

Abstract
Prolonged treatment with the oxazolidinone linezolid is associated with myelosuppression, lactic acidosis, and neuropathies, toxicities likely caused by impairment of mitochondrial protein synthesis (MPS). To evaluate the potential of the novel oxazolidinone tedizolid to cause similar side effects, nonclinical and pharmacokinetic assessments were conducted. In isolated rat heart mitochondria, tedizolid inhibited MPS more potently than did linezolid (average [± standard error of the mean] 50% inhibitory concentration [IC50] for MPS of 0.31 ± 0.02 μM versus 6.4 ± 1.2 μM). However, a rigorous 9-month rat study comparing placebo and high-dose tedizolid (resulting in steady-state area under the plasma concentration-time curve values about 8-fold greater than those with the standard therapeutic dose in humans) showed no evidence of neuropathy. Additional studies explored why prolonged, high-dose tedizolid did not cause these mitochondriopathic side effects despite potent MPS inhibition by tedizolid. Murine macrophage (J774) cell fractionation studies found no evidence of a stable association of tedizolid with eukaryotic mitochondria. Monte Carlo simulations based on population pharmacokinetic models showed that over the course of a dosing interval using standard therapeutic doses, free plasma concentrations fell below the respective MPS IC50 in 84% of tedizolid-treated patients (for a median duration of 7.94 h) and 38% of linezolid-treated patients (for a median duration of 0 h). Therapeutic doses of tedizolid, but not linezolid, may therefore allow for mitochondrial recovery during antibacterial therapy. The overall results suggest that tedizolid has less potential to cause myelosuppression and neuropathy than that of linezolid during prolonged treatment courses. This, however, remains a hypothesis that must be confirmed in clinical studies.
INTRODUCTION
Many drugs, including various antibacterials, adversely affect mitochondrial function (1–3). For instance, oxazolidinones, a class of antibiotics that inhibit bacterial protein synthesis by binding to the 50S ribosomal subunit, can impair mitochondrial protein synthesis (MPS) (4–7) due to structural similarities between mitochondrial and prokaryotic ribosomes (8–10). Inadvertent inhibition of MPS is thought to be the underlying mechanism for several well-known side effects of prolonged use of the oxazolidinone linezolid, such as myelosuppression, lactic acidosis, and peripheral and ocular neuropathies (8, 11–19). Although being generally, but not always, reversible, these toxicities can significantly limit the use of linezolid during long-term therapy (16, 19–24); cautions are included in the official prescribing information for this drug (25).
Tedizolid is a novel oxazolidinone antibacterial with potent activity against a wide range of Gram-positive pathogens, including resistant strains, such as methicillin-resistant Staphylococcus aureus, vancomycin-resistant enterococci, and cfr-positive linezolid-resistant strains (in the absence of certain ribosomal mutations conferring reduced oxazolidinone susceptibility) (7, 26–29). Tedizolid is administered as the prodrug tedizolid phosphate, which is rapidly and extensively converted to the active moiety by endogenous phosphatases (30). In two recent phase 3 trials, tedizolid phosphate (200 mg once daily for 6 days) showed noninferior efficacy relative to that of linezolid (600 mg twice daily for 10 days) for management of acute bacterial skin and skin structure infection (31, 32), a medical condition that generally necessitates only relatively brief antibacterial treatment. In those studies, both agents were well tolerated. Tedizolid, however, had a more favorable gastrointestinal and hematologic profile than that of linezolid (31, 32).
Because oxazolidinones have the potential to cause mitochondrial toxicity, with wide-ranging safety implications, it is important to evaluate whether and to what extent tedizolid might affect mitochondrial function if used to manage infections that necessitate a longer duration of therapy (e.g., osteomyelitis). To this end, we carried out nonclinical studies and analyses based on clinical data, assessing different aspects of the underlying mechanisms or consequences of tedizolid and linezolid mitochondrial toxicity. Our objective was to characterize the potential risk of tedizolid resulting in mitochondrial toxicity-associated adverse events similar to those observed with linezolid during prolonged clinical use.
(Data from murine macrophage cell fractionation studies of tedizolid were presented in part at the 52nd International Conference on Antimicrobial Agents and Chemotherapy, 9 to 12 September 2012, San Francisco, CA [33]. Data from long-term neurotoxicity studies of tedizolid in rats were presented in part at the 53rd International Conference on Antimicrobial Agents and Chemotherapy, 10 to 13 September 2013, Denver, CO [34], and at the 53rd Annual Meeting of the Society of Toxicology, 23 to 27 March 2014, Phoenix, AZ [35].)
MATERIALS AND METHODS
Effects of tedizolid and linezolid on MPS.
An in vitro study using isolated rat heart mitochondria was conducted to compare the tedizolid and linezolid concentrations that inhibit 50% (IC50) of MPS. The assay, sample preparation techniques, and data analysis were previously described in detail (9, 36). In brief, intact, highly coupled mitochondria isolated from normal rat heart were incubated in a medium containing [35S]methionine with tedizolid or linezolid (dissolved at various concentrations in dimethyl sulfoxide as vehicle) and in control medium and vehicle control medium. Each assay was conducted in 6 independent experiments, using 4 independent stock solutions. The variabilities of results within and between stock solutions were similar, and all data from the six independent experiments were thus pooled. The rate of MPS was expressed as picomoles of methionine incorporated per milligram of mitochondrial protein. Dose-response curves were analyzed with Sigma Plot, version 12.5 (Systat Software Inc., San Jose, CA), using the best-fit slope for each concentration of compound tested, expressed as a percentage of the vehicle control best-fit slope, plotted against the test compound concentration (9, 36). Data shown represent means and standard errors of the means (SEM) of the best-fit slopes at each concentration (n = 6); the best-fit hyperbolic decay regression line of these data (using the least-squares method) was used to determine the MPS IC50.
Long-term neurotoxicity study in rats.
