

Abstract
A recent report found that generic parenteral vancomycin products may not have in vivo efficacies equivalent to those of the innovator in a neutropenic murine thigh infection model despite having similar in vitro microbiological activities and murine serum pharmacokinetics. We compared the in vitro and in vivo activities of six of the parenteral vancomycin products available in the United States. The in vitro assessments for the potencies of the vancomycin products included MIC/minimal bactericidal concentration (MBC) determinations, quantifying the impact of human and murine serum on the MIC values, and time-kill studies. Also, the potencies of the vancomycin products were quantified with a biological assay, and the human and mouse serum protein binding rates for the vancomycin products were measured. The in vivo studies included dose-ranging experiments with the 6 vancomycin products for three isolates of Staphylococcus aureus in a neutropenic mouse thigh infection model. The pharmacokinetics of the vancomycin products were assessed in infected mice by population pharmacokinetic modeling. No differences were seen across the vancomycin products with regard to any in vitro evaluation. Inhibitory sigmoid maximal bacterial kill (Emax) modeling of the relationship between vancomycin dosage and the killing of the bacteria in mice in vivo yielded similar Emax and EC50 (drug exposure driving one-half Emax) values for bacterial killing. Further, there were no differences in the pharmacokinetic clearances of the 6 vancomycin products from infected mice. There were no important pharmacodynamic differences in the in vitro or in vivo activities among the six vancomycin products evaluated.
INTRODUCTION
In order for a generic parenteral vancomycin product to be approved by the U.S. Food and Drug Administration (FDA) for sale in the U.S. marketplace, the manufacturer usually is required to file an abbreviated new drug application (ANDA) with the FDA in which a reference listed drug (RLD), which may or may not be the innovator, is identified. The active ingredient, the condition of use, route of administration, dosage form, strength, and labeling are those of the RLD (1, 2). Furthermore, the manufacturer of the generic drug needs to show that its product meets established purity, potency, quality, and identity standards. For the parenteral vancomycin products, the purity and related substance impurities are assessed by quantifying the percentage of the active component of vancomycin (vancomycin factor B) and the individual impurities in the product by a high-pressure liquid chromatography (HPLC) method (3). The potency is measured using the bioassay described by the United States Pharmacopeia (USP<81>) (4, 5). The quality of the vancomycin product must meet the USP standard of having not less than 80% (wt/wt) vancomycin factor B and not more than 9% (wt/wt) of any impurity (6). The ANDA must also provide information regarding the physical and chemical characteristics, composition, method of manufacture, specifications, and stability of the drug substance and the drug product (7, 8). Pharmacokinetic and in vivo efficacy studies may be waived for generic parenteral products (including vancomycin) during the FDA approval process. A generic product that meets all of these criteria for pharmaceutical equivalence and bioequivalence is therapeutically equivalent to the RLD (9, 10).
From 2002 to 2009, Vesga et al. (11) compared the efficacies of the innovator vancomycin product that was manufactured by Eli Lilly & Company (which was withdrawn from the American market in 2004) and three generic vancomycin products for parenteral administration, which included two products that were manufactured in the United States. The vancomycin products showed similar activities in in vitro MIC, minimum bactericidal concentration (MBC), and time-kill studies. The potencies of the vancomycin products were similar when assessed by a microbiological assay. The protein binding rates of the vancomycin products to mouse serum were similar. In a neutropenic mouse thigh model of Staphylococcus aureus infection, the pharmacokinetics of the generic products generated similar or higher areas under the concentration-time curves (AUCs) than did the same dosage of the innovator product as measured by a microbiological assay. However, in the mouse infection model significant differences were noted between the innovator and some of the generic products as well as among the generic products in regard to the amount of bacterial killing that was observed. Further, some of the generic products exhibited an Eagle effect in which the reductions in the bacterial counts in the thigh muscles of mice that were seen with lower dosages of the generic vancomycin products were followed by a paradoxical increase in the bacterial densities at the infection site as higher dosages were used (12). The report by Vesga and colleagues raised concerns about the adequacy of the requirements established by nations for the approval of generic vancomycin products for injection, and potentially all parenteral generics in general (13).
In response to the Vesga et al. report, the FDA's Office of Testing and Research examined six of the parenteral vancomycin products that were available in the United States using ultrahigh-pressure liquid chromatography (UPLC) and high-pressure liquid chromatography (HPLC) (3). The FDA reported that each of the vancomycin products met the marketplace standards for purity established by the USP. Using an HPLC method, the FDA found that all 6 vancomycin products had vancomycin factor B contents of 90 to 95% (wt/wt). The total impurities were 5 to 10%, and the individual impurities accounted for 0.5 to 2% of the product content. By UPLC, the vancomycin factor B content ranged from 89 to 94%, the total impurities ranged from 6 to 11%, and the largest impurity was no higher than 2%. In a companion study, Hadwiger et al. (4) found that the potencies of these six vancomycin products, as measured by the USP bioassay, met the marketplace standards for potency set by the USP. To complete the evaluation of the six vancomycin parenteral products, we repeated many of the in vitro and in vivo tests described by the FDA and/or by Vesga and his colleagues (3, 4, 11). Our studies used the two methicillin-susceptible S. aureus (MSSA) strains that were included in the Vesga et al. report and a methicillin-resistant S. aureus (MRSA) strain purchased from the American Type Culture Collection (Manassas, VA). Specifically, we examined the in vitro microbiological potency of these products by MIC/MBC testing (with and without the addition of pooled mouse and human serum [MS and HS, respectively]), by 24-hour time-kill studies, and with the microbiological assay described in the USP monograph (5). The extents to which the six vancomycin products bound to pooled mouse and human sera were measured by a validated liquid chromatography-tandem mass spectroscopy (LC-MS/MS) method. A population pharmacokinetic evaluation for each vancomycin product was performed in which the vancomycin in murine plasma was measured by LC-MS/MS. Finally, the in vivo potencies of the six vancomycin products were compared using three strains of S. aureus in a neutropenic murine thigh infection model. Different lots of the six vancomycin products than those evaluated in the FDA reports were used in the current studies because the original lots had expired by the time that our studies initiated. Also, Eli Lilly & Company stopped manufacturing the innovator vancomycin product in 2004, and hence, it was not available for use in this project.
MATERIALS AND METHODS
S. aureus strains.
The MSSA strains GRP-0057 and ATCC 29213 were the kind gifts of Omar Vesga (Medellin, Colombia). The methicillin-resistant S. aureus (MRSA) strain ATCC 33591 was purchased from the American Type Culture Collection (Manassas, VA). All isolates were stored at −80°C in 10% glycerol. For the experiments, the bacterial isolates were cultured overnight on blood agar plates to confirm the purity and viability of the microbes. A few colonies were taken from the overnight agar cultures and grown in cation-adjusted Mueller-Hinton II broth (CA-MHB; BBL, Sparks, MD) to mid-log-phase growth. The bacterial suspensions were diluted to the desired concentrations and immediately used. The bacterial densities in the suspensions were confirmed by quantitative cultures.
