

Abstract
We have designed a hybrid peptide by combining sequences of human β-defensin-1 (HBD-1) and θ-defensin, in an attempt to generate a molecule that combines the diversity in structure and biological activity of two different peptides to yield a promising therapeutic candidate. HBD-1 was chosen as it is a natural defensin of humans that is constitutively expressed, but its antibacterial activity is considerably impaired by elevated ionic strength. θ-Defensins are expressed in human bone marrow as a pseudogene and are homologous to rhesus monkey circular minidefensins. Retrocyclins are synthetic human θ-defensins. The cyclic nature of the θ-defensin peptides makes them salt resistant, nonhemolytic, and virtually noncytotoxic in vitro. However, a nonhuman circular molecule developed for clinical use would be less viable than a linear molecule. In this study, we have fused the C-terminal region of HBD-1 to the nonapeptide sequence of a synthetic retrocyclin. Cyclization was achieved by joining the terminal ends of the hybrid peptide by a disulfide bridge. The hybrid peptide with or without the disulfide bridge exhibited enhanced antimicrobial activity against both Gram-negative and Gram-positive bacteria as well as against fungi, including clinical bacterial isolates from eye infections. The peptide retained activity in the presence of NaCl and serum and was nonhemolytic in vitro. Thus, the hybrid peptide generated holds potential as a new class of antibiotics.
INTRODUCTION
Defensins are a group of peptides that are important components of host defense. They have the ability to recognize and neutralize invading microorganisms quickly and specifically (1–3). These peptides are diverse members of a large family of cationic host defense peptides (HDPs) that are widely distributed throughout the animal kingdom (2, 4, 5). These cysteine-rich peptides vary in their length, the spacing of their cysteine residues, and their disulfide connectivities (4, 5). Defensins show antimicrobial activities against Gram-negative and Gram-positive bacterial strains, fungi, as well as some parasites and enveloped viruses (1, 2, 4, 6). Mammalian defensins can be classified into α-, β-, and θ-defensins on the basis of their disulfide connectivities (2, 4, 6–8). Only α- and β-defensins are expressed in humans. θ-Defensin (found in rhesus monkey) displays the same connectivity associated with that of the α-defensins but is not functionally present in humans (7). θ-Defensins are much smaller (18 amino acid residues) than α- and β-defensins (29 to 45 residues), and their antiviral properties are considerably more pronounced than their antibacterial and antifungal effects (9, 10).
β-Defensins are found in epithelial cells that line mucosal surfaces and that provide the first line of defense between an organism and the environment (2, 6). To date, four human β-defensins (human β-defensin-1 [HBD-1] to HBD-4) have been characterized (5, 11–19). HBD-1 is expressed in epithelia that are directly exposed to the environment or microbial flora (e.g., in the eye, lung, mammary gland, salivary gland, kidney, pancreas, and prostate) (16, 17, 20, 21). HBD-1 shows antibacterial activity at micromolar concentrations against some Gram-negative bacteria (i.e., Escherichia coli, Pseudomonas aeruginosa, and Klebsiella pneumoniae), as well as the yeast Candida albicans (22). When tested in vitro, HBD-1 is relatively less potent against the Gram-positive bacterium Staphylococcus aureus (12, 17, 23), but these microbicidal activities are inhibited at high concentrations of NaCl (23).
θ-Defensins have been purified only from the leukocytes and bone marrow of rhesus macaques (7, 8, 24). These are circular octadecapeptides with six cysteines pairing to form a ladder-like disulfide array that connects their two antiparallel β sheets (7). Human bone marrow expresses θ-defensin homologues, but the human retrocyclin gene does not produce a functional product owing to a premature stop codon (7, 9). Six rhesus macaque θ-defensin isoforms (RTD-1 to RTD-6) are expressed in neutrophils, where they are packaged in cytoplasmic granules. Retrocyclins are synthetic humanized θ-defensins whose structures are based on human multiple θ-defensin pseudogenes (7). They are octadecapeptides that contain two linked nonapeptides that may be identical or different. The N terminus of one nonapeptide forms a peptide bond with the C terminus of another nonapeptide, resulting in a cyclic 18-residue peptide with three intramolecular disulfide bonds (7, 8). θ-Defensins have antibacterial activity against both Gram-positive and Gram-negative bacteria (8, 25) as well as antifungal (8) and antiviral (9, 26) activities. Retrocyclins have been shown to protect human cells from infection by HIV-1 (9) and have been evaluated as a topical anti-HIV agent for the prevention of HIV transmission (10, 27). θ-Defensins have also been shown to inactivate germinating anthrax spores and act as a competitive inhibitor of anthrax lethal factor protease (28).
The emergence of resistance to antibiotics (29, 30) has led to a growing interest in exploiting the biological activities of host defense peptides as a new class of therapeutic agents to target multidrug-resistant (MDR) pathogens. The main advantage of these peptides as factors of innate immunity is that they can function without either high specificity or memory (2).
The C-terminal cationic segment of HBD-1 (Phd-1) with a single disulfide bond exhibits antimicrobial activity (31). However, the peptide is inactive in the presence of high concentrations of NaCl. With a view to generate a peptide with improved antimicrobial activity, we have designed a hybrid peptide composed of the C-terminal segment of HBD-1 and the RIGRRIC segment of θ-defensin. The hybrid peptide has greater number of R residues than K residues, which could enhance binding to the microbial cell surface. We have observed that the hybrid peptide shows antimicrobial activity against Gram-negative and Gram-positive bacteria, fungi, as well as multidrug-resistant bacterial strains isolated from patients with eye infections. Antimicrobial activity was not attenuated in the presence of physiological concentrations of NaCl. Antimicrobial activity against all organisms tested except E. coli was also observed in the presence of serum.
MATERIALS AND METHODS
9-Fluorenylmethoxycarbonyl (Fmoc)-protected amino acids were purchased from Novabiochem (Merck). Fmoc-l-NovaSyn-cysteine-TGA resin was obtained from Novabiochem (Merck). N-Hydroxybenzotriazole hydrate (HOBT) and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were from Advanced Chemtech (Louisville, KY). Piperidine was from Loba-Chemie Pvt. Ltd. (India). Reagents for deprotection of peptides were purchased from Sigma Chemical Co. (St. Louis, MO), and 5(6)-carboxyfluorescein (CF) was from Molecular Probes, Life Technologies.
Peptide synthesis.