Because neuropathy can be a serious oxazolidinone side effect potentially caused by impairment of mitochondrial function, an in vivo study in rats evaluated the potential neurotoxicity of long-term administration of tedizolid at drug exposures higher than that achieved at the human-equivalent therapeutic dose. Animals were approximately 9 weeks old at the start of dosing and were given either active drug or control for up to 9 months. Tedizolid in vehicle was administered via oral gavage to Long Evans pigmented rats (n = 84) once daily at dose levels of up to 30 mg/kg of body weight (males) and 10 mg/kg (females). Additional matched groups of animals (n = 34) received a placebo control. Different doses were used in males and females because of sex-specific differences in tedizolid metabolism (only known to be the case in rats, not other species) that result in approximately 3-fold higher total tedizolid plasma concentrations in female rats than in male rats (30). Neurotoxicology groups consisted of 12 males and 12 females each for the control and tedizolid dose groups. Toxicokinetic groups consisted of five males and five females each for control groups and nine males and nine females each for the tedizolid dose groups. Neurotoxicity evaluations included functional observational battery and locomotor activity assessments (each in 6 animals/sex/group) and ophthalmic evaluation (in all 12 animals/sex/group) at baseline and near the end of the 9-month period. In addition, at the end of the 9-month period, detailed microscopic neuropathological examinations were conducted in 10 animals/sex each in the control and high-dose tedizolid groups, on multiple regions of the central nervous system and several central and peripheral nerves (cervical spinal, lumbar spinal, optic [retrobulbar and intracranial], peroneal, sciatic, sural, tibial, and trigeminal nerves, lumbar and cervical dorsal root ganglia, and dorsal and ventral root fibers), and the results were reviewed by a second pathologist. Tedizolid exposure was assessed throughout the study in the animals of the toxicokinetic groups: blood samples (3 animals/sex/group/time point) were collected before dosing and 1, 2, 4, 8, and 24 h after dosing at weeks 0 and 39. Control group blood samples were only collected 2 h after dosing. Tedizolid concentrations in plasma were analyzed by use of a validated liquid chromatography-tandem mass spectrometry method (M. J. Schlosser, H. Hosako, A. Radovsky, M. T. Butt, D. Draganov, J. Vija, and F. Oleson, submitted for publication). Continuous functional observational battery data were analyzed by parametric one-way analysis of variance (ANOVA) to determine intergroup differences; if significant intergroup variance was found, the Dunnett test was used to compare treated with control groups. Repeated-measures ANOVA was used to compare total and ambulatory locomotor activities. Scalar and descriptive data (i.e., remaining functional observational battery parameters and neuropathological findings) were analyzed using the Fisher exact test. Animals were maintained in accordance with the Guide for the Care and Use of Laboratory Animals (61). The animal facilities at WIL Research are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Cell fractionation studies.
Previous studies have shown that tedizolid concentrates approximately 10-fold more than linezolid in macrophages (37, 38). Cell fractionation and subcellular localization studies were therefore conducted to determine whether intracellular tedizolid would be associated with mitochondria, which might represent a possible toxicity hazard. Murine J774 macrophages were selected because these cells can easily be fractionated and their mitochondria adequately separated from other cell organelles by centrifugation (39). The methods were previously described in detail and validated to determine the subcellular localization of various antibiotics, such as aminoglycosides, fluoroquinolones, and macrolides (40) as well as oxazolidinones (39). Cells were incubated for 2 h with 20 mg/liter tedizolid before collection. Subsequently, the pericellular membranes were disrupted by homogenization using a Dounce tissue grinder, and subcellular organelles were separated by subjecting the resulting suspension to one of two types of centrifugation: (i) differential centrifugation (centrifugation at increasing speeds [39], separating organelles mainly on the basis of size) or (ii) isopycnic centrifugation (centrifugation through sucrose gradients [160,000 × g for 16 h], in which organelles are separated based on their buoyant densities). Fractions were then assayed for enzyme markers of the cell components of interest and for tedizolid content. Marker enzymes included N-acetyl-β-hexosaminidase for lysosomes, cytochrome c oxidase for mitochondria, and lactate dehydrogenase for cytosol. Tedizolid was extracted from cell fractions by phase partition and protein precipitation and quantified by liquid chromatography (reverse phase) coupled to mass spectrometry, using an LTQ-Orbitrap mass spectrometer (Thermo Scientific, Waltham, MA). Linezolid was used as an internal standard, with linear responses obtained in the 10- to 1,000-ng/ml range.
PK analyses.
Human free plasma concentration-time data for tedizolid and linezolid in relation to the MPS IC50 of each drug were evaluated to compare the potential for mitochondrial recovery between tedizolid and linezolid given at therapeutic doses; pharmacokinetic (PK) data were derived from two sources: (i) extensively sampled data from healthy subjects (25, 41) and (ii) Monte Carlo simulations. Simulations were conducted based on previously presented population PK models for tedizolid and linezolid (42, 43). Two sets of 2,500 virtual patients were generated by resampling the characteristics of patients in the combined population from phase 3 studies comparing tedizolid and linezolid against acute bacterial skin and skin structure infections. One set was simulated to receive 200 mg tedizolid phosphate orally once daily for 6 days and the other to receive 600 mg linezolid twice daily orally for 10 days. Simulated drug concentrations at 10-min increments over the dosing interval were obtained based on the respective final population PK models, including covariate effects. Ideal body weight and total bilirubin were significant covariates in the tedizolid population PK model, and weight and age were significant covariates in the linezolid population PK model (42, 43). Thus, these covariates were randomly resampled from the vectors of patient characteristics from the combined population of phase 3 patients in order to maintain the covariance structure inherent between covariates. Per the original publications, neither model included covariance between PK parameters. Free plasma drug concentrations were calculated by assuming 80% protein binding for tedizolid (30) and 31% protein binding for linezolid (25). Time below the MPS IC50 was calculated for each virtual tedizolid patient on day 3 and for each virtual linezolid patient on day 5, and the results of these calculations were summarized across the 2,500 patients.
RESULTS
Effects of tedizolid and linezolid on MPS.
Time course data showed that the rate of [35S]methionine incorporation into mitochondrial protein, reflecting the rate of protein synthesis, was linear over time with all evaluated tedizolid and linezolid concentrations in both studies. Dose-response curves (Fig. 1) indicated that the average (±SEM) MPS IC50 was 0.31 ± 0.02 μM for tedizolid and 6.4 ± 1.2 μM for linezolid. A control experiment confirmed that the solvent used to dissolve both drugs did not affect the assay. These results suggest that tedizolid and linezolid readily cross the inner mitochondrial membrane and inhibit MPS and that tedizolid is a more potent MPS inhibitor.
FIG 1.
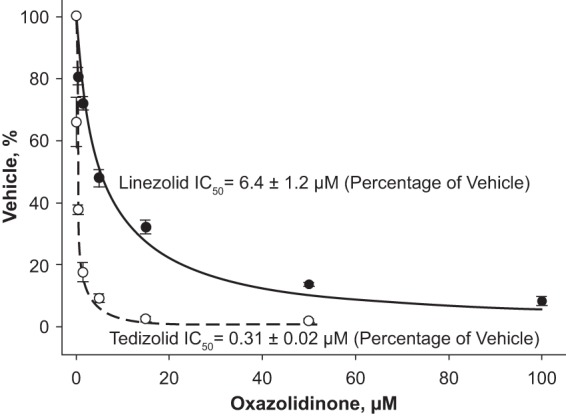
Concentration-response effects of tedizolid and linezolid on MPS. Highly coupled rat heart mitochondria were incubated with [35S]methionine in the presence of increasing concentrations of tedizolid (open circles) or linezolid (closed circles). Data are the means (± standard errors of the means) from six independent experiments. IC50 is defined as the concentration of drug causing a 50% reduction of the value for the control (vehicle only), with its value calculated using best-fit hyperbolic decay regression.
