford D, Nixon G, Asadollahy E, Park BK, Maggs JL, Roberts P, Stocks PA, Biagini G, Bray PG, Davies J, Berry N, Hall C, Rimmer K, Winstanley PA, Hindley S, Bambal RB, Davis CB, Bates M, Gresham SL, Brigandi RA, Gomez-de-Las-Heras FM, Gargallo DV, Parapini S, Vivas L, Lander H, Taramelli D, Ward SA. 2009. Synthesis, antimalarial activity, and preclinical pharmacology of a novel series of 4′-fluoro and 4′-chloro analogues of amodiaquine; identification of a suitable “back-up” compound for N-tert-butyl isoquine. J Med Chem 52:1828–1844. doi: 10.1021/jm8012757. [DOI] [PubMed] [Google Scholar]
- 25.Araujo FG, Huskinson J, Remington JS. 1991. Remarkable in vitro and in vivo activities of the hydroxynaphthoquinone 566C80 against tachyzoites and tissue cysts of Toxoplasma gondii. Antimicrob Agents Chemother 35:293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rozman RS, Molek NA, Koby R. 1978. The absorption, distribution, and excretion in mice of the antimalarial mefloquine, erythro-2,8-bis(trifluoromethyl)-alpha-(2-piperidyl)-4-quinolinemethanol hydrochloride. Drug Metab Dispos 6:654–658. [PubMed] [Google Scholar]
- 27.Moore BR, Page-Sharp M, Stoney JR, Ilett KF, Jago JD, Batty KT. 2011. Pharmacokinetics, pharmacodynamics, and allometric scaling of chloroquine in a murine malaria model. Antimicrob Agents Chemother 55:3899–3907. doi: 10.1128/AAC.00067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore BR, Batty KT, Andrzejewski C, Jago JD, Page-Sharp M, Ilett KF. 2008. Pharmacokinetics and pharmacodynamics of piperaquine in a murine malaria model. Antimicrob Agents Chemother 52:306–311. doi: 10.1128/AAC.00878-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winstanley P, Edwards G, Orme M, Breckenridge A. 1987. The disposition of amodiaquine in man after oral administration. Br J Clin Pharmacol 23:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frisk-Holmberg M, Bergqvist Y, Termond E, Domeij-Nyberg B. 1984. The single dose kinetics of chloroquine and its major metabolite desethylchloroquine in healthy subjects. Eur J Clin Pharmacol 26:521–530. doi: 10.1007/BF00542151. [DOI] [PubMed] [Google Scholar]
- 31.Chen YC, Fleckenstein L. 2001. Improved assay method for the determination of pyronaridine in plasma and whole blood by high-performance liquid chromatography for application to clinical pharmacokinetic studies. J Chromatogr. B Analyt Technol Biomed Life Sci 752:39–46. [DOI] [PubMed] [Google Scholar]
- 32.Dorkenoo MA, Barrette A, Agbo YM, Bogreau H, Kutoati S, Sodahlon YK, Morgah K. 2012. Surveillance of the efficacy of artemether-lumefantrine and artesunate-amodiaquine for the treatment of uncomplicated Plasmodium falciparum among children under five in Togo, 2005-2009. Malar J 11:338. doi: 10.1186/1475-2875-11-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faye B, Offianan AT, Ndiaye JL, Tine RC, Toure W, Djoman K, Sylla K, Ndiaye PS, Penali L, Gaye O. 2010. Efficacy and tolerability of artesunate-amodiaquine (Camoquin plus) versus artemether-lumefantrine (Coartem) against uncomplicated Plasmodium falciparum malaria: multisite trial in Senegal and Ivory Coast. Trop Med Int Health 15:608–613. [DOI] [PubMed] [Google Scholar]
- 34.Ndounga M, Mayengue PI, Casimiro PN, Loumouamou D, Basco LK, Ntoumi F, Brasseur P. 2013. Artesunate-amodiaquine efficacy in Congolese children with acute uncomplicated falciparum malaria in Brazzaville. Malar J 12:53. doi: 10.1186/1475-2875-12-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ratcliff A, Siswantoro H, Kenangalem E, Maristela R, Wuwung RM, Laihad F, Ebsworth EP, Anstey NM, Tjitra E, Price RN. 2007. Two fixed-dose artemisinin combinations for drug-resistant falciparum and vivax malaria in Papua, Indonesia: an open-label randomised comparison. Lancet 369:757–765. doi: 10.1016/S0140-6736(07)60160-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hatton CS, Peto TE, Bunch C, Pasvol G, Russell SJ, Singer CR, Edwards G, Winstanley P. 1986. Frequency of severe neutropenia associated with amodiaquine prophylaxis against malaria. Lancet i:411–414. [DOI] [PubMed] [Google Scholar]
- 37.Neftel KA, Woodtly W, Schmid M, Frick PG, Fehr J. 1986. Amodiaquine induced agranulocytosis and liver damage. Br Med J (Clin Res Ed) 292:721–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naisbitt DJ, Williams DP, O'Neill PM, Maggs JL, Willock DJ, Pirmohamed M, Park BK. 1998. Metabolism-dependent neutrophil cytotoxicity of amodiaquine: a comparison with pyronaridine and related antimalarial drugs. Chem Res Toxicol 11:1586–1595. [DOI] [PubMed] [Google Scholar]
- 39.Ruscoe JE, Tingle MD, O'Neill PM, Ward SA, Park BK. 1998. Effect of disposition of mannich antimalarial agents on their pharmacology and toxicology. Antimicrob Agents Chemother 42:2410–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Neill PM, Park BK, Shone AE, Maggs JL, Roberts P, Stocks PA, Biagini GA, Bray PG, Gibbons P, Berry N, Winstanley PA, Mukhtar A, Bonar-Law R, Hindley S, Bambal RB, Davis CB, Bates M, Hart TK, Gresham SL, Lawrence RM, Brigandi RA, Gomez-delas-Heras FM, Gargallo DV, Ward SA. 2009. Candidate selection and preclinical evaluation of N-tert-butyl isoquine (GSK369796), an affordable and effective 4-aminoquinoline antimalarial for the 21st century. J Med Chem 52:1408–1415. doi: 10.1021/jm8012618. [DOI] [PubMed] [Google Scholar]
- 41.Burrows JN, van Huijsduijnen RH, Möhrle JJ, Oeuvray C, Wells TNC. 2013. Designing the next generation of medicines for malaria control and eradication. Malar J 12:187. doi: 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.