The hybrid peptide hBTD-1 was manually synthesized by solid-phase methods, using Fmoc-l-Cys-(trityl)-NovaSyn-TGA resin and 9-fluorenylmethoxycarbonyl chemistry as described earlier (31). The peptide was cleaved from the resin using trifluoroacetic acid (TFA) containing thioanisole, meta-cresol, and ethanedithiol (10:1:1:0.5, vol/vol). Formation of the disulfide bond was accomplished by air oxidation in 20% (vol/vol) aqueous dimethyl sulfoxide (32) at a concentration of 0.5 mg/ml for 24 h at room temperature. The peptide was purified by high-pressure liquid chromatography on a reversed-phase C18 column (Hi-pore reversed-phase column, 4.6 mm by 250 mm) using gradients of solvent A, which consisted of 0.1% (vol/vol) TFA in H2O, and solvent B, which consisted of 0.1% (vol/vol) TFA in CH3CN. Purified peptide was characterized by matrix-assisted laser desorption ionization–time of flight mass spectrometry on an Applied Biosystems 4800 instrument in the Proteomics Facility of CSIR-Centre for Cellular and Molecular Biology using recrystallized α-cyano-4-hydroxycinnamic acid as the matrix. The theoretical mass calculated was 2,737.32, and the observed m/z was 2,736.67.
Labeling of hBTD-1 with CF at the free amino group of the N-terminal amino acid was carried out by treating 10 mg of resin-bound peptide with 0.8 ml of dimethylformamide containing CF and activating agents as described earlier (33). The deprotection of CF-labeled peptide from the resin, purification, and characterization by mass spectrometry were carried out as described earlier (31).
Antibacterial activity.
The bacterial strains used were Escherichia coli (MG 1655), Staphylococcus aureus (ATCC 8530), and Pseudomonas aeruginosa (NCTC 6751). The clinical strains tested were obtained from the Jhaveri Microbiology Centre, L. V. Prasad Eye Institute, Hyderabad, India. All strains were isolated from clinical samples from patients with ocular infection. The bacterial isolates tested included MDR Pseudomonas aeruginosa, methicillin-resistant Staphylococcus aureus (MRSA), Burkholderia cepacia, Corynebacterium amycolatum, and Staphylococcus epidermidis. All isolates were tested for susceptibility to a large number of antibiotics by the Kirby-Bauer disc diffusion method, and susceptibilities were interpreted per CLSI guidelines.
The antibacterial activity of the peptide was examined in sterile 96-well plates at a final volume of 100 μl, as follows. Bacteria were grown in nutrient broth (Difco Bacto nutrient broth) to mid-log phase and diluted to 106 CFU/ml in 10 mM sodium phosphate buffer (pH 7.4). Bacteria were incubated with different concentrations of peptide for 2 h at 37°C, and suitably diluted aliquots were spread on nutrient agar plates. After the plates were incubated at 37°C for 18 h, the colonies formed were counted. The lethal concentration (LC) was the concentration of the peptide at which no viable colonies were formed. Cell survival was expressed as a percentage of the control. Percent killing was calculated as follows: [(number of colonies from control cells − number of colonies from treated cells)/number of colonies from control cells] × 100. The LC determined was the average of three independent experiments done in duplicate. In control experiments, cells were incubated with only buffer.
The activity of the peptide analog was also tested in the presence of 1 mM dithiothreitol (DTT). The disulfide bridge was broken by incubation with DTT at 37°C for 1 h. An antimicrobial assay was performed with reduced peptide in the presence of 1 mM DTT. In all the experiments, untreated peptide and cells in the presence of 1 mM DTT were used as controls.
To determine the effect of salt on antibacterial activity, different concentrations of NaCl were included in the incubation buffer with the peptide analog at its LC. Different concentrations of divalent cations Ca2+ and Mg2+ (as their chloride salts) were included in the buffer to determine their effect on activity at the lethal concentration of the peptide. In control experiments, cells were incubated with only buffer. Data are presented as the mean ± standard deviation computed from three independent replicates.
Antifungal activity.
The fungal strains used were Candida albicans and Saccharomyces cerevisiae. The minimum fungicidal concentration (MFC) of the peptide was determined by growing the fungi aerobically in yeast extract-peptone-dextrose (YEPD) medium at 30°C. After 20 h, 0.5 ml from this suspension was subcultured for 2 h in 20 ml of YEPD broth to obtain a mid-log-phase culture. Cells were harvested by centrifugation, washed with 10 mM phosphate buffer (PB), pH 7.4, and resuspended in the same buffer, and the concentration was adjusted to 106 cells/ml. Aliquots of diluted cells were incubated with peptide in 100-μl volumes at 30°C for 2 h. Cell suspensions were diluted and spread onto YEPD agar plates, and the plates were incubated for 24 h at 30°C.
Colonies were counted, and the concentration of the peptide at which no viable colonies were formed was taken as the MFC. Cell survival was expressed as a percentage of the control. Percent killing was calculated as [(number of colonies from control cells − number of colonies from treated cells)/number of colonies from control cells] × 100. The average of the results from three independent experiments done with duplicate samples was taken for calculation of the MFC. In control experiments, cells were incubated with only buffer.
To determine the effect of salt on antifungal activity, different concentrations of NaCl were included in the incubation buffer with the peptide analog at its LC. Different concentrations of divalent cations Ca2+ and Mg2+ (as their chloride salts) were included in the buffer to determine their effect on activity at the lethal concentration of the peptide. In control experiments, cells were incubated with only buffer. Data are presented as the mean ± standard deviation computed from three independent replicates.
Kinetics of killing.
The kinetics of microbial killing of Gram-negative and Gram-positive bacteria and fungi were determined. Mid-log-phase bacteria (106 CFU/ml) were incubated with the LC of the peptide in 10 mM sodium phosphate buffer (pH 7.4) in a final volume of 100 μl. Aliquots of 20 μl were removed at fixed intervals and spread on nutrient agar plates. The number of CFUs was counted after incubations of 18 h at 37°C and 24 h at 28°C in the case of bacteria and fungi, respectively. Data are presented as the mean ± standard deviation computed from three independent replicates.
Serum sensitivity.
To study the effect of serum components on the antimicrobial activity of the peptide, serum was isolated from human blood after the removal of erythrocytes by centrifugation. Approval was obtained from the Institutional Ethical Committee of the Centre for Cellular and Molecular Biology. The serum isolated was used for the study without heat inactivation. For the antimicrobial activity, 20% serum was included in the buffer before the addition of the peptide. Cells with only serum were used as a control.
CD.
Circular dichroism (CD) spectra were recorded in 10 mM phosphate buffer (pH 7.4), trifluoroethanol (TFE), and 10 mM sodium dodecyl sulfate (SDS) micelles on a Jasco J-715 automatic recording spectropolarimeter at 25°C using a quartz cell with a path length of 1 mm. Each spectrum (185 to 250 nm) was an average of six scans. Data are represented as mean residue ellipticities.
Hemolysis.
The hemolytic activity of hBTD-1 was determined using human erythrocytes, as described earlier (34). Briefly, erythrocytes were obtained by the centrifugation (800 × g) of heparinized blood and were washed thrice with 5 mM HEPES (pH 7.4) containing 150 mM NaCl. Aliquots containing 107 red blood cells/ml were incubated in the presence of different peptide concentrations in 0.5-ml tubes containing a lower volume of 100 μl for 30 min at 37°C with gentle mixing. The samples were centrifuged, and the absorbance of the supernatants at 540 nm was measured. Erythrocyte lysis occurring in deionized water was taken as maximal lysis.