Long-term neurotoxicity study in rats.
Toxicokinetic analyses showed that tedizolid exposure increased proportionally to dose, with little accumulation over time. The tedizolid steady-state total area under the concentration-time curve (AUC) exposure at 9 months for the high-dose group was 222 μg · h/ml for male rats and 189 μg · h/ml for female rats (i.e., approximately 8-fold greater, on average, than those observed in humans with the therapeutic dose).
After 9 months, no tedizolid-related effects were observed on functional observational battery testing, locomotor activity assessment, ophthalmic examination, or macroscopic and microscopic neuropathological examination. In both control and treated animals, axonal degeneration was observed in several of the nerves evaluated; however, when present, this degeneration was only minimal to mild. Neuropathology findings were similar between treated animals and controls: at 9 months, an average of 3.7 axonal degenerations/animal was observed in rats in the high-dose tedizolid group, compared with an average of 3.6 axonal degenerations/animal in the control group. Therefore, the degenerations most likely were related to aging and were not a treatment effect. The distributions of axonal degenerations across the evaluated nerves were also similar between the high-dose tedizolid and control groups after a full 9 months (Fig. 2); we observed no degeneration in optic nerves. There were no significant differences in axonal degenerations between male and female animals.
FIG 2.
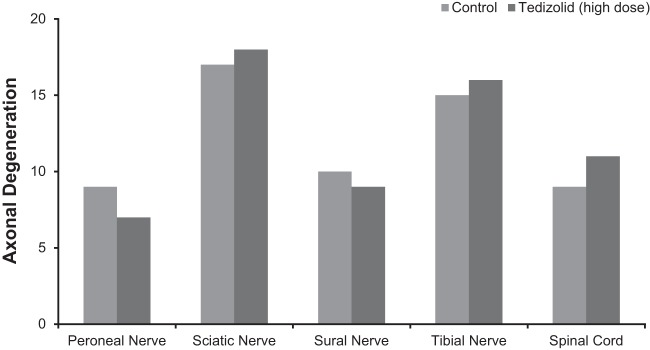
Neuropathology results from a long-term neurotoxicity study comparing control rats (n = 20) to rats administered tedizolid (n = 20) at a dose with a mean total plasma exposure equivalent to approximately 8 times the total AUC exposure achieved with the human therapeutic dose.
Cell fractionation studies.
After differential centrifugation, 87% of the cell-associated tedizolid was recovered in the high-speed supernatant (Fig. 3A), along with 71% of lactate dehydrogenase (marker for the cytosol) and only 0.25% of cytochrome c oxidase (marker for mitochondria). Conversely, 69% of cytochrome c oxidase and 62% of N-acetyl-β-hexosaminidase (marker for lysosomes) were found in the granule fraction, which contained only 2.7% of the tedizolid. Overall, these results suggested that tedizolid was recovered from the cytosol and was not associated in a stable fashion with mitochondria or other organelles.
FIG 3.

Results of cell fractionation studies conducted by differential centrifugation (A) and isopycnic centrifugation (B). For differential centrifugation, the cell homogenate was subjected to centrifugation at increasing speeds to collect organelles of decreasing size and a final supernatant. Results are shown as the percentage of each constituent recovered in each fraction. For isopycnic centrifugation, a postnuclear supernatant (cell homogenate minus nuclei and unbroken cells) was deposited on top of a linear sucrose gradient, which was centrifuged at high speed to allow subcellular organelles to move to and equilibrate at their buoyant densities. Results are shown as frequency distribution histograms (fractional amount recovered/density increment versus the density span of the gradient) (see the work of Lemaire et al. [37] for details). Cyt-oxidase, cytochrome c oxidase; Hex B, N-acetyl-β-hexosaminidase; LDH, lactate dehydrogenase.
After isopycnic centrifugation of the postnuclear fraction (homogenate minus the nuclear and unbroken cell fraction), 99% of tedizolid was recovered in the top three fractions (Fig. 3B), indicating no association with cell organelles, which move further into the gradient and equilibrate at their respective buoyant densities. The separation of tedizolid from mitochondria was strikingly complete, with only 7% of cytochrome c oxidase activity recovered in the top three fractions and the remaining activity equilibrating in the bottom fractions. For N-acetyl-β-hexosaminidase, 27% of the activity was recovered in the top three fractions (most likely corresponding to enzyme released from damaged lysosomes), with the remaining activity spreading at larger densities throughout the gradient, consistent with the heterogeneous character of lysosomes. For lactate dehydrogenase, 93% of the activity was recovered in the top three fractions. There was a modest move of lactate dehydrogenase into the gradient, resulting in the partial dissociation of its distribution from that of tedizolid due to the large molecular weight of this protein in conjunction with the high centrifugal field used in these experiments. As with differential centrifugation, these results also suggested that tedizolid was free in the cytosol and not associated in a stable fashion with subcellular organelles.
Pharmacokinetic analyses.
Figure 4 shows the steady-state free drug exposure levels for linezolid and tedizolid based on current dosing strategies (600 mg twice daily and 200 mg once daily, respectively), as previously reported in healthy subjects (25, 41), in relation to the mean MPS IC50 of each agent (Table 1). These data from healthy subjects with extensive, serial sampling over time are believed to be more reliable PK data and are reflective of patient PK data obtained with sparse sampling. While the free plasma concentration of linezolid remained above its MPS IC50 for the duration of the dosing interval, free tedizolid plasma concentrations fell below the corresponding MPS IC50 about 18 h into the 24-h dosing interval. The population PK model-based Monte Carlo simulations yielded similar results, specifically for patients treated with tedizolid or linezolid for serious bacterial skin infections. The values for the time below the MPS IC50 and the percentage of patients with values consistently greater than the IC50, as estimated by these simulations, are shown in Table 1. In most tedizolid subjects (84%), there were periods (median duration, 8 h) during which tedizolid free plasma concentrations fell below the tedizolid MPS IC50. In contrast, in most linezolid subjects (62%), linezolid free plasma concentrations remained above the linezolid MPS IC50 for the duration of the dosing interval. The model-derived minimum observed free drug concentration in plasma (Cmin), maximum observed free drug concentration in plasma (Cmax), and AUC values are shown for comparison in Table 2.
FIG 4.