Antibiotics.
The six vancomycin HCl products for injection were purchased directly from the U.S. marketplace through local pharmacies and supplied to investigators at the University of Florida College of Medicine by the FDA in a blinded manner, labeled A through F. The investigators remained blinded to the manufacturers of the drugs until the studies were completed and the draft of the manuscript was written. Products A through E were supplied as vials of lyophilized sterile powders. Listed in alphabetical order, the manufacturers were APP Pharmaceuticals, Inc.; Hospira, Inc.; Mylan, Inc. (formerly Bioniche Pharma USA, LLC); Pfizer, Inc. (formerly Akorn Strides LLC); and Sandoz, Inc. Each vial contained 1 g of vancomycin HCl for injection. These vials were stored at room temperature. Vancomycin product F (Baxter Healthcare Corp.) was a brand-name product that was provided as a premixed, ready-to-use solution for intravenous (i.v.) use and, hence, was functionally not blinded. It was provided as pharmaceutical i.v. bags, each containing 1 g of vancomycin HCl in 200 ml of frozen solution. Product F was delivered frozen and was kept at −20°C until use. Analysis of the purity of the vancomycin products was completed by the FDA prior to shipment using the HPLC method previously described (3). The results of the analysis are shown in Table 1. Also shown in Table 1 are the manufacturers of the vancomycin products, which became known to the investigators after the experiments and analysis of the results were completed. For this project, the dosages and concentrations of vancomycin used in the in vivo and in vitro studies, respectively, were based on the labeled weights of 1 g per container. We did not adjust for the purity measurements derived from the analyses conducted by the FDA when preparing the solutions for the in vitro and in vivo experiments. All of the vancomycin products contained no excipients other than sodium hydroxide and hydrochloric acid used by the manufacturers for pH adjustment in some preparations.
TABLE 1.
Percentages of vancomycin factor B (purity) and impurities in six of the vancomycin products licensed for parenteral use in the United States measured by the FDA by HPLCa
| VAN product | Manufacturer | Lot no. | % |
||
|---|---|---|---|---|---|
| VAN factor B | Total impurities | Largest impurity | |||
| A | Mylan, Inc.b | 12L37772A | 92.5 | 7.5 | 2.1 |
| B | Hospira, Inc. | 204628E04 | 92.9 | 7.1 | 1.1 |
| C | Pfizer, Inc.c | 7601646 | 91.6 | 8.4 | 1.2 |
| D | APP Pharmaceutics, LLC | 6105175 | 92.2 | 7.8 | 2.2 |
| E | Sandoz, Inc. | BU6345 | 92.5 | 7.5 | 1.5 |
| F | Baxter Healthcare Corp. | 2G3552/NC077974 | 94.6 | 5.4 | 2.4 |
The data for the vancomycin (VAN) products were provided blinded (labeled products A through F) by the FDA to the University of Florida researchers. The names of the manufacturers of the vancomycin products were provided to the University of Florida researchers only after the experiments and analysis of the data were completed and the first draft of the manuscript was written.
Mylan, Inc., purchased Bioniche Pharma USA, LLC.
Pfizer, Inc., purchased Akorn Strides, LLC.
Vancomycin products A through E were reconstituted with 20 ml of sterile water for injection (Hospira, Inc., Lake Forest, IL) to a concentration of 50 mg/ml. The drugs were then diluted with sterile normal saline for injection (Hospira, Inc.) to the desired concentrations for use in animals or CA-MHB for the in vitro experiments. The vials of reconstituted drugs and the i.v. bag of defrosted product F were stored at 4°C for up to 14 days before they were discarded. The (working) solutions administered to the animals or that were evaluated in the in vitro experiments were used immediately or were stored at 4°C and were used within 24 h.
Mouse and human sera.
Pooled female CD-1 mouse serum and pooled human serum were purchased from Gemini Bio-Products (Sacramento, CA). CD-1 mouse serum was selected because this strain of mouse was used in the investigations reported by Vesga and colleagues (11). The mouse serum was delivered frozen within 24 h after collection from mice. Frozen human serum was delivered to the investigators within 1 week after collection (after the screenings for blood-borne pathogens were completed). The sera were stored at −20°C upon receipt. The mouse and human sera were used for the susceptibility testing and protein binding studies within 48 h of receipt. Only for the susceptibility studies, a portion of the sera were briefly heated to inactivate complement.
Susceptibility studies.
The MICs for the six vancomycin products were determined for the three S. aureus isolates in CA-MHB using the broth microdilution method specified by the CLSI (14, 15). The MICs were read after the cultures had incubated at 35°C for 24 h. Subsequently, broth microdilution susceptibility studies were conducted simultaneously in CA-MHB with and without 50% and 80% complement-inactivated mouse and human sera in order to determine the effect of protein binding on the biological activities of the vancomycin products. The susceptibility studies were conducted at least 6 times. MBC values were determined by subculturing 0.01 ml of the bacterial suspensions onto blood agar plates. The MBC was defined as the lowest concentration of vancomycin that reduced the total colony count by at least 3 log CFU/ml in 24 h.
Protein binding studies.
The protein binding rates for the six vancomycin products to mouse and human serum were determined by an ultracentrifugation method using Centrifree Ultrafiltration devices (Millipore, Carrigtwohill, Cork, Ireland). The protein binding studies were conducted in duplicate on 2 days (for four trials) for 1-, 60-, and 300-mg/liter concentrations of the vancomycin products. The concentrations of the drugs in the serum and the supernatants were measured with a validated LC-MS/MS assay.
Vancomycin bioassay.
The relative activities of the vancomycin products were assessed with the bioassay method using Bacillus subtilis ATCC 6633 and medium 8 as described by the USP (5). The vancomycin B standard was obtained from USP. Initial studies were conducted with the bioassay to identify the concentrations of the USP vancomycin standard that produced a linear relationship between drug concentrations and the diameters of the zones of growth inhibition. The concentration of the vancomycin standard that was near the middle of the linear standard curve was 25.6 mg/liter. Next, the potencies of the vancomycin products were assessed. Briefly, 40 μl of different concentrations of the vancomycin standard was added to three of six cylinders that were placed onto the surface of each agar plate. One of the cylinders on each agar plate received a 25.6-mg/liter solution of the vancomycin standard. Forty microliters of a 25.6-mg/liter solution of one of the other vancomycin products was added to the remaining 3 wells. After 16 h of incubation at 35°C, the diameters of the zones of inhibition were measured with a caliper. The potencies of vancomycin products A through F were calculated using the standard curve that was produced with the data generated with the vancomycin standard and by directly comparing the zones of inhibition of the 25.6-mg/liter concentration of the vancomycin standard with the other vancomycin product inoculated onto that agar plate. The bioassay was conducted three times each day for 3 days. The calculated concentrations of products A through F were expressed as a percentage relative to the biological activity of the vancomycin standard. The results of the studies were averaged. Analysis of variance (ANOVA) was applied to the data to determine if there was a statistical difference in the activities of the vancomycin products.