MTT assay.
The toxicity effect of hBTD-1 on cells of the human cell line HEK 293 was analyzed at the enzymatic level by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells (1 × 105) were seeded in 96-well cell culture plates (Costar) and allowed to adhere to the surface for 12 to 14 h. After settling of the cells, fresh serum-free medium containing various concentrations of hBTD-1 was added to the wells. In the control, no test solution was added. The cells were allowed to grow for 24 h, and 10 μl of MTT reagent (5-mg/ml stock concentration) was added to each well to give final concentrations of 0.5 mg/ml. The reaction was stopped after observing purple precipitates in the control wells. The media were carefully removed without disturbing the cell layers and dried. Dimethyl sulfoxide was added to the dried wells, and the plates were incubated overnight to solubilize the precipitates. The absorbance of the solubilized material at 550 nm was taken using a Versa Max microplate reader (Molecular Devices, Sunnyvale, CA). The experiments were performed in triplicate.
Outer membrane permeabilization assay.
The ability of peptide to permeabilize the outer membranes of E. coli MG 1655 cells was investigated using an N-phenyl-1-napthylamine (NPN) uptake assay (35, 36). Briefly, cells at an optical density at 600 nm (OD600) of 0.1 were suspended in 5 mM HEPES (pH 7.4) with 10 mM NPN. After 15 min of incubation, peptide was added, and the fluorescence of NPN was monitored. The excitation wavelength used was 350 nm, and the emission wavelength was 420 nm. The experiment was carried out at 25°C.
Inner membrane permeabilization assay.
The ability of the peptide to permeabilize the inner membranes of bacteria was studied using E. coli GJ2455 (a lacI lacZ+ strain derived from E. coli GJ2544 at the Centre for Cellular and Molecular Biology), which constitutively expresses β-galactosidase in its cytoplasm. o-Nitrophenyl-β-d-galactopyranoside (ONPG) was used as the substrate for β-galactosidase in the assay (37). Late-logarithmic-phase (OD600, 0.5 to 0.6) cells were washed and diluted to an OD600 of 0.03 in 10 mM sodium phosphate buffer (pH 7.4), and 0.53 mM ONPG was added. After the addition of different peptide concentrations, measurements of the OD550 and the OD420 were made every 5 min, and absorbance calculations [A420 − (1.75 × A550)] were taken as a measure of the β-galactosidase activity. The production of o-nitrophenyl was monitored at 420 nm. The value 1.75 × A550 represents the light scattering by cell debris at 420 nm. Bacterial cells without any peptide and without ONPG were used as a control.
Confocal microscopy.
Localization of the peptide was analyzed by treating E. coli, C. albicans, and S. cerevisiae with CF-labeled hBTD-1, FM4-64 (for E. coli), and propidium iodide (PI) (for fungi) for 15 min. Organisms were grown overnight in nutrient broth for E. coli and yeast extract-peptone-dextrose medium for C. albicans and S. cerevisiae and washed with 10 mM phosphate buffer (pH 7.4), and the concentrations were adjusted to 1 × 107 cells/ml in 10 mM phosphate buffer. These cells were treated with a sublethal concentration of peptide for different time intervals. The peptide was incubated for 5 min, 10 min, 20 min, and 30 min at 37°C, to capture different stages of peptide entry, localization on the membrane, and killing. The cells were examined with a Zeiss LSM 510 Meta confocal microscope. Optical sectioning was done at 1 airy unit by using the 488- and 543-nm-wavelength laser lines with a ×100 oil lens objective. Emission data were collected using 500- to 530-nm band-pass and 565- to 615-nm band-pass filters for CF and PI, respectively, in the multitrack mode. z-sections were acquired at 0.35-μm intervals and projected using LSM-FCS software (version 3.2). The bright-field images were obtained simultaneously using a transmitted-light detector. Images shown are representative of the different events (initial interaction with the membrane and subsequent translocation into the cells) that could be captured.
RESULTS AND DISCUSSION
Peptide design.
All mammalian defensins are cationic (2, 4, 38, 39). However, the R/K ratio is variable, as is the number of R and K residues. R residues play an important role in modulating the activity of defensins (40–43). Hence, a hybrid peptide containing the C-terminal K-rich segment of HBD-1 and the R-rich domain of θ-defensin was designed and synthesized. The sequences of the peptides are shown in Table 1 (25, 44, 45). Since defensin analogs with a single disulfide bridge do show activity, only two cysteines were incorporated so that a disulfide bond can be formed. The hybrid peptide has an R/K ratio of >1. We designed the hybrid peptide to improve the activity spectrum and stability of HBD-1, which is active against certain Gram-negative bacteria and Candida albicans (22). The antibacterial activity of HBD-1 is attenuated in the presence of physiological concentrations of NaCl (23). The antimicrobial activities of HBD-1 and RTD-1 to RTD-3 are shown in Table 2. RTD-1 to RTD-3 have been shown to be active against the reported microorganisms at very low concentrations (25). Hence, we attempted to design an analog of HBD-1 with improved activity and stability by fusing its C-terminal sequence with the sequence from human retrocyclin. Cyclization was achieved by joining the terminal ends of the hybrid peptide by a disulfide bridge.
TABLE 1.
Primary structure of HBD-1, human retrocyclin, and the hybrid peptide
| Peptide | Sequencea | Net charge |
|---|---|---|
| HBD-1 | DHYNC1VSSGGQC2LYSAC3PIFTKIQGTC2YRGKAKC1C3K | +5 |
| θ-Defensin (retrocyclin) | c(RC1IC2GRRIC2-RC1IC3GRRIC3) | +6 |
| hBTD-1 | AC1PIFTKIQGTYRGKAKRIGRRIC1 | +7 |
The numbers of disulfide bridges are shown by superscript numbers adjacent to cysteines in bold.
TABLE 2.
Antimicrobial activities of HBD-1 and RTD-1 to RTD-3
Antimicrobial activity.
We examined the antimicrobial activity of the hybrid peptide against Gram-negative and Gram-positive bacteria and fungi, such as C. albicans and S. cerevisiae. HBD-1 is constitutively expressed by corneal and conjunctival epithelial cells (20, 46). It is rendered inactive by the high salt concentrations present in tear fluid (47). Retrocyclin-2 has been explored as an antiviral therapeutic agent in a murine model of herpes simplex virus 1 keratitis (48). Hence, we examined the activity of the hybrid peptide against clinical strains isolated from the human eye.
The antimicrobial activity of the peptide against representative microorganisms is summarized in Table 3. The lethal concentration of the peptide was found to be 1 μM against the Gram-negative bacterial strains tested and 2 μM against the Gram-positive bacterial strain tested, and in the case of the fungal strains, the minimum fungicidal concentration (MFC) was 2 μM. Activity was not lost when the disulfide bridge was broken by DTT (data not shown).