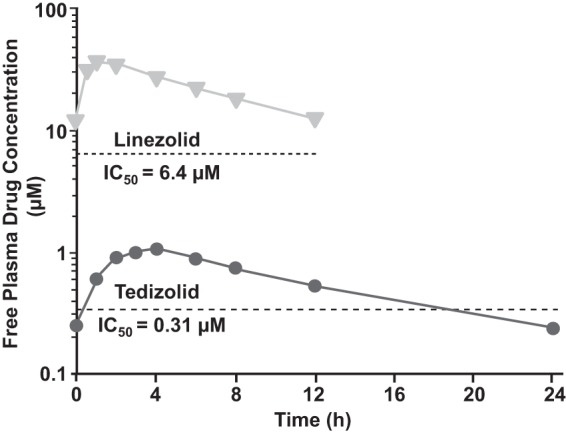
Mean free (unbound) drug plasma exposure concentrations at steady state for therapeutic-dose tedizolid (200 mg once daily; circles) and linezolid (600 mg twice daily; triangles) over the course of the dosing interval, based on published values (25, 41), in relation to the MPS IC50 of each agent.
TABLE 1.
Population pharmacokinetic simulations with tedizolid and linezolid
| Parametera | Value |
|
|---|---|---|
| Tedizolidb | Linezolidc | |
| Mean (SE) MPS IC50 (μM) | 0.31 (0.02) | 6.4 (1.2) |
| Time below MPS IC50 (h) | ||
| Mean (SD) | 7.62 (5.49) | 3.17 (5.29) |
| Median | 7.94 | 0 |
| 25th–75th percentiles | 2.48–11.92 | 0–4.93 |
| % of patients with all free drug concentrations above the IC50 | 16 | 62 |
IC50, 50% inhibitory concentration; MPS, mitochondrial protein synthesis.
Administered as 200 mg once daily; protein binding of 80% was assumed.
Administered as 600 mg twice daily; protein binding of 31% was assumed.
TABLE 2.
Summary statistics of model-derived exposure measures for tedizolid and linezolid at the respective midpoints of the treatment durations evaluated in two recent phase 3 studies of acute bacterial skin and skin structure infections
| Exposure measure | Value |
|
|---|---|---|
| Tedizolid on day 3 | Linezolid on day 5 | |
| fAUC(0–24) (μg · h/ml) | ||
| Mean (SD) | 4.47 (1.44) | 129.88 (67.08) |
| 90% confidence interval | (4.43, 4.52) | (127.67, 132.08) |
| Median | 4.29 | 115 |
| Minimum, maximum | 0.8, 10.8 | 15.4, 502.0 |
| n | 2,500 | 2,500 |
| fCmax (μg/ml) | ||
| Mean (SD) | 0.38 (0.11) | 7.59 (3.39) |
| 90% confidence interval | (0.3730, 0.3801) | (7.48, 7.70) |
| Median | 0.38 | 7.03 |
| Minimum, maximum | 0.0, 0.8 | 0.7, 23.8 |
| n | 2,500 | 2,500 |
| fCmin (μg/ml) | ||
| Mean (SD) | 0.07 (0.04) | 3.45 (2.63) |
| 90% confidence interval | (0.0703, 0.0733) | (3.37, 3.54) |
| Median | 0.06 | 2.82 |
| Minimum, maximum | 0.0, 0.3 | 0.0, 20.0 |
| n | 2,500 | 2,500 |
DISCUSSION
This article presents the first account of prospective, nonclinical studies aiming to assess and compare oxazolidinone mitochondrial toxicities in the context of their anticipated therapeutic exposures (toxicodynamics) by using linezolid, considered a reference oxazolidinone, and tedizolid, a novel oxazolidinone antibacterial. Because impairment of MPS seems to be the primary trigger of certain clinical adverse effects that can occur with long-term linezolid treatment, such as myelosuppression, neuropathy, and lactic acidosis (8, 11–19), we initially conducted an in vitro study to compare the abilities of linezolid and tedizolid to inhibit MPS. The results clearly demonstrated dose- and time-dependent inhibition of MPS, which was more pronounced with tedizolid than with linezolid. The observation that tedizolid is a more potent MPS inhibitor than linezolid is not surprising, since tedizolid has additional target site interactions (7), as reflected in its lower MICs than those of linezolid against Gram-positive pathogens (7, 27, 28). Because bacterial and mitochondrial ribosomes share an evolutionary origin and structural similarities, differential binding and ensuing protein synthesis inhibition by oxazolidinones would affect both types of ribosomes (bacterial and mitochondrial) similarly. Considered independently, this result would predict that tedizolid would cause more mitochondrial toxicity than linezolid during prolonged clinical use. However, this was not found in a rigorous 9-month study in rats that was looking for neuropathological effects after long-term administration of tedizolid at multiples of the human therapeutic exposure. The results of our additional analyses might offer an explanation and suggest that tedizolid might in fact cause fewer mitochondriopathic effects than linezolid at human therapeutic exposures. This hypothesis, however, requires formal evaluation in clinical trials of sufficient duration.
In our first additional analysis, using cells incubated with tedizolid for a short period, we found no evidence of stable association of the drug with eukaryotic mitochondria. Although this does not preclude the potential for reversible MPS inhibition with tedizolid, it does suggest a rapid dissociation and avoidance of prolonged effects once the drug concentration is lowered. Second, two analyses were conducted to compare free (unbound) plasma exposures of tedizolid and linezolid with the MPS IC50 of each drug. These comparisons suggested that at therapeutic doses, tedizolid, but not linezolid, allows partial mitigation of mitochondrial impairment over the course of a dosing interval, which might have important implications for long-term therapeutic use of these agents.
Several groups of researchers have suggested the trough concentration (within certain ranges) as the exposure measure that best predicts the safety of oxazolidinones (44–48). The hypothesis that plasma trough levels above the MPS IC50 of an oxazolidinone will result in permanently inhibited MPS was first advanced by Garrabou et al. (12). It is believed that patients with trough levels dropping below the MPS IC50 might have some mitochondrial recovery and that the duration of this recovery period parallels the duration that plasma levels remain under this IC50 over the course of a dosing interval (12). Therefore, protein synthesis inhibitors maintaining antibacterial efficacy at systemic exposures that allow for a sufficient period of mitochondrial recovery might result in fewer related adverse events than agents for which this is not the case. Therapeutic efficacies of linezolid and tedizolid are driven by their respective ratios of the area under the concentration-time curve over the dosing interval (τ) at steady state for free drug to the MIC (fAUC0–τ/MIC) (49, 50). Because the PK properties of tedizolid allow for once-daily dosing while maintaining an fAUC/MIC sufficient for activity (30, 43), the therapeutically effective daily dose of tedizolid can be reduced to 200 mg, which is 6 times lower than that of linezolid (1,200 mg). The resulting differences in drug exposures are magnified further by the greater protein binding and distribution of tedizolid in the body (30), resulting in free exposures that are 19 times higher for linezolid than for tedizolid based on the fAUC, 33 times higher based on the Cmax, and 49 times higher based on the Cmin, based on previously published data for both agents (25, 41). Assuming that mitochondrion-related toxicities are driven by the time during which the free Cmin remains higher than the MPS IC50 of a drug (12, 48), our data help to explain why tedizolid may be safer than linezolid for clinical use, despite tedizolid having a lower MPS IC50 (by molar value).