In vitro time-kill studies.
S. aureus GRP-0057 in CA-MHB was incubated at 35°C in a water shaker bath to achieve mid-log-phase growth. The bacterial suspension was diluted with fresh medium to 106 CFU/ml. The bacterial suspension was dispensed into flasks and was incubated at 35°C in a water shaker bath. A vancomycin product was added to the flasks to achieve vancomycin concentrations that were 0, 1, 2, 4, 10, and 20 times the MIC for the microbe. At 0, 2, 4, 6, 8, and 24 h of incubation, a sample was collected from each flask. The samples were washed twice with sterile saline to prevent drug carryover before they were quantitatively cultured on blood agar plates. After the cultures had incubated for 24 h at 35°C, the colonies were enumerated.
In a separate series of studies, single multiples of a vancomycin MIC (i.e., 1, 2, 4, 10, or 20 times MIC) for the six vancomycin products were examined simultaneously in time-kill studies in order to eliminate interday variability. These time-kill studies were executed using the protocol described in the paragraph above.
Mice.
Female CD-1 mice (22 to 25 g) were purchased from Charles River Laboratories (Frederick, MD). CD-1 mice were chosen since this strain was used in the studies reported by Vesga and colleagues (11). Neutropenia was induced by treating each mouse with 150 mg/kg of body weight of cyclophosphamide (Sigma-Aldrich Inc., St. Louis, MO) via the intraperitoneal (i.p.) route 4 days prior to bacterial inoculation and 100 mg/kg of cyclophosphamide given i.p. 1 day prior to bacterial challenge. This regimen resulted in neutropenia in mice (neutrophil count of <100 cells/ml) for at least 5 days from the time that the second dose of cyclophosphamide was administered (data not shown). All experimental methods using mice were approved by the University of Florida's Institutional Animal Care and Use Committee.
Dose-range studies and inhibitory sigmoid Emax (maximal effect of the antibiotic in reducing bacterial density) effect modeling.
The six vancomycin products were evaluated simultaneously in the comparative in vivo dose-range experiments for each of the three S. aureus strains. Transient neutropenia in mice was induced with cyclophosphamide using the protocol described above. One day after the second dose of cyclophosphamide was administered, the mice were injected in each posterior thigh muscle with 3 × 104 CFU of an S. aureus isolate in 0.1 ml of CA-MHB. The bacterial inoculum was confirmed by quantitative cultures. Two mice per time point were sacrificed immediately after bacterial inoculation and 2 h later. Both posterior thigh muscles were collected from each mouse and were separately homogenized and quantitatively cultured in order to determine the bacterial density in thigh muscles at baseline and to confirm that the bacteria were in log-phase growth prior to the start of therapy. The remaining infected mice were divided into six treatment groups for each of the simultaneously tested vancomycin products. An additional group of mice served as the untreated control arm. For strain GRP-0057, doses ranged from 18.75 mg/kg/day through 1,200 mg/kg/day to replicate the range of vancomycin dosages examined by Vesga et al. in their murine studies with S. aureus GRP-0057 (11). The doses studied for ATCC 33591 ranged from 4.69 to 300 mg/kg/day, and those for ATCC 29213 ranged from 9.88 to 300 mg/kg/day. The vancomycin products were given subcutaneously (s.c.) as four equally divided doses every 6 h. The different dosages of the vancomycin products were selected for examination for the three S. aureus strains based on the results of pilot dose-range studies that were conducted using 0 to 1,200 mg/kg/day of vancomycin product B (Fig. 1). This vancomycin product had the highest purity of the blinded generic drugs (91.6% to 92.9%). Product F was not used in the pilot study because larger volumes of the premixed solution needed to be used to administer the higher dosages. The dosages selected for further evaluation were those that were predicted to lie on the steep portion of the dose-response curve based on the pilot experiments. At hour 26 (24 h after initiation of treatment), two mice per group were sacrificed. Both posterior thigh muscles were collected from each mouse and were separately homogenized and quantitatively cultured. After the cultures had incubated for 24 h, the colonies were enumerated. The comparative vancomycin dose-range studies for the six vancomycin products were performed twice for each bacterial strain.
FIG 1.
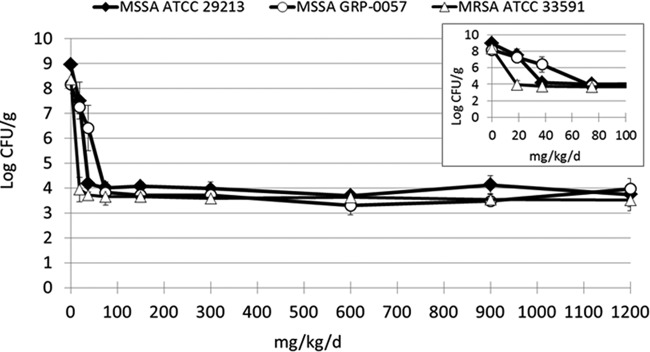
Pilot dose-range studies for the three S. aureus strains using vancomycin product B in the neutropenic mouse thigh infection model. The two MSSA (GRP-0057 and ATCC 29213) strains had vancomycin MICs of 1 mg/liter. The MRSA strain ATCC 33591 had a MIC of 2 mg/liter. The error bars represent 1 standard deviation. The inset shows the effect of the vancomycin product on the three S. aureus strains at the lower dosages.
An inhibitory sigmoid Emax effect model was fitted to the data: E = Econ − [(Emax × doseH)/(doseH + EC50H)], where E is microbial effect, Econ is the microbial density in the control arm at 24 h after therapy initiation (26 hours after infection), Emax is the maximal effect of the antibiotic in reducing the bacterial density in logs, dose is the drug exposure for 24 h, EC50 is the exposure for which there is 50% of maximal kill, and H is the Hill constant. Comparisons across groups for a bacterial isolate were made by examining the point estimates of Emax and EC50 and their 95% confidence intervals, as these parameter values relate drug exposure to microbial kill.
The inhibitory sigmoid Emax effect model employed inverse of the observation variance weighting as the best approximation to the homoscedastic assumption. The ADAPT V package of programs (16) was employed to obtain point estimates of parameter values and their attendant 95% confidence intervals.
Single-dose pharmacokinetic studies for the vancomycin products.
Neutropenic mice were challenged in each posterior thigh muscle with 3 × 104 CFU of MSSA strain GRP-0057. Two hours later, mice were given a single dose of a vancomycin product at 10, 20, or 40 mg/kg via the s.c. route. At 20, 40, 60, 80, 100, 120, 140, and 160 min after drug administration, 3 mice per group were sacrificed. Blood was collected from individual mice by cardiac puncture and was placed on ice in vials containing EDTA. The plasma was frozen at −80°C. The concentration of drug in plasma was measured by LC-MS/MS. All pharmacokinetic data were population modeled with the program Non-Parametric Adaptive Grid (NPAG) (17) using a two-compartment model with first-order input. Cohorts of animals at a specific dose and at a specific time point had their concentration data averaged. The variances were employed in an inverse-observed-variance weighting scheme to give the best approximation to the homoscedastic assumption. As the dynamic driver for vancomycin bacterial kill for S. aureus is AUC/MIC ratio (18, 19), the point estimate of clearance and its attendant 95% confidence interval were employed as the metric for comparing the six different vancomycin products.