TABLE 3.
Antimicrobial activity of hBTD-1a
| Organism | Antibacterial activity |
Antifungal activity |
||
|---|---|---|---|---|
| LC (μM) | LC (μM) in 20% serum | MFC (μM) | MFC (μM) in 20% serum | |
| E. coli | 1 | IA | ||
| P. aeruginosa | 1 | 4 | ||
| S. aureus | 2 | 4 | ||
| C. albicans | 2 | 5 | ||
| S. cerevisiae | 2 | 5 | ||
The values reported are averages of those from three independent experiments done in duplicate. The variations observed were ∼2%. IA, inactive.
Reports of the in vitro activity of θ-defensin against drug-resistant strains have been made. Lamers and coworkers (49) have characterized an analog of retrocyclin (RC-101) that is preventative of S. aureus nasal carriage at a minimum concentration of 2.5 μM. Similarly, another group has shown RTD-1 to be active against antibiotic-resistant S. aureus and P. aeruginosa at 3 μM (50). However, there are no reported values for the activity of HBD-1 against multidrug-resistant bacterial strains in vitro. Hence, we checked the activity of the hybrid peptide against a few multidrug-resistant bacteria. The data on activity against clinical strains are summarized in Table 4. The peptide was found to be active against MDR Pseudomonas aeruginosa, MRSA, Corynebacterium amycolatum, and Staphylococcus epidermidis, for which the LC was 2 μM (Table 4). However, the hybrid peptide was not active when tested against Burkholderia cepacia. The resistance of the B. cepacia strain to the peptide is possibly due to the unusual structure of its lipopolysaccharide (LPS). It has been reported that the LPS of the B. cepacia complex lowers the anionic charge of the cell surface, which inhibits the binding and subsequent effects of cationic antibiotics (51).
TABLE 4.
Antimicrobial activity of hBTD-1 against clinical strains
| Bacterial strain | Clinical diagnosis | Type of sample | Resistance phenotypea | LC (μM) |
|---|---|---|---|---|
| Corynebacterium amycolatum | Microbial keratitis | Corneal scraping | CHL, CIP, MXF | 2 |
| MDR P. aeruginosa | Microbial keratitis | Corneal scraping | AMK, AMX, AMC, CAZ, CRO, CXM, CTX, CHL, CIP, COT, GAT, GEN, IPM, MEM, MXF, OFX, PIP, TZP, TIC, TOB | 2 |
| MRSA | Microbial keratitis | Corneal scraping | CFZ, FOX, CTX, CXM, CIP, MXF, OFX | 2 |
| Staphylococcus epidermidis | Postoperative endophthalmitis | Vitreous biopsy specimen | None | 2 |
| Burkholderia cepacia | Microbial keratitis | Corneal scraping | AMK, AMX, AMC, CFZ, CXM, CHL, CIP, CST, GAT, GEN, MEM, MXF, OFX, TIC, TOB | IAb |
AMK, amikacin; AMX, amoxicillin; AMC, amoxicillin-clavulanic acid; CAZ, ceftazidime; CXM, cefuroxime; CHL, chloramphenicol; CIP, ciprofloxacin; CST, colistin; CFZ, cefazolin; CTX, cefotaxime; COT, co-trimoxazole; CRO, ceftriaxone; FOX, cefoxitin; CAZ, ceftazidime; GAT, gatifloxacin; GEN, gentamicin; IPM, imipenem; MEM, meropenem; MXF, moxifloxacin; OFX, ofloxacin; PIP, piperacillin; TZP, piperacillin-tazobactam; TOB, tobramycin; TIC, ticarcillin.
IA, inactive.
The hybrid peptide was active against different microorganisms in the presence of serum. However, a slight increase in the LC was observed (Table 3). Despite being rich in R and K, the peptide is active in the presence of serum, suggesting that it is refractory to the action of proteases and binding to serum components. Vogel and coworkers (52) have shown that peptide cyclization by disulfide formation results in increased serum stability and the microbicidal activity of the peptides.
The hybrid peptide was inactive in the presence of serum when tested against E. coli. A similar result was observed in the case of RTD-3, which exhibited activity against S. aureus, whereas against E. coli its activity was lost (25). In certain E. coli strains, a high-molecular-weight polysaccharide, O antigen, is produced and is not agglutinated by the serum unless heated (53). It is possible that this O antigen binds to the peptide and inactivates it.
Kinetics of killing.
Killing of bacteria and fungi (reduction in the log number of CFU/ml) as a function of time at the LC is shown in Fig. 1. The rates of killing of the microbes tested were comparable. Killing was observed at earlier time points. However, complete killing (100%) was observed after 30 min.
FIG 1.

Kinetics of killing of the hybrid peptide hBTD-1. Both bacteria and fungi (106 CFU/ml) at mid-log phase were incubated with peptides at the LC for different times. Data are the means from three independent experiments. Standard deviation values ranged from 0.002 to 0.3. Symbols: E. coli (■), P. aeruginosa (●), S. aureus (▲), C. albicans (▼), S. cerevisiae (⧫).
Salt sensitivity.
In the presence of NaCl, in the case of Gram-negative bacteria, peptide activity was only marginally attenuated at 100 and 150 mM concentrations of NaCl (Fig. 2A), while in the case of Gram-positive bacteria and fungi, the peptide exhibited a decrease in activity (Fig. 2A and B). In contrast, in the case of Gram-negative bacteria, the peptide showed a complete loss of activity even at low concentrations of the divalent cations Ca2+ and Mg2+ (Fig. 2A), whereas in the case of Gram-positive bacteria and fungi, the activity of the peptide was only marginally attenuated (Fig. 2A and B).
FIG 2.

Effect of NaCl and divalent cations on antibacterial and antifungal activity of the hybrid peptide. Both bacteria and fungi (106 CFU/ml) at mid-log phase were incubated with peptides at the LC in the absence and presence of the indicated concentrations of NaCl and divalent cations. (A) Antibacterial activity of E. coli (■), P. aeruginosa (□), S. aureus (▩); (B) antifungal activity of C. albicans (■) and S. cerevisiae (□). The data are mean values from three independent experiments, and the error bars represent the standard deviations of the measurements. Standard deviation values ranged from 0.3 to 1.5.
Our results suggest that a stretch of K residues followed by R residues results in a favorable electrostatic interaction with Gram-negative bacteria even in the presence of NaCl. A loss of antibacterial activity against Gram-negative bacteria in the presence of Ca2+ and Mg2+ arises, as these divalent cations stabilize the LPS network, preventing binding of the peptide to the surface of Gram-negative bacteria.
Cell lysis.