An additional factor that may contribute to fewer mitochondrial effects with tedizolid than with linezolid might be the substantial drug accumulation (and increased plasma concentrations) with repeated administration of linezolid (25, 51, 52). In contrast, most tedizolid-treated patients stay within the desired exposure window, allowing mitochondrial recovery because of narrow PK variability and a lack of significant accumulation (43, 52). It is speculated that accumulation of linezolid is caused by impairment of hepatic mitochondria, leading to autoinhibition of its metabolism (53). The particular linezolid population PK model applied in this analysis used a conservative approach with no drug accumulation; linezolid accumulation would adversely affect the proportion of patients experiencing the benefits of mitochondrial recovery. Other linezolid models published in the literature used either parallel linear and Michaelis-Menten elimination or a kinetics that was linear initially and subsequently saturable (53–55). Lack of mitochondrial recovery is thought to be a problem largely in patients who receive prolonged courses of oxazolidinone therapy (16, 19–23). In these patients, the differences in mitochondrial toxicity between tedizolid and linezolid might have important implications for care.
In the context of clinically relevant mitochondrial effects, the potential for neuropathy is of particular importance. In contrast with the 9-month rat study with tedizolid that is presented herein, linezolid was previously shown to have neurotoxic potential when administered to rats for 6 months (i.e., 3 months fewer than in our rat study) at linezolid exposure levels comparable to those in humans (25). In contrast, tedizolid was tested at much higher (approximately 8-fold) systemic exposures than would be achieved in humans at the therapeutic dose. In the previous linezolid study, minimal to mild sciatic nerve degeneration was observed as early as 3 months into the study, and minimal to moderate optic nerve degeneration was observed after 6 months of drug administration (25). The noticeable differences in neurotoxicity profiles between linezolid and tedizolid suggest that the risk for neuropathy during long-term treatment is lower with tedizolid. The data presented herein are also consistent with previous studies conducted in rats and dogs for 1 month or 3 months. In those studies, tedizolid was also without hematopoietic toxicity at exposures up to 4 to 7 times the human-equivalent exposure, whereas linezolid toxicity was evident at drug levels comparable to the corresponding human-equivalent exposure (25, 56, 57). Hematopoietic effects of linezolid are believed to be the result of MPS inhibition in bone marrow cells (2, 25, 56, 58). In the clinical setting, results from two phase 3 trials conducted in patients (n = 1,333) with acute bacterial skin and skin structure infections indicated that tedizolid given at 200 mg once daily for 6 days resulted in a lower incidence of adverse changes in hematologic parameters, in particular platelets, than that with linezolid given at 600 mg twice daily for 10 days. This outcome was observed independently of the differences in exposure times (31, 32). Pooled data from these clinical trials suggested a potentially clinically relevant difference in terms of adverse platelet outcomes between linezolid and tedizolid, but the authors concluded that longer-term studies were necessary to ascertain this possibility (59).
Mitochondrial gene mutations play an important role in increasing the risk for adverse effects resulting from MPS inhibition with various antibacterials that act as protein synthesis inhibitors. Genetic polymorphism is one reason that some individuals are more susceptible to antibacterial-associated mitochondrial toxicity (2, 60), helping to explain why only a minority of patients receiving linezolid develop clinically relevant adverse effects via this mechanism. Certain comorbidities, such as diabetes mellitus and rare, inherited mitochondrial disorders, might also place patients at greater risk for mitochondriopathic effects. The same factors might apply to tedizolid but could not be evaluated in the context of the studies presented herein. Additional limitations of our studies should be pointed out. The population PK analyses used total plasma concentrations. Although oxazolidinone plasma levels are proportional to tissue concentrations, they are not equivalent to actual drug levels at tissue sites of potential toxicity, and this issue is compounded by the fact that tedizolid and linezolid penetrate different body tissues to different extents. Another limitation is the lack of head-to-head studies directly comparing tedizolid and linezolid in terms of subcellular distribution and long-term neurotoxic potential in animals. Finally, we must point out the absence of clinical studies in which tedizolid was administered for more than 3 weeks. Before any statements on clinically relevant differences between tedizolid and linezolid in terms of mitochondrial impairment can be made, actual clinical experience with prolonged tedizolid therapy is necessary.
In conclusion, the overall results suggest that although tedizolid is a more potent inhibitor of MPS than linezolid on a molar basis, its potential to cause mitochondrion-related adverse events (such as myelosuppression, neuropathy, and lactic acidosis) in vivo, in terms of frequency and severity, may be less than that of linezolid when each drug is assessed in the context of its respective therapeutic dosage. This illustrates that PK parameters must be taken fully into account to correctly translate the results of in vitro toxicity studies to clinical practice, leading the way to true toxicodynamic assessment, an approach that may be used for the study of the safety of other antibiotics and drugs. The studies reported here also add to the growing body of evidence suggesting that tedizolid may have a more favorable overall tolerability profile than that of linezolid. However, the hypothesis that long-term antibacterial therapy with tedizolid may be better tolerated than that with linezolid first requires verification in prospective clinical trials before conclusive statements can be made.
ACKNOWLEDGMENTS
We thank Ken Bartizal for his efforts in study conception and guidance on data interpretation, Michael J. Schlosser for designing the long-term animal neurotoxicity study and providing guidance on interpreting the study data, and Axel Lambert and Giulio Muccioli for performing the liquid chromatography-tandem mass spectrometry assays of tedizolid in cell fractions. Medical writing support was provided by Dominik Wolf, an employee of Cubist Pharmaceuticals.
The MPS inhibition study and the long-term animal neurotoxicity study were funded by Trius Therapeutics (now part of Cubist Pharmaceuticals). The PK analyses were funded by Cubist Pharmaceuticals.
All authors had full access to the data. The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development, and had final responsibility for the decision to submit for publication.
Shawn Flanagan and Philippe Prokocimer are employees of Cubist Pharmaceuticals. Edward E. McKee has received research funding from Trius Therapeutics (now part of Cubist Pharmaceuticals). Debaditya Das was a postdoctoral fellow of the Belgian Fonds de la Recherche Scientifique Médicale (FRSM) and has been a consultant to Trius Therapeutics (now part of Cubist Pharmaceuticals). He is presently an employee of Novartis Healthcare Private Limited, Hyderabad, India. Paul M. Tulkens is an emeritus and unpaid invited professor at the Université Catholique de Louvain, Brussels, Belgium, and has received research grants from and been a consultant to Trius Therapeutics (now part of Cubist Pharmaceuticals). Hiromi Hosako and Ann Radovsky conducted the long-term animal neurotoxicity study in the course of their employment, and their employer (WIL Research) received compensation from Cubist Pharmaceuticals. Julie Passarell and Jill Fiedler-Kelly conducted the pharmacokinetic analyses in the course of their employment, and their employer (Cognigen) received compensation from Cubist Pharmaceuticals.