LC-MS/MS assay.
Frozen CD-1 mouse and human sera were obtained from Gemini Bio (West Sacramento, CA), thawed, and aliquoted into vials and stored at −20°C until further use. HPLC-MS-grade water, acetonitrile, formic acid, and methanol were obtained from Fisher Scientific (Pittsburgh, PA). Vancomycin HCl hydrate was obtained from Sigma-Aldrich (St. Louis, MO). Linezolid as an internal standard was purchased from Curascript, Inc. (Orlando, FL), in an i.v. bag of 2-mg/ml solution.
The samples (vancomycin in plasma) were treated as follows: 25 ml of internal standard solution was added to a 50-ml sample aliquot prior to adding acetonitrile (200 ml) for protein precipitation. The resulting solutions were centrifuged, and 200 ml of the supernatant was dried and reconstituted with 100 ml HPLC water for analysis by LC-MS/MS.
The HPLC-MS/MS assay was done using a Thermo TSQ Vantage mass spectrometer coupled with a Shimadzu Prominence UFLC system. Chromatographic separation was performed using a Thermo Scientific Hypersil Gold C18 column, 50 by 2.1 mm, 3-μm pore diameter, at a flow rate of 0.450 ml/min using a linear gradient of 0.1% (vol/vol) formic acid in water and 0.1% (vol/vol) formic acid in acetonitrile. Vancomycin concentrations were calculated by monitoring the vancomycin MS/MS transition from m/z 725 → m/z 144 and linezolid from m/z 338.0 → m/z 235.1. The total analysis run time was 6.0 min. The concentration range was split into lower and upper curves, from 0.28 to 90 mg/liter and 90 to 450 mg/liter, respectively. Quality controls showed that overall accuracy and precision ranged from 81 to 125% and 0.20 to 6.53%, respectively. The lower limit of quantitation was 0.28 mg/liter. The lower limit of detectability was 0.028 mg/liter.
RESULTS
MIC and MBC values and impact of serum protein binding.
The susceptibility studies were performed at least 6 times per strain for each vancomycin product. The impacts of 50 and 80% human serum (HS) and murine serum (MS) on the MIC values were also determined. The average MIC and MBC values of six vancomycin products for the three S. aureus isolates are displayed in Table 2. The data are shown as vancomycin A through F to reflect the blinding of the products at the time that the experiments were conducted. For the two MSSA strains GRP-0057 and ATCC 29213, the MICs for the six vancomycin products were 1 mg/liter when the susceptibility studies were conducted in CA-MHB. The MBC values in CA-MHB were 2-fold higher than the MIC values. The addition of 50% and 80% human serum and 50% mouse serum to the CA-MHB had no effect on the MIC and MBC values for these S. aureus strains. However, the addition of 80% mouse serum to the CA-MHB increased the MIC and MBC values for the six vancomycin products by 2-fold.
TABLE 2.
MICs and MBCs for 6 vancomycin products against three strains of S. aureus by a broth microdilution method using cation-adjusted Mueller-Hinton broth alone and with 50% and 80% complement-inactivated pooled human serum and mouse seruma
| Vancomycin product | Condition | Geometric mean value, mg/liter (range) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
S. aureus ATCC 29213 |
S. aureus GRP-0057 |
S. aureus ATCC 33591 |
||||||||
| MIC | MBC | MBC/MIC | MIC | MBC | MBC/MIC | MIC | MBC | MBC/MIC | ||
| A | MHB alone | 1.17 (1–2) | 1.5 (1–2) | 1.28 (1–2) | 1.08 (1–2) | 1 (1–1) | 0.93 (0.5–1) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) |
| 50% HS | 1 (1–1) | 2.5 (1–4) | 2.5 (1–4) | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 80% HS | 1 (1–1) | 2 (2–2) | 2 (2–2) | 1 (1–1) | 1 (1–1) | 1 (1–1) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 50% MS | 1 (1–1) | 2 (2–2) | 2 (2–2) | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) | |
| 80% MS | 2 (2–2) | 4 (4–4) | 2 (2–2) | 2 (2–2) | 2 (2–2) | 1 (1–1) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) | |
| B | MHB alone | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) |
| 50% HS | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 1 (1–1) | 1 (1–1) | 1 (1–1) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 80% HS | 1 (1–1) | 2 (2–2) | 2 (2–2) | 1 (1–1) | 1 (1–1) | 1 (1–1) | 1.5 (1–2) | 3 (2–4) | 2 (2–4) | |
| 50% MS | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 1.17 (1–2) | 2 (2–2) | 1.71 (1–2) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 80% MS | 2 (2–2) | 2 (2–2) | 1 (1–1) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) | |
| C | MHB alone | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 1.08 (1–2) | 1.5 (1–2) | 1.39 (1–2) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) |
| 50% HS | 1 (1–1) | 1 (1–1) | 1 (1–1) | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 80% HS | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 1 (1–1) | 1 (1–1) | 1 (1–1) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 50% MS | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 1.17 (1–2) | 2.5 (1–4) | 2.14 (1–4) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 80% MS | 2 (2–2) | 3 (2–4) | 1.5 (1–2) | 1.5 (1–2) | 2 (2–2) | 1.33 (1–2) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) | |
| D | MHB alone | 1 (1–1) | 2 (2–2) | 2 (2–2) | 1.25 (1–2) | 1 (1–1) | 0.8 (0.5–1) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) |
| 50% HS | 1 (1–1) | 1 (1–1) | 1 (1–1) | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 80% HS | 1 (1–1) | 2 (2–2) | 2 (2–2) | 1 (1–1) | 1 (1–1) | 1 (1–1) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 50% MS | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 1.17 (1–2) | 1 (1–1) | 0.85 (0.5–1) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 80% MS | 2 (2–2) | 4 (4–4) | 2 (2–2) | 2 (2–2) | 4 (4–4) | 2 (2–2) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) | |
| E | MHB alone | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 1 (1–1) | 1 (1–1) | 1 (1–1) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) |
| 50% HS | 1 (1–1) | 1 (1–1) | 1 (1–1) | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 80% HS | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 50% MS | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 1.33 (1–2) | 3 (2–4) | 2.26 (2–4) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 80% MS | 2 (2–2) | 3 (2–4) | 1.5 (1–2) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) | |
| F | MHB alone | 1 (1–1) | 1.5 (1–2) | 1.5 (1–2) | 1 (1–1) | 1 (1–1) | 1 (1–1) | 2 (2–2) | 2 (2–2) | 2 (2–2) |
| 50% HS | 1 (1–1) | 1 (1–1) | 1 (1–1) | 1 (1–1) | 1 (1–1) | 1 (1–1) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 80% HS | 1 (1–1) | 2 (2–2) | 2 (2–2) | 1 (1–1) | 1 (1–1) | 1 (1–1) | 2 (2–2) | 2 (2–2) | 1 (1–1) | |
| 50% MS | 1 (1–1) | 2 (2–2) | 2 (2–2) | 1.33 (1–2) | 3 (2–4) | 2.26 (2–4) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) | |
| 80% MS | 2 (2–2) | 4 (4–4) | 2 (2–2) | 2 (2–2) | 2 (2–2) | 1 (1–1) | 2 (2–2) | 3 (2–4) | 1.5 (1–2) | |
Abbreviations: MHB, Mueller-Hinton broth; HS, human serum; MS, murine serum; VAN, vancomycin.