The hemolytic activity of the peptide against human erythrocytes was determined as a major measure of peptide toxicity toward higher eukaryotic cells. The hybrid peptide hBTD-1 did not exhibit lysis even at a 100 μM concentration (Table 5), which is 100 times more than its LC against the microorganisms.
TABLE 5.
Mean OD values of MTT and hemolytic assays
| Assay | OD at the following hBTD-1 concn (μM)a: |
||||
|---|---|---|---|---|---|
| 0 | 5 | 10 | 50 | 100 | |
| MTT | 0.9920 ± 0.027 | 0.9885 ± 0.067 | 0.9787 ± 0.021 | 0.9755 ± 0.041 | 0.9710 ± 0.014 |
| Hemolysis | 0 | 0.0012 ± 0.001 | 0.0020 ± 0.001 | 0.0040 ± 0.002 | 0.0053 ± 0.001 |
The values reported are mean ± standard deviation of the optical density recorded from three independent experiments done in duplicate.
The viability of HEK 293 cells was analyzed through an MTT assay (Table 5). The data indicated no significant difference in cell viability among the different concentrations of hBTD-1 (10, 50, and 100 μM), which is comparable to the findings for the control (not treated with peptide). The result suggests that the peptide does not have any effect on the viability of human cell lines.
CD.
In order to examine whether any secondary structural feature is responsible for the antimicrobial activity of hBTD-1, CD spectra (Fig. 3A) were recorded in buffer, SDS micelles, and the structure promoting solvent trifluoroethanol (TFE) (54). The spectrum in buffer showed a minimum of ∼198 nm and a crossover of <190 nm, suggesting a predominantly unordered conformation (55). In 10 mM SDS and TFE, the spectra showed a negative band at ∼208 nm, a shoulder at ∼220 nm, a crossover at ∼200 nm, and a positive band at ∼195 nm. These features are characteristic of peptide in a helical conformation (55). The lower negative ellipticity value suggests a lower helical content in SDS than in TFE. However, it is clear that the peptide has the propensity to fold into a helical conformation on encountering negatively charged hydrophobic molecules like those in bacterial or fungal cell surfaces and also in medium with a low dielectric constant. The helical wheel diagram of hBTD-1 (AC1PIFTKIQGTYRGKAKRIGRRIC1, where the numbers of disulfide bridges are shown by superscript numbers adjacent to cysteines in bold) is shown in Fig. 3B. While the peptide is not amphipathic, the positioning of cationic residues in a helical conformation could facilitate effective interaction with negatively charged surfaces. Since the peptide is not amphipathic, it does not lyse erythrocytes and cultured cells, a characteristic feature of several cationic helical amphipathic peptides (56, 57). Specificity towards the microbial membranes and the lack of hemolytic properties highlight the potential of the peptide to be used as a therapeutic agent or a drug scaffold.
FIG 3.

(A) Circular dichroism spectra of hBTD-1. CD spectra were recorded in 10 mM phosphate buffer (■), 10 mM SDS (●), and TFE (▲). [θ]MRE, mean residue ellipticity. (B) Helical wheel representation of hBTD-1. Circles, hydrophilic residues; diamonds, hydrophobic residues; triangles, potentially negatively charged residues; pentagons, potentially positively charged residues. The peptide does not show amphipathicity.
Outer and inner membrane permeabilization.
NPN, a neutral hydrophobic probe, has been used to examine the permeabilization of the outer membranes of Gram-negative bacteria. The peptide permeabilized the outer membrane of E. coli in a concentration-dependent manner, as observed by an increase in NPN fluorescence (Fig. 4A). When the ability of the peptide to permeabilize the inner membrane of E. coli was examined by the ONPG assay, an influx of ONPG was observed, suggesting that the peptide was effective in permeabilizing the inner membrane (Fig. 4B).
FIG 4.

Membrane permeabilization assays. (A) Outer membrane permeabilization of E. coli MG 1655 by hBTD-1. Permeabilization of the outer membrane was monitored as an increase in the NPN fluorescence intensity in the presence of increasing concentrations of peptide (0 to 5 μM), as indicated adjacent to the traces. (B) Inner membrane permeabilization of E. coli GJ2544 by hBTD-1 as a function of time at different concentrations (0 to 5 μM) and 37°C. The hydrolysis of ONPG by β-galactosidase was used to monitor inner membrane permeabilization by determination of the absorbance at 420 nm.
The preferential binding of the hybrid peptide to anionic membranes rather than zwitterionic membranes correlates with the high level of activity against microbes and low hemolytic properties. It also indicates that electrostatic attractions between the cationic peptide and the anionic bacterial membrane are important for the antibacterial activity.
Confocal microscopy.
Interaction of the peptide with E. coli, C. albicans, and S. cerevisiae was examined by confocal microscopy using CF-labeled hBTD-1. The antimicrobial activity of the CF-labeled peptide was comparable to that of the unlabeled peptide. The cellular localization of the peptide was investigated using CF-labeled hBTD-1 and confocal microscopy analysis.
In the case of bacteria, the labeled peptide was seen to be uniformly localized on the membrane, as indicated by the ring-like green fluorescence all along the membrane (Fig. 5A). Subsequently, a diffuse intracellular green fluorescence, which indicates the translocation of the peptide into the cells, was observed (Fig. 5B). The images show the localization of the peptide on the cell surface followed by internalization. The images do not indicate colocalization with the FM4-64-stained inner membrane. The peptide appears to be bound to the outer membrane. After initial localization on the outer membrane, the peptide translocates into the cytosol. Though the peptide traverses through the inner membrane, the lack of diffuse staining indicates that it is rendered leaky but not solubilized. All the cells in the frame are not permeabilized, as a sublethal concentration of peptide was used for the study.
FIG 5.

Confocal microscope images of E. coli in the presence of CF-labeled hBTD-1. Bacterial cells (1 × 107) were treated with CF-labeled peptide and FM4-64 and were incubated for different times. (A) The initial time point when the peptide was found to be spread uniformly on the bacterial membrane; (B) a subsequent time point when the peptide was found to be uniformly distributed inside the cell (the increased green fluorescence is shown by an arrow).
In the case of C. albicans, the peptide initially binds to the fungal cell surface (Fig. 6A and 7). This is followed by entry into the cytoplasm (Fig. 6B). The peptide then completely translocates into the cells, as indicated by the increased green fluorescence (Fig. 6C). The staining by PI and the peptide indicates extensive intracellular damage (Fig. 6D). The cells whose membranes are compromised take up PI, and the diffuse red staining is due to the spreading of the nuclear material in the dead cells. Control cells showed negative staining for PI. S. cerevisiae is killed by a similar mechanism. Permeabilization and entry are more rapid in the case of S. cerevisiae (Fig. 7).
FIG 6.