REFERENCES
- 1.Barnhill AE, Brewer MT, Carlson SA. 2012. Adverse effects of antimicrobials via predictable or idiosyncratic inhibition of host mitochondrial components. Antimicrob Agents Chemother 56:4046–4051. doi: 10.1128/AAC.00678-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen BH, Saneto RP. 2012. Mitochondrial translational inhibitors in the pharmacopeia. Biochim Biophys Acta 1819:1067–1074. doi: 10.1016/j.bbagrm.2012.02.023. [DOI] [PubMed] [Google Scholar]
- 3.Scatena R. 2012. Mitochondria and drugs. Adv Exp Med Biol 942:329–346. doi: 10.1007/978-94-007-2869-1_15. [DOI] [PubMed] [Google Scholar]
- 4.Shinabarger D. 1999. Mechanism of action of the oxazolidinone antibacterial agents. Expert Opin Invest Drugs 8:1195–1202. doi: 10.1517/13543784.8.8.1195. [DOI] [PubMed] [Google Scholar]
- 5.Lin AH, Murray RW, Vidmar TJ, Marotti KR. 1997. The oxazolidinone eperezolid binds to the 50S ribosomal subunit and competes with binding of chloramphenicol and lincomycin. Antimicrob Agents Chemother 41:2127–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colca JR, McDonald WG, Waldon DJ, Thomasco LM, Gadwood RC, Lund ET, Cavey GS, Mathews WR, Adams LD, Cecil ET, Pearson JD, Bock JH, Mott JE, Shinabarger DL, Xiong L, Mankin AS. 2003. Cross-linking in the living cell locates the site of action of oxazolidinone antibiotics. J Biol Chem 278:21972–21979. doi: 10.1074/jbc.M302109200. [DOI] [PubMed] [Google Scholar]
- 7.Shaw KJ, Poppe S, Schaadt R, Brown-Driver V, Finn J, Pillar CM, Shinabarger D, Zurenko G. 2008. In vitro activity of TR-700, the antibacterial moiety of the prodrug TR-701, against linezolid-resistant strains. Antimicrob Agents Chemother 52:4442–4447. doi: 10.1128/AAC.00859-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leach KL, Swaney SM, Colca JR, McDonald WG, Blinn JR, Thomasco LM, Gadwood RC, Shinabarger D, Xiong L, Mankin AS. 2007. The site of action of oxazolidinone antibiotics in living bacteria and in human mitochondria. Mol Cell 26:393–402. doi: 10.1016/j.molcel.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 9.McKee EE, Ferguson M, Bentley AT, Marks TA. 2006. Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrob Agents Chemother 50:2042–2049. doi: 10.1128/AAC.01411-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagiec EE, Wu L, Swaney SM, Chosay JG, Ross DE, Brieland JK, Leach KL. 2005. Oxazolidinones inhibit cellular proliferation via inhibition of mitochondrial protein synthesis. Antimicrob Agents Chemother 49:3896–3902. doi: 10.1128/AAC.49.9.3896-3902.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palenzuela L, Hahn NM, Nelson RP Jr, Arno JN, Schobert C, Bethel R, Ostrowski LA, Sharma MR, Datta PP, Agrawal RK, Schwartz JE, Hirano M. 2005. Does linezolid cause lactic acidosis by inhibiting mitochondrial protein synthesis? Clin Infect Dis 40:e113–e116. doi: 10.1086/430441. [DOI] [PubMed] [Google Scholar]
- 12.Garrabou G, Soriano A, López S, Guallar JP, Giralt M, Villarroya F, Martínez JA, Casademont J, Cardellach F, Mensa J, Miró O. 2007. Reversible inhibition of mitochondrial protein synthesis during linezolid-related hyperlactatemia. Antimicrob Agents Chemother 51:962–967. doi: 10.1128/AAC.01190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerson SL, Kaplan SL, Bruss JB, Le V, Arellano FM, Hafkin B, Kuter DJ. 2002. Hematologic effects of linezolid: summary of clinical experience. Antimicrob Agents Chemother 46:2723–2726. doi: 10.1128/AAC.46.8.2723-2726.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Javaheri M, Khurana RN, O'Hearn TM, Lai MM, Sadun AA. 2007. Linezolid-induced optic neuropathy: a mitochondrial disorder? Br J Ophthalmol 91:111–115. doi: 10.1136/bjo.2006.102541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joshi L, Taylor SR, Large O, Yacoub S, Lightman S. 2009. A case of optic neuropathy after short-term linezolid use in a patient with acute lymphocytic leukemia. Clin Infect Dis 48:e73–e74. doi: 10.1086/597298. [DOI] [PubMed] [Google Scholar]
- 16.Metaxas EI, Falagas ME. 2009. Update on the safety of linezolid. Expert Opin Drug Saf 8:485–491. doi: 10.1517/14740330903049706. [DOI] [PubMed] [Google Scholar]
- 17.Narita M, Tsuji BT, Yu VL. 2007. Linezolid-associated peripheral and optic neuropathy, lactic acidosis, and serotonin syndrome. Pharmacotherapy 27:1189–1197. doi: 10.1592/phco.27.8.1189. [DOI] [PubMed] [Google Scholar]
- 18.Apodaca AA, Rakita RM. 2003. Linezolid-induced lactic acidosis. N Engl J Med 348:86–87. doi: 10.1056/NEJM200301023480123. [DOI] [PubMed] [Google Scholar]
- 19.De Vriese AS, Coster RV, Smet J, Seneca S, Lovering A, Van Haute LL, Vanopdenbosch LJ, Martin JJ, Groote CC, Vandecasteele S, Boelaert JR. 2006. Linezolid-induced inhibition of mitochondrial protein synthesis. Clin Infect Dis 42:1111–1117. doi: 10.1086/501356. [DOI] [PubMed] [Google Scholar]
- 20.Sotgiu G, Centis R, D'Ambrosio L, Alffenaar JW, Anger HA, Caminero JA, Castiglia P, De Lorenzo S, Ferrara G, Koh WJ, Schecter GF, Shim TS, Singla R, Skrahina A, Spanevello A, Udwadia ZF, Villar M, Zampogna E, Zellweger JP, Zumla A, Migliori GB. 2012. Efficacy, safety and tolerability of linezolid containing regimens in treating MDR-TB and XDR-TB: systematic review and meta-analysis. Eur Respir J 40:1430–1442. doi: 10.1183/09031936.00022912. [DOI] [PubMed] [Google Scholar]
- 21.Vinh DC, Rubinstein E. 2009. Linezolid: a review of safety and tolerability. J Infect 59(Suppl 1):S59–S74. doi: 10.1016/S0163-4453(09)60009-8. [DOI] [PubMed] [Google Scholar]
- 22.Chao CC, Sun HY, Chang YC, Hsieh ST. 2008. Painful neuropathy with skin denervation after prolonged use of linezolid. J Neurol Neurosurg Psychiatry 79:97–99. doi: 10.1136/jnnp.2007.127910. [DOI] [PubMed] [Google Scholar]