For the MRSA strain ATCC 33591, the MIC in CA-MHB was 2 mg/liter for all of the vancomycin products. The MBCs were the same as or 2-fold higher than the MICs. For this bacterial strain, the addition of human and mouse serum to the CA-MHB did not affect the MIC or MBC values.
Microbiologic assay.
The 25.6-mg/liter concentration for vancomycin products A through F produced zones of inhibition that were 100.2 ± 4.5, 101.6 ± 3.7, 102.1 ± 4.2, 99.7 ± 3.7, 99.3 ± 4.7, and 100.6 ± 4.6%, respectively, of the zone of inhibition for this concentration of the USP vancomycin standard. Based on the microbiological data, there was no statistical difference between the potencies of vancomycin products A through F (P = 0.64 by one-sided ANOVA).
Protein binding rates.
The protein binding rates for vancomycin products A through F in pooled mouse serum ranged between 41.8 and 47.3% (Table 3). In pooled human serum, the protein binding rates for the vancomycin products ranged between 33.8 and 36.8%.
TABLE 3.
Protein binding rates for the six vancomycin products in pooled mouse and human seraa
| Serum | Vancomycin concn (mg/liter) | Binding rate (% ± 1 SD) for vancomycin product: |
|||||
|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | ||
| Mouse | 1 | 43.1 ± 9.9 | 46.8 ± 4.3 | 43.0 ± 8.3 | 44.7 ± 4.4 | 40.9 ± 3.7 | 40.6 ± 6.5 |
| 60 | 43.6 ± 6.9 | 44.0 ± 5.5 | 47.9 ± 2.8 | 35.7 ± 4.2 | 46.9 ± 3.2 | 43.1 ± 3.4 | |
| 300 | 47.5 ± 11.0 | 40.3 ± 13.4 | 50.9 ± 5.7 | 45.1 ± 4.5 | 48.4 ± 4.0 | 45.9 ± 5.6 | |
| Mean binding rate | 44.7 ± 9.3 | 43.7 ± 7.7 | 47.3 ± 5.6 | 41.8 ± 4.4 | 45.4 ± 3.7 | 43.2 ± 5.2 | |
| Human | 1 | 40.1 ± 6.4 | 35.6 ± 9.2 | 42.4 ± 3.1 | 37.4 ± 8.7 | 33.1 ± 6.1 | 35.1 ± 5.7 |
| 60 | 41.3 ± 7.1 | 37.0 ± 1.6 | 35.6 ± 4.2 | 33.3 ± 4.4 | 35.7 ± 2.8 | 32.2 ± 5.5 | |
| 300 | 27.9 ± 6.2 | 33.3 ± 8.4 | 32.4 ± 4.8 | 30.7 ± 8.1 | 34.6 ± 2.6 | 42.9 ± 5.5 | |
| Mean binding rate | 36.4 ± 6.5 | 35.3 ± 6.4 | 36.8 ± 4.1 | 33.8 ± 7.1 | 34.5 ± 3.9 | 36.7 ± 5.6 | |
The protein binding experiments were conducted in duplicate on two separate days (four samples for each vancomycin product and dose).
Time-kill studies.
The MIC was 1 mg/liter for all of the vancomycin products for the MSSA isolate GRP-0057. The exposure-response relationship for vancomycin product A in the time-kill study is shown in Fig. 2A for this S. aureus isolate. All of the other vancomycin products performed similarly, with regrowth observed with the 1× MIC and reduction in the bacterial density with the 2× to 20× MICs (data not shown). The 4× to 20× MIC time-kill curves overlapped each other. In a separate series of time-kill experiments, the 1× MICs for the six vancomycin products were examined simultaneously, followed by time-kill studies evaluating the 2×, 4×, 10×, and then the 20× concentrations. Figure 2B through F shows the antimicrobial effects of individual multiples of MICs for time-kill studies using MSSA strain GRP-0057. For each multiple of MIC, the six vancomycin products performed similarly.
FIG 2.

Time-kill studies for MSSA strain GRP-0057 for the six vancomycin products with exposures ranging from 1× MIC through 20× MIC. (A) All the multiples of MICs conducted in a single study for vancomycin product A. The results were similar for the other vancomycin products (data not shown). (B to F) Studies in which the microbiological activities of the six vancomycin products were examined simultaneously for a single multiple of the MIC. The MIC for the six vancomycin products for this S. aureus strain was 1 mg/liter.
Population pharmacokinetic analyses by vancomycin product.
The concentration-time profiles for 10, 20, and 40 mg/kg given s.c. for the six vancomycin products are shown in Fig. 3. There was a delay in the initial distribution of the highest dosage of vancomycin product A (Fig. 3C) and the Cmax (maximum concentration of drug in serum) values of this dose of vancomycin product A and the 20-mg/kg dose of vancomycin product C were lower than the values for the other vancomycin products. Otherwise, the pharmacokinetic profiles for the vancomycin products were similar. Population pharmacokinetic analysis of the data derived from the single-dose pharmacokinetic studies showed that the 95% confidence intervals for the clearances of vancomycin in the six products overlapped, indicating that the clearances of vancomycin for the products were not statistically different (Fig. 4).
FIG 3.

Concentration-time curves for single doses of the six vancomycin products delivered to mice with S. aureus thigh infections at 10 mg/kg (A), 20 mg/kg (B), and 40 mg/kg (C) via the s.c. route. The error bars represent 1 standard deviation.
FIG 4.
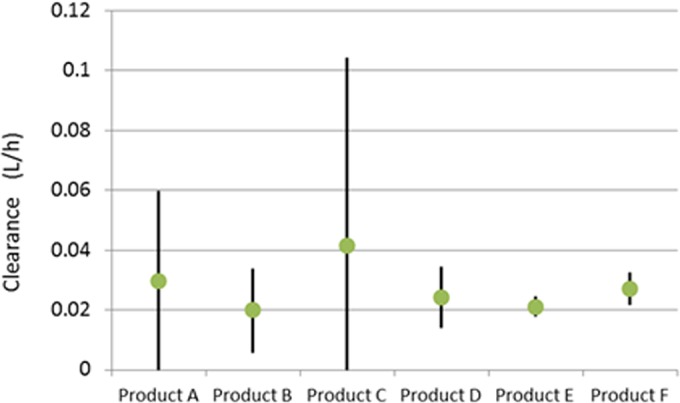
Mean plasma clearances (liters/h) and 95% confidence intervals for six vancomycin products as determined by a population pharmacokinetic model in mice with thigh infections due to MSSA strain GRP-0057.