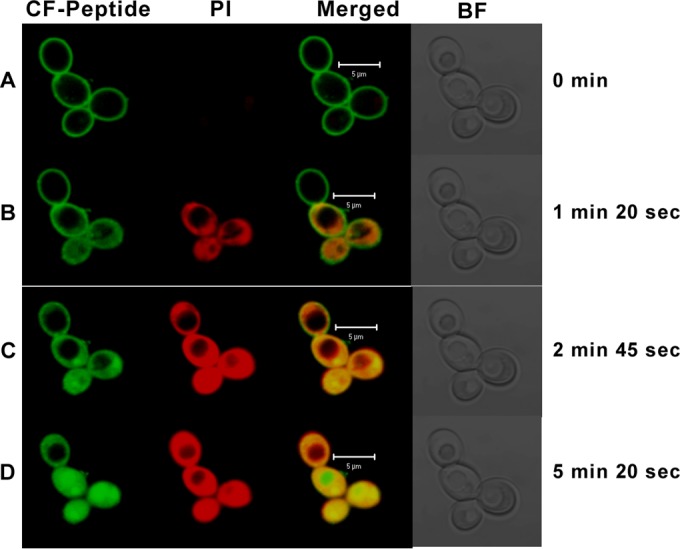
Confocal microscope images of C. albicans in the presence of CF-labeled hBTD-1. C. albicans cells (1 × 107) were treated with CF-labeled peptide at a sublethal concentration and with 2 μg/ml of PI. The treated cells were incubated for different times to capture different events during the process of peptide entry into the cell. (A) An early time point when the peptide was found to be uniformly distributed over the membrane; (B) stages when the peptide diffuses into the cells (green) and the cells whose membranes are compromised are seen taking up PI stain (red); (C) an increased amount of peptide diffusion and PI spread at a subsequent time point; (D) dead cells (yellow in the merged panel). Green fluorescence, the diffused peptide; red fluorescence, diffused nuclear material. BF, bright field.
FIG 7.

Confocal microscope images of S. cerevisiae in the presence of CF-labeled hBTD-1. S. cerevisiae cells (1 × 107) were treated with a sublethal concentration of CF-labeled peptide and with 2 μg/ml of PI. The treated cells were incubated for different times to capture different events during the process of peptide entry into the cell. (A) An early time point when the peptide was found to be uniformly spread over the membrane; (B) stages when the peptide diffuses into the cells (green) and the cells whose membranes are compromised are seen taking up PI stain (red); (C) increased amount of peptide diffusion and PI spread at a subsequent time point; (D) dead cells (yellow in the merged panel). Green fluorescence, the diffused peptide; red fluorescence, diffused nuclear material. BF, bright field.
The mechanism of bacterial and fungal killing involves initial binding of the peptide to the cell surface, resulting in destabilization, thereby providing a pathway for entry into the cytoplasm, where the peptide interferes with the metabolic process, resulting in cell death. The confocal images exclude the possibility of a detergent-type lysis. We propose that such a mechanism would render the development of resistance difficult or impossible, as the metabolic cost for the microorganisms would be very high, as demonstrated recently in amphotericin B-resistant C. albicans (58). Even in the case of magainin-resistant strains, it has been shown that the resistance mechanism is complex and involves several metabolic pathways, although the peptide presumably acts by permeabilizing membranes (59).
Linear peptides with a minimal length and without multiple disulfide bridges would be attractive candidates as therapeutic agents. Another approach that would reduce the costs of peptide therapeutics is improving peptide stability and pharmacokinetics. In the present study, the hybrid peptide that we have designed is a molecule that has activity at <5 μM, kills fairly rapidly, and exhibits stability under physiological conditions. Moreover, it is not toxic to human cells. Although cysteines were introduced, a disulfide bridge does not appear to be necessary for antimicrobial activity. The findings from the present work suggest that the production of β- and θ-defensin in the form of a hybrid peptide is a promising strategy for the development of clinically relevant molecules for therapeutic applications and may lead to a cost-effective solution for the large-scale production of antimicrobial peptides.
ACKNOWLEDGMENTS
This work was supported by funding from the WOS-A Scheme of the Department of Science and Technology, India. R.N. is the recipient of a J. C. Bose fellowship from the Department of Science and Technology, India.
We acknowledge the Hyderabad Eye Research Foundation, Hyderabad, India, for providing the clinical strains. Our special thanks go to Dorairajan Balasubramanian from the L. V. Prasad Eye Institute, India, for his guidance and support with carrying out the work. We also acknowledge Savitri Sharma and Sanhita Roy from the L. V. Prasad Eye Institute, India, for their help with the work on clinical strains.
REFERENCES
- 1.Bals R. 2000. Epithelial antimicrobial peptides in host defense against infection. Respir Res 1:141–150. doi: 10.1186/rr25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ganz T. 2003. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol 3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 3.Yeung AT, Gellatly SL, Hancock RE. 2011. Multifunctional cationic host defence peptides and their clinical applications. Cell Mol Life Sci 68:2161–2176. doi: 10.1007/s00018-011-0710-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selsted ME, Ouellette AJ. 2005. Mammalian defensins in the antimicrobial immune response. Nat Immunol 6:551–557. doi: 10.1038/ni1206. [DOI] [PubMed] [Google Scholar]
- 5.Pazgier M, Hoover DM, Yang D, Lu W, Lubkowski J. 2006. Human beta-defensins. Cell Mol Life Sci 63:1294–1313. doi: 10.1007/s00018-005-5540-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lehrer RI. 2004. Primate defensins. Nat Rev Microbiol 2:727–738. doi: 10.1038/nrmicro976. [DOI] [PubMed] [Google Scholar]
- 7.Selsted ME. 2004. Theta-defensins: cyclic antimicrobial peptides produced by binary ligation of truncated alpha-defensins. Curr Protein Pept Sci 5:365–371. doi: 10.2174/1389203043379459. [DOI] [PubMed] [Google Scholar]
- 8.Tang YQ, Yuan J, Osapay G, Osapay K, Tran D, Miller CJ, Ouellette AJ, Selsted ME. 1999. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated alpha-defensins. Science 286:498–502. doi: 10.1126/science.286.5439.498. [DOI] [PubMed] [Google Scholar]
- 9.Cole AM, Hong T, Boo LM, Nguyen T, Zhao C, Bristol G, Zack JA, Waring AJ, Yang OO, Lehrer RI. 2002. Retrocyclin: a primate peptide that protects cells from infection by T- and M-tropic strains of HIV-1. Proc Natl Acad Sci U S A 99:1813–1818. doi: 10.1073/pnas.052706399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallo SA, Wang W, Rawat SS, Jung G, Waring AJ, Cole AM, Lu H, Yan X, Daly NL, Craik DJ, Jiang S, Lehrer RI, Blumenthal R. 2006. Theta-defensins prevent HIV-1 Env-mediated fusion by binding gp41 and blocking 6-helix bundle formation. J Biol Chem 281:18787–18792. doi: 10.1074/jbc.M602422200. [DOI] [PubMed] [Google Scholar]