- 23.Beekmann SE, Gilbert DN, Polgreen PM, IDSA Emerging Infections Network . 2008. Toxicity of extended courses of linezolid: results of an Infectious Diseases Society of America Emerging Infections Network survey. Diagn Microbiol Infect Dis 62:407–410. doi: 10.1016/j.diagmicrobio.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 24.Azamfirei L, Copotoiu SM, Branzaniuc K, Szederjesi J, Copotoiu R, Berteanu C. 2007. Complete blindness after optic neuropathy induced by short-term linezolid treatment in a patient suffering from muscle dystrophy. Pharmacoepidemiol Drug Saf 16:402–404. doi: 10.1002/pds.1320. [DOI] [PubMed] [Google Scholar]
- 25.Pharmacia and Upjohn Co. 2014. Zyvox (linezolid) injection, tablets and oral suspension (prescribing information). Pharmacia and Upjohn Co, Division of Pfizer, Inc, New York, NY. [Google Scholar]
- 26.Yum JH, Choi SH, Yong D, Chong Y, Im WB, Rhee DK, Lee K. 2010. Comparative in vitro activities of torezolid (DA-7157) against clinical isolates of aerobic and anaerobic bacteria in South Korea. Antimicrob Agents Chemother 54:5381–5386. doi: 10.1128/AAC.00728-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown SD, Traczewski MM. 2010. Comparative in vitro antimicrobial activities of torezolid (TR-700), the active moiety of a new oxazolidinone, torezolid phosphate (TR-701), determination of tentative disk diffusion interpretive criteria, and quality control ranges. Antimicrob Agents Chemother 54:2063–2069. doi: 10.1128/AAC.01569-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prokocimer P, Bien P, Deanda C, Pillar CM, Bartizal K. 2012. In vitro activity and microbiological efficacy of tedizolid (TR-700) against gram-positive clinical isolates from a phase 2 study of oral tedizolid phosphate (TR-701) in patients with complicated skin and skin structure infections. Antimicrob Agents Chemother 56:4608–4613. doi: 10.1128/AAC.00458-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Locke JB, Morales G, Hilgers M, Kedar GC, Rahawi S, Picazo JJ, Shaw KJ, Stein JL. 2010. Elevated linezolid resistance in clinical cfr-positive Staphylococcus aureus isolates is associated with co-occurring mutations in ribosomal protein L3. Antimicrob Agents Chemother 54:5352–5355. doi: 10.1128/AAC.00714-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ong V, Flanagan S, Fang E, Dreskin H, Locke JB, Bartizal K, Prokocimer P. 2014. Absorption, distribution, metabolism, and excretion of the novel oxazolidinone prodrug tedizolid phosphate. Drug Metab Dispos 42:1275–1284. doi: 10.1124/dmd.113.056697. [DOI] [PubMed] [Google Scholar]
- 31.Prokocimer P, De Anda C, Fang E, Mehra P, Das A. 2013. Tedizolid phosphate vs linezolid for treatment of acute bacterial skin and skin structure infections: the ESTABLISH-1 randomized trial. JAMA 309:559–569. doi: 10.1001/jama.2013.241. [DOI] [PubMed] [Google Scholar]
- 32.Moran GJ, Fang E, Corey GR, Das AF, De Anda C, Prokocimer P. 2014. Tedizolid for 6 days versus linezolid for 10 days for acute bacterial skin and skin-structure infections (ESTABLISH-2): a randomised, double-blind, phase 3, non-inferiority trial. Lancet Infect Dis 14:696–705. doi: 10.1016/S1473-3099(14)70737-6. [DOI] [PubMed] [Google Scholar]
- 33.Flanagan S, McKee EE, Das D, Tulkens PM, Hosako H, Fiedler-Kelly J, Passarell J, Radovsky A, Prokocimer P. 2012. Abstr 52nd Int Conf Antimicrob Agents Chemother, 9 to 12 September 2012, San Francisco, CA, abstr A-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flanagan S, McKee EE, Das D, Tulkens PM, Hosako H, Fiedler-Kelly J, Passarell J, Radovsky A, Prokocimer P. 2013. Abstr 53rd Int Conf Antimicrob Agents Chemother, 10 to 13 September 2013, Denver, CO, abstr A-017b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flanagan S, McKee EE, Das D, Tulkens PM, Hosako H, Fiedler-Kelly J, Passarell J, Radovsky A, Prokocimer P. 2014. Abstr 53rd Annu Meet Soc Toxicol, 23 to 27 March 2014, Phoenix, AZ, abstr 1634. [Google Scholar]
- 36.McKee EE, Grier BL, Thompson GS, McCourt JD. 1990. Isolation and incubation conditions to study heart mitochondrial protein synthesis. Am J Physiol 258:E492–E502. [DOI] [PubMed] [Google Scholar]
- 37.Lemaire S, Van Bambeke F, Appelbaum PC, Tulkens PM. 2009. Cellular pharmacokinetics and intracellular activity of torezolid (TR-700): studies with human macrophage (THP-1) and endothelial (HUVEC) cell lines. J Antimicrob Chemother 64:1035–1043. doi: 10.1093/jac/dkp267. [DOI] [PubMed] [Google Scholar]
- 38.Housman ST, Pope JS, Russomanno J, Salerno E, Shore E, Kuti JL, Nicolau DP. 2012. Pulmonary disposition of tedizolid following administration of once-daily oral 200-milligram tedizolid phosphate in healthy adult volunteers. Antimicrob Agents Chemother 56:2627–2634. doi: 10.1128/AAC.05354-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lemaire S, Tulkens PM, Van Bambeke F. 2010. Cellular pharmacokinetics of the novel biaryloxazolidinone radezolid in phagocytic cells: studies with macrophages and polymorphonuclear neutrophils. Antimicrob Agents Chemother 54:2540–2548. doi: 10.1128/AAC.01723-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tulkens PM. 1991. Intracellular distribution and activity of antibiotics. Eur J Clin Microbiol Infect Dis 10:100–106. doi: 10.1007/BF01964420. [DOI] [PubMed] [Google Scholar]
- 41.Flanagan S, Minassian SL, Muñoz KA, Dreskin H, Fang E, Prokocimer P. 2013. Lack of pharmacokinetic drug interaction of tedizolid phosphate with pseudoephedrine in healthy subjects, poster 921. 23rd Eur Congr Clin Microbiol Infect Dis, Berlin, Germany. [Google Scholar]