We chose to present the pharmacokinetic analysis as total clearances because the pharmacodynamic driver for vancomycin for bacterial kill is the AUC/MIC ratio (18, 19). As the MIC is a fixed value, differences would need to be explained by AUC, which is calculated using the equation AUC = dose/clearance (CL). Since the vancomycin in the six parenteral products does not differ in total clearance, any mg/kg dose of the vancomycin products is expected to generate similar AUC values.
Comparative dose-range effects of the six vancomycin products in a neutropenic mouse thigh infection model.
The comparative dose-range studies for MSSA strain GRP-0057, the S. aureus strain used by Vesga et al. (11) in their in vitro and in vivo evaluations of several vancomycin products, are shown in Fig. 5. Overall, in our comparative experiments, the six vancomycin products produced similar amounts and rates of reduction of the bacterial counts per dosage (Fig. 5 and accompanying inset). Also, all of the vancomycin products had similar maximal killing effects, reducing the bacterial density for this S. aureus strain by approximately 5 log CFU/ml. Vancomycin product A had an outlier for the mean quantitative culture value for the 150-mg/kg/day dose. However, the value was statistically similar to the quantitative culture results for the same dose of the other vancomycin products (P = 0.61 by ANOVA). An Eagle effect was not observed for the higher dosages, which included the same total daily dosages examined by Vesga and colleagues (11).
FIG 5.
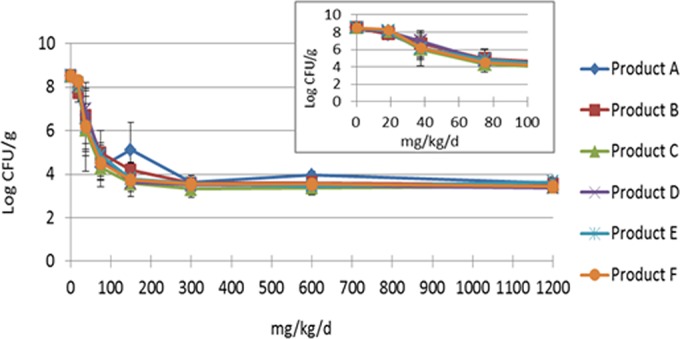
Dose-range study for six vancomycin products in a neutropenic murine thigh infection model with the total daily dose given as four equally divided doses administered every 6 h. The mice were infected with S. aureus strain GRP-0057. The results are the averages of two experiments. The error bars represent 1 standard deviation. The outlier data point for 150 mg/kg/day for vancomycin product A was not statistically different from the quantitative culture results for the same dose of the other vancomycin products (P > 0.05). The inset shows the overlapping of the antimicrobial activities for the steep portion of the dose-response curves, which underscores the similarity of antimicrobial effects of the vancomycin products. The inhibitory sigmoid Emax analysis of all of the data derived from the dose-response studies for this strain and two other S. aureus strains are shown in Table 4.
Inhibitory sigmoid Emax modeling was applied to the comparative dose-range data for data shown in Fig. 5 for MSSA strain GRP-0057 and for the two other S. aureus strains. All of the data generated from the dose-range studies were used in the modeling. Table 4 shows the estimated Emax values and EC50s and the respective 95% confidence intervals derived from the inhibitory sigmoid Emax modeling of the dose-response effects for the six vancomycin products when they were examined simultaneously in mice that were infected with the three S. aureus strains. In addition, the coefficients of determination (r2) for the overall fit of the inhibitory sigmoid Emax model to the data for each of the 18 dose-range studies (three bacterial isolates and 6 vancomycin generic products) are shown.
TABLE 4.
Emax and EC50 values and respective 95% confidence intervals derived by fitting the inhibitory sigmoid Emax equation to all of the dose-response data for the three Staphylococcus aureus isolates for six of the parenteral vancomycin products available in the United States
| Bacterial isolate | Vancomycin product | Emaxa (95% CIc) | EC50b (95% CI) | r2d |
|---|---|---|---|---|
| GRP-0057 | A | 4.425 (2.822 to 6.027) | 37.31 (17.89 to 56.72) | 0.947 |
| B | 4.977 (4.232 to 5.721) | 50.70 (36.96 to 64.44) | 0.999 | |
| C | 5.217 (4.917 to 5.517) | 39.75 (32.97 to 46.52) | 0.995 | |
| D | 4.892 (4.611 to 5.172) | 45.15 (38.96 to 51.33) | 0.999 | |
| E | 4.920 (4.530 to 5.311) | 38.29 (36.63 to 39.96) | 0.975 | |
| F | 4.985 (4.698 to 5.271) | 43.43 (25.50 to 61.35) | 0.989 | |
| ATCC 33591 | A | 4.981 (4.754 to 5.209) | 22.33 (14.17 to 30.50) | 0.985 |
| B | 5.349 (3.720 to 6.979) | 24.88 (−5.339 to 55.10) | 0.966 | |
| C | 5.372 (3.670 to 7.073) | 18.97 (−3.333 to 41.26) | 0.847 | |
| D | 5.093 (4.001 to 6.185) | 17.50 (10.80 to 24.20) | 0.989 | |
| E | 5.440 (5.145 to 5.736) | 22.59 (15.85 to 29.34) | 0.985 | |
| F | 4.934 (4.460 to 5.409) | 18.34 (14.74 to 21.93) | 0.950 | |
| ATCC 29213 | A | 5.675 (5.318 to 6.032) | 17.10 (13.43 to 20.77) | 0.922 |
| B | 5.728 (5.250 to 6.206) | 14.87 (8.064 to 21.67) | 0.961 | |
| C | 5.369 (4.852 to 5.886) | 18.21 (15.20 to 21.22) | 0.621 | |
| D | 5.893 (5.462 to 6.324) | 27.35 (10.59 to 44.10) | 0.996 | |
| E | 6.078 (4.650 to 7.507) | 29.37 (4.692 to 54.05) | 0.861 | |
| F | 5.898 (5.284 to 6.511) | 29.47 (12.92 to 46.02) | 0.967 |
Emax is the maximal cell kill generated by the product.
EC50 is the vancomycin product dose that produces half the maximal cell kill.
CI, confidence interval.
r2 value for inhibitory sigmoid Emax relationship.
For the three S. aureus isolates examined, the 95% confidence intervals were overlapping for both Emax and EC50 estimates for the six vancomycin products tested for the three S. aureus strains (Fig. 6). This indicates that the six vancomycin products produced Emax bacterial killing effects that were not statistically different for each bacterium, and the dose producing half-maximal bacterial kill was also not different. None of the data points were excluded from the analysis.