- 11.Fulton C, Anderson GM, Zasloff M, Bull R, Quinn AG. 1997. Expression of natural peptide antibiotics in human skin. Lancet 350:1750–1751. doi: 10.1016/S0140-6736(05)63574-X. [DOI] [PubMed] [Google Scholar]
- 12.Harder J, Bartels J, Christophers E, Schroder JM. 1997. A peptide antibiotic from human skin. Nature 387:861. doi: 10.1038/43088. [DOI] [PubMed] [Google Scholar]
- 13.Harder J, Bartels J, Christophers E, Schroder JM. 2001. Isolation and characterization of human beta-defensin-3, a novel human inducible peptide antibiotic. J Biol Chem 276:5707–5713. doi: 10.1074/jbc.M008557200. [DOI] [PubMed] [Google Scholar]
- 14.Liu AY, Destoumieux D, Wong AV, Park CH, Valore EV, Liu L, Ganz T. 2002. Human beta-defensin-2 production in keratinocytes is regulated by interleukin-1, bacteria, and the state of differentiation. J Investig Dermatol 118:275–281. doi: 10.1046/j.0022-202x.2001.01651.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhao C, Wang I, Lehrer RI. 1996. Widespread expression of beta-defensin hBD-1 in human secretory glands and epithelial cells. FEBS Lett 396:319–322. doi: 10.1016/0014-5793(96)01123-4. [DOI] [PubMed] [Google Scholar]
- 16.Bensch KW, Raida M, Magert HJ, Schulz-Knappe P, Forssmann WG. 1995. hBD-1: a novel beta-defensin from human plasma. FEBS Lett 368:331–335. doi: 10.1016/0014-5793(95)00687-5. [DOI] [PubMed] [Google Scholar]
- 17.Valore EV, Park CH, Quayle AJ, Wiles KR, McCray PB Jr, Ganz T. 1998. Human beta-defensin-1: an antimicrobial peptide of urogenital tissues. J Clin Invest 101:1633–1642. doi: 10.1172/JCI1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia JR, Krause A, Schulz S, Rodriguez-Jimenez FJ, Kluver E, Adermann K, Forssmann U, Frimpong-Boateng A, Bals R, Forssmann WG. 2001. Human beta-defensin 4: a novel inducible peptide with a specific salt-sensitive spectrum of antimicrobial activity. FASEB J 15:1819–1821. doi: 10.1096/fj.00-0865fje. [DOI] [PubMed] [Google Scholar]
- 19.Garcia JR, Jaumann F, Schulz S, Krause A, Rodriguez-Jimenez J, Forssmann U, Adermann K, Kluver E, Vogelmeier C, Becker D, Hedrich R, Forssmann WG, Bals R. 2001. Identification of a novel, multifunctional beta-defensin (human beta-defensin 3) with specific antimicrobial activity. Its interaction with plasma membranes of Xenopus oocytes and the induction of macrophage chemoattraction. Cell Tissue Res 306:257–264. doi: 10.1007/s004410100433. [DOI] [PubMed] [Google Scholar]
- 20.Haynes RJ, Tighe PJ, Dua HS. 1999. Antimicrobial defensin peptides of the human ocular surface. Br J Ophthalmol 83:737–741. doi: 10.1136/bjo.83.6.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehmann OJ, Hussain IR, Watt PJ. 2000. Investigation of beta defensin gene expression in the ocular anterior segment by semiquantitative RT-PCR. Br J Ophthalmol 84:523–526. doi: 10.1136/bjo.84.5.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schroder JM. 1999. Epithelial antimicrobial peptides: innate local host response elements. Cell Mol Life Sci 56:32–46. doi: 10.1007/s000180050004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM. 1997. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 88:553–560. doi: 10.1016/S0092-8674(00)81895-4. [DOI] [PubMed] [Google Scholar]
- 24.Garcia AE, Osapay G, Tran PA, Yuan J, Selsted ME. 2008. Isolation, synthesis, and antimicrobial activities of naturally occurring theta-defensin isoforms from baboon leukocytes. Infect Immun 76:5883–5891. doi: 10.1128/IAI.01100-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tran D, Tran P, Roberts K, Osapay G, Schaal J, Ouellette A, Selsted ME. 2008. Microbicidal properties and cytocidal selectivity of rhesus macaque theta defensins. Antimicrob Agents Chemother 52:944–953. doi: 10.1128/AAC.01090-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Penberthy WT, Chari S, Cole AL, Cole AM. 2011. Retrocyclins and their activity against HIV-1. Cell Mol Life Sci 68:2231–2242. doi: 10.1007/s00018-011-0715-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munk C, Wei G, Yang OO, Waring AJ, Wang W, Hong T, Lehrer RI, Landau NR, Cole AM. 2003. The theta-defensin, retrocyclin, inhibits HIV-1 entry. AIDS Res Hum Retroviruses 19:875–881. doi: 10.1089/088922203322493049. [DOI] [PubMed] [Google Scholar]
- 28.Wang W, Mulakala C, Ward SC, Jung G, Luong H, Pham D, Waring AJ, Kaznessis Y, Lu W, Bradley KA, Lehrer RI. 2006. Retrocyclins kill bacilli and germinating spores of Bacillus anthracis and inactivate anthrax lethal toxin. J Biol Chem 281:32755–32764. doi: 10.1074/jbc.M603614200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levy SB, Marshall B. 2004. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med 10:S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 30.Neu HC. 1992. The crisis in antibiotic resistance. Science 257:1064–1073. doi: 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- 31.Krishnakumari V, Singh S, Nagaraj R. 2006. Antibacterial activities of synthetic peptides corresponding to the carboxy-terminal region of human beta-defensins 1-3. Peptides 27:2607–2613. doi: 10.1016/j.peptides.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 32.Tam JP, Wu CR, Liu W, Zhang JW. 1991. Disulfide bond formation in peptides by dimethyl sulfoxide. Scope and applications. J Am Chem Soc 113:6657–6662. doi: 10.1021/ja00017a044. [DOI] [Google Scholar]
- 33.Weber PJ, Bader JE, Folkers G, Beck-Sickinger AG. 1998. A fast and inexpensive method for N-terminal fluorescein-labeling of peptides. Bioorg Med Chem Lett 8:597–600. doi: 10.1016/S0960-894X(98)00084-5. [DOI] [PubMed] [Google Scholar]
- 34.Varkey J, Nagaraj R. 2005. Antibacterial activity of human neutrophil defensin HNP-1 analogs without cysteines. Antimicrob Agents Chemother 49:4561–4566. doi: 10.1128/AAC.49.11.4561-4566.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sedgwick EG, Bragg PD. 1987. Distinct phases of the fluorescence response of the lipophilic probe N-phenyl-1-naphthylamine in intact cells and membrane vesicles of Escherichia coli. Biochim Biophys Acta 894:499–506. doi: 10.1016/0005-2728(87)90129-0. [DOI] [PubMed] [Google Scholar]