- 42.Abe S, Chiba K, Cirincione B, Grasela TH, Ito K, Suwa T. 2009. Population pharmacokinetic analysis of linezolid in patients with infectious disease: application to lower body weight and elderly patients. J Clin Pharmacol 49:1071–1078. doi: 10.1177/0091270009337947. [DOI] [PubMed] [Google Scholar]
- 43.Flanagan S, Passarell J, Lu Q, Fiedler-Kelly J, Ludwig E, Prokocimer P. 2014. Tedizolid population pharmacokinetics, exposure response, and target attainment. Antimicrob Agents Chemother 58:6462–6470. doi: 10.1128/AAC.03423-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsumoto K, Shigemi A, Takeshita A, Watanabe E, Yokoyama Y, Ikawa K, Morikawa N, Takeda Y. 2014. Analysis of thrombocytopenic effects and population pharmacokinetics of linezolid: a dosage strategy according to the trough concentration target and renal function in adult patients. Int J Antimicrob Agents 44:242–247. doi: 10.1016/j.ijantimicag.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 45.Dong HY, Xie J, Chen LH, Wang TT, Zhao YR, Dong YL. 2014. Therapeutic drug monitoring and receiver operating characteristic curve prediction may reduce the development of linezolid-associated thrombocytopenia in critically ill patients. Eur J Clin Microbiol Infect Dis 33:1029–1035. doi: 10.1007/s10096-013-2041-3. [DOI] [PubMed] [Google Scholar]
- 46.Cattaneo D, Orlando G, Cozzi V, Cordier L, Baldelli S, Merli S, Fucile S, Gulisano C, Rizzardini G, Clementi E. 2013. Linezolid plasma concentrations and occurrence of drug-related haematological toxicity in patients with gram-positive infections. Int J Antimicrob Agents 41:586–589. doi: 10.1016/j.ijantimicag.2013.02.020. [DOI] [PubMed] [Google Scholar]
- 47.Pea F, Viale P, Cojutti P, Del Pin B, Zamparini E, Furlanut M. 2012. Therapeutic drug monitoring may improve safety outcomes of long-term treatment with linezolid in adult patients. J Antimicrob Chemother 67:2034–2042. doi: 10.1093/jac/dks153. [DOI] [PubMed] [Google Scholar]
- 48.Brown AN, Louie A, Adams J, Jambunathan K, Baluya D, Hafner R, Drusano GL. 2014. Relationship between linezolid (LZD) exposure profiles and toxicity in the hollow fiber infection model (HFIM) system, poster A-026b. Abstr 54th Intersci Conf Antimicrob Agents Chemother, 5 to 9 September 2014, Washington, DC. [Google Scholar]
- 49.Louie A, Liu W, Kulawy R, Drusano GL. 2011. In vivo pharmacodynamics of torezolid phosphate (TR-701), a new oxazolidinone antibiotic, against methicillin-susceptible and methicillin-resistant Staphylococcus aureus strains in a mouse thigh infection model. Antimicrob Agents Chemother 55:3453–3460. doi: 10.1128/AAC.01565-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andes D, van Ogtrop ML, Peng J, Craig WA. 2002. In vivo pharmacodynamics of a new oxazolidinone (linezolid). Antimicrob Agents Chemother 46:3484–3489. doi: 10.1128/AAC.46.11.3484-3489.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morata L, Cuesta M, Rojas JF, Rodriguez S, Brunet M, Casals G, Cobos N, Hernandez C, Martínez JA, Mensa J, Soriano A. 2013. Risk factors for a low linezolid trough plasma concentration in acute infections. Antimicrob Agents Chemother 57:1913–1917. doi: 10.1128/AAC.01694-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flanagan SD, Bien PA, Muñoz KA, Minassian SL, Prokocimer PG. 2014. Pharmacokinetics of tedizolid following oral administration: single and multiple dose, effect of food, and comparison of two solid forms of the prodrug. Pharmacotherapy 34:240–250. doi: 10.1002/phar.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Plock N, Buerger C, Joukhadar C, Kljucar S, Kloft C. 2007. Does linezolid inhibit its own metabolism? Population pharmacokinetics as a tool to explain the observed nonlinearity in both healthy volunteers and septic patients. Drug Metab Dispos 35:1816–1823. doi: 10.1124/dmd.106.013755. [DOI] [PubMed] [Google Scholar]
- 54.Meagher AK, Forrest A, Rayner CR, Birmingham MC, Schentag JJ. 2003. Population pharmacokinetics of linezolid in patients treated in a compassionate-use program. Antimicrob Agents Chemother 47:548–553. doi: 10.1128/AAC.47.2.548-553.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Antal E, Grasela T, Bergstrom T, Bruss J, Wong E. 2000. The role of population PK/PD analysis during the implementation of a bridging strategy for linezolid, poster P-16 36th Drug Info Assoc (DIA) Meet, Hong Kong, People's Republic of China, 16 to 19 November 2000. [Google Scholar]
- 56.FDA. 2000. FDA briefing package. Anti-Infective Drugs Advisory Committee 68th meeting. New drug applications 21-130, 21-131, 21-132 ZyvoxTM (linezolid). Appendix A, preclinical summary, part II. FDA, Silver Spring, MD. [Google Scholar]
- 57.Cubist Pharmaceuticals. 2014. NDA 205435 NDA 205436. Tedizolid phosphate for the treatment of acute bacterial skin and skin structure infections. Anti Infect Drugs Advis Comm Meet, 31 March 2014. [Google Scholar]
- 58.Bernstein WB, Trotta RF, Rector JT, Tjaden JA, Barile AJ. 2003. Mechanisms for linezolid-induced anemia and thrombocytopenia. Ann Pharmacother 37:517–520. doi: 10.1345/aph.1C361. [DOI] [PubMed] [Google Scholar]
- 59.Lodise TP, Fang E, Minassian SL, Prokocimer P. 22 September 2014. Platelet profile in patients with acute bacterial skin and skin structure infections receiving tedizolid or linezolid: findings from the phase 3 ESTABLISH clinical trials. Antimicrob Agents Chemother doi: 10.1128/AAC.03509-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pacheu-Grau D, Gómez-Durán A, Iglesias E, López-Gallardo E, Montoya J, Ruiz-Pesini E. 2013. Mitochondrial antibiograms in personalized medicine. Hum Mol Genet 22:1132–1139. doi: 10.1093/hmg/dds517. [DOI] [PubMed] [Google Scholar]
- 61.National Research Council. 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington, DC. [Google Scholar]