FIG 6.

Emax and EC50 values and the respective 95% confidence intervals (CIs) derived by fitting the inhibitory sigmoid Emax equation to the dose-response data for the three S. aureus isolates for six of the parenteral vancomycin products available in the United States.
DISCUSSION
In this project, we did not find significant differences in the in vitro and in vivo microbiological activities among six of the vancomycin products for parenteral use, which were marketed in the United States. Vesga et al. (11) performed an extensive evaluation of the innovator product (vancomycin manufactured by Eli Lilly & Company) and three generic vancomycin products, two of which were manufactured in the United States. They found no in vitro differences in the antimicrobial activities among the four products based on MIC and MBC measures, time-kill studies, and a microbiological assay. Furthermore, they reported that the serum protein binding rates and the vancomycin serum pharmacokinetics of the different products in infected mice were similar. However, these investigators found major differences between the innovator and generic products in the amount of bacterial killing generated in a neutropenic murine thigh infection model. Indeed, there were differences noted among generics as well as between generics and the innovator product. Surprisingly, in some instances the amount of bacterial killing by some of the vancomycin products reached a maximum and then declined with increasing drug exposure. Such behavior is worrisome, as many patients have life-threatening S. aureus infections. Given that guidelines for the use of vancomycin now recommend exposures that would push into the range where less activity was seen in the Vesga et al. paper (11), such drug behavior would increase the risk of patient mortality.
The findings reported by Vesga and colleagues prompted the FDA to evaluate the quality and potency of the six vancomycin products for injection that were available for clinical use in the United States at the time that those studies were initiated. The FDA measured the purity (percentage of the active component of vancomycin [vancomycin factor B]) and the amount of impurities in these vancomycin preparations using UPLC and HPLC methods (3). No differences were found in the amounts of active drug substance, vancomycin factor B, in the products. The purity of the vancomycin products ranged from 90 to 95% by HPLC and 89 to 94% by UPLC, levels which met the marketplace standards for vancomycin factor B of not less than 80%. Also, none of the individual impurities in the vancomycin products exceeded the USP maximum standard of 9%. Importantly, the amount of crystalline degradation product 1 (CDP-1), a degradation by-product of vancomycin factor B that might decrease the activity of vancomycin factor B (11), was no more than 2% in any of the vancomycin products. Spiking the six vancomycin products with 9% CDP-1 decreased the potency of the six vancomycin products from 97 to 112% to 98 to 105% (4). These values are well within the USP potency criteria for vancomycin products of 90 to 115% (6). Thus, the amount of CDP-1 measured in the vancomycin products was unlikely to have an impact on the activity of these products.
To complete this evaluation, our laboratory investigated both the in vitro and in vivo activities of the six vancomycin products that were assessed by the FDA. Concordant with the findings of Vesga et al. (11), we did not detect differences in the in vitro microbiological activities of any of the vancomycin products as measured by MIC and MBC. The addition of 50% or 80% human serum or murine serum to the test system had similar effects on the MICs and MBCs of the vancomycin products. In the time-kill studies, the six vancomycin products performed similarly. There was regrowth at 1× MIC. Higher multiples of MICs produced sustained bacterial killing over the 24-hour experiments. There was little difference in the amount of bacterial killing between the 4×, 10×, and 20× MIC arms. An Eagle effect was not observed with any of the concentrations examined. Also, the pharmacokinetic studies that were conducted in infected mice demonstrated that there was also no difference in the clearances (and, hence, AUCs) between the vancomycin products.
In contrast to Vesga et al., we did not find a statistical difference for Emax or EC50s across any of the vancomycin products evaluated in the mouse thigh infection model. Examination of the 95% confidence intervals about the point estimates demonstrates overlap for all products for an individual bacterial isolate for both parameter estimates. These estimates are the measures of bacterial kill in vivo. In the evaluations for strain GRP-0057, all the coefficients of determinations (r2) were greater than 0.9, indicating an acceptable fit of the model to the data. For isolate ATCC 33591, five of six had r2 values in excess of 0.9. Generic product C had an r2 of 0.847. This value was caused by one outlier point (observed value of 6.749 logs; predicted value of 4.633 logs). We did not remove the point, and it did not affect the estimation of Emax. As we were seeking to exclude a process like that seen by Vesga et al. (11), where higher exposures generated less cell kill, we felt that it was unwise to exclude data points that the model “did not fit.” The highest exposure produced an effect that was accurately predicted by the model and showed no tendency toward the loss of activity that was reported by Vesga et al. for some of the generic vancomycin products that they evaluated. For isolate ATCC 29213, four of six evaluations had r2 values in excess of 0.9. Product C had a value of 0.621, while product E had a value of 0.861. For product C, there were two outliers in the middle of the exposure range, with observed values of 6.616 logs and 6.045 logs being predicted as 3.418 and 3.057 logs, respectively. The last two exposures produced effects well predicted by the model. Finally, for product E, we saw a single outlier, where the observed value was 6.216 logs and the predicted was 3.818 logs. This was in the middle of the exposure range, and subsequent higher exposures had effects well predicted. Indeed, examining the confidence intervals for Emax surrounding the four outlier outcomes demonstrates that they were all well estimated.
The innovator vancomycin product is no longer produced by Eli Lilly & Company and, therefore, was not available for use in the current project. However, in the studies in which we infected mice with the same S. aureus GRP-0057 strain that was used by Vesga et al., the amount of bacterial killing by the vancomycin products used in our in vivo studies approximated those for the innovator in the murine experiments reported by Vesga et al. Furthermore, for mice infected with the S. aureus strain GRP-0057, we did not observe the paradoxical regrowth with the same higher dosages of vancomycin that were evaluated by Vesga et al. Hence, we are confident that the loss of activity seen in the Vesga et al. paper (11) did not occur in the current evaluation.
Recently, Tattevin et al. (13) compared the activities of six generic vancomycin products for injection in a rabbit model of staphylococcal endocarditis. Two of the vancomycin products were purchased in the United States, two in France, and one each in Spain and Switzerland. They found no difference in the potency of the drugs in time-kill studies. The pharmacokinetics of the products were similar, as were the amounts of killing of the bacteria in the cardiac vegetations and spleens of rabbits with experimental S. aureus endocarditis and bacteremia.