- 36.Loh B, Grant C, Hancock RE. 1984. Use of the fluorescent probe 1-N-phenylnaphthylamine to study the interactions of aminoglycoside antibiotics with the outer membrane of Pseudomonas aeruginosa. Antimicrob Agents Chemother 26:546–551. doi: 10.1128/AAC.26.4.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehrer RI, Barton A, Ganz T. 1988. Concurrent assessment of inner and outer membrane permeabilization and bacteriolysis in E. coli by multiple-wavelength spectrophotometry. J Immunol Methods 108:153–158. doi: 10.1016/0022-1759(88)90414-0. [DOI] [PubMed] [Google Scholar]
- 38.Hoover DM, Chertov O, Lubkowski J. 2001. The structure of human beta-defensin-1: new insights into structural properties of beta-defensins. J Biol Chem 276:39021–39026. doi: 10.1074/jbc.M103830200. [DOI] [PubMed] [Google Scholar]
- 39.Lehrer RI, Ganz T. 2002. Defensins of vertebrate animals. Curr Opin Immunol 14:96–102. doi: 10.1016/S0952-7915(01)00303-X. [DOI] [PubMed] [Google Scholar]
- 40.Schmidt NW, Mishra A, Lai GH, Davis M, Sanders LK, Tran D, Garcia A, Tai KP, McCray PB, Ouellette AJ, Selsted ME, Wong GC. 2011. Criterion for amino acid composition of defensins and antimicrobial peptides based on geometry of membrane destabilization. J Am Chem Soc 133:6720–6727. doi: 10.1021/ja200079a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt NW, Tai KP, Kamdar K, Mishra A, Lai GH, Zhao K, Ouellette AJ, Wong GC. 2012. Arginine in alpha-defensins: differential effects on bactericidal activity correspond to geometry of membrane curvature generation and peptide-lipid phase behavior. J Biol Chem 287:21866–21872. doi: 10.1074/jbc.M112.358721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zou G, de Leeuw E, Li C, Pazgier M, Zeng P, Lu WY, Lubkowski J, Lu W. 2007. Toward understanding the cationicity of defensins. Arg and Lys versus their noncoded analogs. J Biol Chem 282:19653–19665. doi: 10.1074/jbc.M611003200. [DOI] [PubMed] [Google Scholar]
- 43.Olli S, Rangaraj N, Nagaraj R. 2013. Effect of selectively introducing arginine and d-amino acids on the antimicrobial activity and salt sensitivity in analogs of human beta-defensins. PLoS One 8:e77031. doi: 10.1371/journal.pone.0077031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krishnakumari V, Packiyanathan KK, Nagaraj R. 2013. Human-beta-defensins-1-3 and analogs do not require proton motive force for antibacterial activity against Escherichia coli. FEMS Microbiol Lett 348:52–57. doi: 10.1111/1574-6968.12242. [DOI] [PubMed] [Google Scholar]
- 45.Krishnakumari V, Rangaraj N, Nagaraj R. 2009. Antifungal activities of human beta-defensins HBD-1 to HBD-3 and their C-terminal analogs Phd1 to Phd2. Antimicrob Agents Chemother 53:256–260. doi: 10.1128/AAC.00470-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Narayanan S, Miller WL, McDermott AM. 2003. Expression of human beta-defensins in conjunctival epithelium: relevance to dry eye disease. Invest Ophthalmol Vis Sci 44:3795–3801. doi: 10.1167/iovs.02-1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang LC, Jean D, Proske RJ, Reins RY, McDermott AM. 2007. Ocular surface expression and in vitro activity of antimicrobial peptides. Curr Eye Res 32:595–609. doi: 10.1080/02713680701446653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brandt CR, Akkarawongsa R, Altmann S, Jose G, Kolb AW, Waring AJ, Lehrer RI. 2007. Evaluation of a theta-defensin in a murine model of herpes simplex virus type 1 keratitis. Invest Ophthalmol Vis Sci 48:5118–5124. doi: 10.1167/iovs.07-0302. [DOI] [PubMed] [Google Scholar]
- 49.Lamers RP, Eade CR, Waring AJ, Cole AL, Cole AM. 2011. Characterization of the retrocyclin analogue RC-101 as a preventative of Staphylococcus aureus nasal colonization. Antimicrob Agents Chemother 55:5338–5346. doi: 10.1128/AAC.00619-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tai KP, Kamdar K, Yamaki J, Le VV, Tran D, Tran P, Selsted ME, Ouellette AJ, Wong-Beringer A. 17 December 2013. Microbicidal effects of alpha- and theta-defensins against antibiotic-resistant Staphylococcus aureus and Pseudomonas aeruginosa. Innate Immun doi: 10.1177/1753425913514784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vinion-Dubiel AD, Goldberg JB. 2003. Lipopolysaccharide of Burkholderia cepacia complex. J Endotoxin Res 9:201–213. doi: 10.1177/09680519030090040101. [DOI] [PubMed] [Google Scholar]
- 52.Nguyen LT, Chau JK, Perry NA, de Boer L, Zaat SA, Vogel HJ. 2010. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS One 5:e12684. doi: 10.1371/journal.pone.0012684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goldman RC, White D, Orskov F, Orskov I, Rick PD, Lewis MS, Bhattacharjee AK, Leive L. 1982. A surface polysaccharide of Escherichia coli O111 contains O-antigen and inhibits agglutination of cells by O-antiserum. J Bacteriol 151:1210–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buck M. 1998. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q Rev Biophys 31:297–355. [DOI] [PubMed] [Google Scholar]
- 55.Woody RW. 1995. Circular dichroism. Methods Enzymol 246:34–71. doi: 10.1016/0076-6879(95)46006-3. [DOI] [PubMed] [Google Scholar]
- 56.Shai Y. 1999. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta 1462:55–70. doi: 10.1016/S0005-2736(99)00200-X. [DOI] [PubMed] [Google Scholar]
- 57.Sitaram N, Nagaraj R. 1999. Interaction of antimicrobial peptides with biological and model membranes: structural and charge requirements for activity. Biochim Biophys Acta 1462:29–54. doi: 10.1016/S0005-2736(99)00199-6. [DOI] [PubMed] [Google Scholar]
- 58.Meadows R. 2013. A welcome chink in drug resistance. PLoS Biol 11:e1001693. doi: 10.1371/journal.pbio.1001693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maria-Neto S, Candido Ede S, Rodrigues DR, de Sousa DA, da Silva EM, de Moraes LM, Otero-Gonzalez Ade J, Magalhaes BS, Dias SC, Franco OL. 2012. Deciphering the magainin resistance process of Escherichia coli strains in light of the cytosolic proteome. Antimicrob Agents Chemother 56:1714–1724. doi: 10.1128/AAC.05558-11. [DOI] [PMC free article] [PubMed] [Google Scholar]