Is there an explanation for the discordance with the Vesga et al. paper? In order to minimize differences between the two laboratories, two of the three S. aureus strains (GRP-0057 and ATCC 29213) evaluated in the current project were kindly provided by O. Vesga. We also used the same gender and strain of mice employed by Vesga et al. (female CD-1 mice), albeit from a different colony of mice. Although the innovator product is no longer manufactured by Eli Lilly & Company and, hence, was not available for examination in the current project, the amount of killing of S. aureus GRP-0057 by the vancomycin products examined in our project approximated the degree of killing reported by Vesga and colleagues. Vesga et al. did examine different vancomycin products (and lots) than our laboratory did. The findings of Hadwiger et al. (4), in which the addition of 9% CDP-1 to the six vancomycin products available in the United States had no effect on the drug potency, strongly suggest that CDP-1 was not responsible for the discordant results. Finally, and, perhaps most importantly, the studies conducted by Vesga et al. (11) stretched over 7 years. In 2005 and 2009, two new vancomycin products that entered the marketplace were examined by Vesga and colleagues. These vancomycin products were manufactured in the United States by Baxter Healthcare Corp. and Hospira, Inc., using the protocols of the innovator, Eli Lilly & Company. In the in vitro and in vivo experiments conducted by Vesga et al., these products performed as well as the innovator (11). The vancomycin products manufactured by Baxter Healthcare Corp. and Hospira, Inc., were included in our project. It may be that the time frame of evaluation and change in products over time may be important.
We felt that the current study was required to document the relative in vitro and in vivo activities of the vancomycin products currently available in the United States. The findings of Hadwiger et al. and Nambiar et al. (3, 4), which examined the quality and potency of six of the vancomycin products sold in the U.S. marketplace today, together with the in vitro/in vivo pharmacodynamic analysis described in the current report, support the conclusion that there is no concern regarding the effectiveness of the parenteral vancomycin products in use today in the United States.
ACKNOWLEDGMENTS
This work was supported by grant R01AI090802 from the National Institutes of Health, National Institute of Allergy and Infectious Diseases.
The opinions expressed in this paper are those of the authors and may not be reflective of those of NIAID or the FDA. The findings and conclusions of this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any Agency determination or policy.
We thank Omar Vesga for providing two of the S. aureus strains used in this study.
The authors have no conflicts to disclose.
REFERENCES
- 1.Code of Federal Regulations. 2011. Title 21. Food and drugs. Chapter 1. Food and Drug Administration. Subchapter D. Drugs for human use. Part 314. Applications for FDA approval to market a new drug. 21 CFR 314.94(a)(3). [Google Scholar]
- 2.Code of Federal Regulations. 2011. Title 21. Food and drugs. Chapter 1. Food and Drug Administration. Subchapter D. Drugs for human use. Part 314. Applications for FDA approval to market a new drug. 21 CFR 314.94(a). [Google Scholar]
- 3.Nambiar S, Madurawe RD, Zuk SM, Khan SR, Ellison CD, Faustino PJ, Mans DJ, Trehy ML, Hadwiger ME, Boyne MT II, Biswas K, Cox EM. 2012. Product quality of parenteral vancomycin products in the United States. Antimicrob Agents Chemother 56:2819–2823. doi: 10.1128/AAC.05344-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hadwiger ME, Sommers CD, Mans DJ, Patel V, Boyne MT II. 2012. Quality assessment of U.S. marketplace vancomycin for injection products using high-resolution liquid chromatography-mass spectroscopy and potency assays. Antimicrob Agents Chemother 56:2824–2830. doi: 10.1128/AAC.00164-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.USP. 2011. Antibiotics—microbial assays. United States pharmacopeia and national formulary. USP 34-NF 29. USP, Rockville, MD. [Google Scholar]
- 6.USP. 2011. Vancomycin HCl for injection. United States pharmacopeia and national formulary. USP 34-NF 29. USP, Rockville, MD. [Google Scholar]
- 7.Code of Federal Regulations. 2011. Title 21. Food and drugs. Chapter 1. Food and Drug Administration. Subchapter D. Drugs for human use. Part 314. Applications for FDA approval to market a new drug. 21 CFR 314.94(a)(9)(iii). [Google Scholar]
- 8.Code of Federal Regulations. 2011. Title 21. Food and drugs. Chapter 1. Food and Drug Administration. Subchapter D. Drugs for human use. Part 314. Applications for FDA approval to market a new drug. 21 CFR 314.94(a)(9). [Google Scholar]
- 9.Code of Federal Regulations. 2011. Title 21. Food and drugs. Chapter 1. Food and Drug Administration. Subchapter D. Drugs for human use. Part 320. Bioavailability and bioequivalence requirements. 21 CFR 320.22. [Google Scholar]
- 10.Lee SL, Yu LX, Cai B, Johnsons GR, Rosenberg AS, Cherney BW, Guo W, Raw AS. 2011. Scientific considerations for generic synthetic salmon calcitonin nasal spray. AAPS J 13:14–19. doi: 10.1208/s12248-010-9242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vesga O, Agudelo M, Salazar BE, Rodriguez CA, Zuluaga AF. 2010. Generic vancomycin products fail in vivo despite being pharmaceutical equivalents of the innovator. Antimicrob Agents Chemother 54:3271–3279. doi: 10.1128/AAC.01044-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eagle H, Musselman AD. 1948. The rate of bactericidal action of penicillin in vitro as a function of its concentration, and its paradoxically reduced activity at high concentrations against certain organisms. J Exp Med 88:99–131. doi: 10.1084/jem.88.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tattevin P, Saleh-Mghir A, Davido B, Ghout I, Massias L, Garcia de la Maria C, Miró JM, Perronne C, Laurent F, Crémieux AC. 2013. Comparison of six generic vancomycin products for treatment of methicillin-resistant Staphylococcus aureus experimental endocarditis in rabbits. Antimicrob Agents Chemother 57:1157–1162. doi: 10.1128/AAC.01669-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clinical and Laboratory Standards Institute. 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standards, 8th ed. CLSI publication M07-A8. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 15.Clinical and Laboratory Standards Institute. 2009. Performance standards for antimicrobial susceptibility testing, approved standard M100-S19. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 16.D'Argenio DZ, Schumitzky A, Wang X. 2009. ADAPT 5 user's guide: pharmacokinetic/pharmacodynamic systems analysis software. Biomedical Simulations Resource, Los Angeles, CA. [Google Scholar]
- 17.Leary R, Jelliffe R, Schumitzky A, Van Guilder M. 2001. An adaptive grid non-parametric approach to pharmacokinetic and dynamic (PK/PD) models, p 389–394. In Proceedings of the 14th IEEE Symposium on Computer-Based Medical Systems. IEEE Computer Society, Bethesda, MD. [Google Scholar]
- 18.Craig WA. 2003. Basic pharmacodynamics of antibacterials with clinical applications to the use of beta-lactams, glycopeptides, and linezolid. Infect Dis Clin North Am 17:479–501. doi: 10.1016/S0891-5520(03)00065-5. [DOI] [PubMed] [Google Scholar]
- 19.Nicasio AM, Bulitta JB, Lodise TP, D'Hondt RE, Kulawy R, Louie A, Drusano GL. 2012. Evaluation of once-daily vancomycin against methicillin-resistant Staphylococcus aureus in a hollow fiber infection model. Antimicrob Agents Chemother 56:682–668. doi: 10.1128/AAC.05664-11. [DOI] [PMC free article] [PubMed] [Google Scholar]