

Abstract
Negligible in vivo growth of enterococci and high-level dispersion of data have led to inaccurate estimations of antibiotic pharmacodynamics (PD). Here we improved an in vivo model apt for PD studies by optimizing the in vitro culture conditions for enterococci. The PD of vancomycin (VAN), ampicillin-sulbactam (SAM), and piperacillin-tazobactam (TZP) against enterococci were determined in vivo, comparing the following different conditions of inoculum preparation: aerobiosis, aerobiosis plus mucin, and anaerobiosis plus mucin. Drug exposure was expressed as the ratio of the area under the concentration-time curve for the free, unbound fraction of the drug to the MIC (fAUC/MIC) (VAN) or the time in a 24-h period that the drug concentration for the free, unbound fraction exceeded the MIC under steady-state pharmacokinetic conditions (fT>MIC) (SAM and TZP) and linked to the change in log10 CFU/thigh. Only anaerobiosis plus mucin enhanced the in vivo growth, yielding significant PD parameters with all antibiotics. In conclusion, robust in vivo growth of enterococci was crucial for better determining the PD of tested antibacterial agents, and this was achieved by optimizing the procedure for preparing the inoculum.
INTRODUCTION
Enterococci are commensal organisms in the gastrointestinal tracts of many species, from insects to humans (1), but are also the third leading cause of hospital infections (2, 3). They display intrinsic and acquired resistance to almost all antibiotics in clinical use, and no single agent is able to kill more than 3 log10 CFU/g in vivo (4). Animal models of infection are invaluable tools for anti-infective pharmacology, and numerous in vivo enterococcal models have been used to test old and new drugs, but their validation for assessing the efficacy of antimicrobial agents is frequently not reported (5). The enterococcal endocarditis rabbit model is perhaps used the most, but it is also more expensive, cumbersome, and centered on a tissue with complex pharmacokinetics (i.e., cardiac valves), limiting its relevance for other systemic infections (6). Additionally, the usual lack of a full dose-response curve hinders the determination of pharmacokinetic/pharmacodynamic (PK/PD) indices necessary to translate the results to humans (5, 7). Moreover, the bacterial growth in all enterococcal models is either not quantified (e.g., peritonitis model) (8), poor (∼1 log10 CFU/g), or even negative (e.g., thigh infection) (9, 10), and the variability is high (e.g., rabbit endocarditis models) (11), particularly with vancomycin (VAN)-resistant enterococcus (VRE) strains (12). Negligible growth and high-level dispersion may lead to inaccurate estimations of antibiotic PD, as has been the case with daptomycin (13).
We optimized the in vitro culture conditions for enterococci by using anaerobic incubation and mucin supplementation, aiming to enhance the in vivo growth of vancomycin-susceptible enterococcus (VSE) and VRE strains and to determine its impact on the PD of three antibiotics: VAN, ampicillin-sulbactam (SAM), and piperacillin-tazobactam (TZP).
(Different aspects of this work were presented at the 3rd ASM International Conference on Enterococci and at the 51st and 53rd ICAAC [14–16].)
MATERIALS AND METHODS
Bacterial strains, antibiotics, and susceptibility testing.
The wild-type strains Enterococcus faecalis ATCC 29212 and E. faecium ATCC 19434 and the vancomycin-resistant strains E. faecalis ATCC 51299 (VanB) and E. faecium ATCC 51559 (VanA) were used from vials stored at −70°C. VRE strains were tested after optimizing the experimental conditions with VSE. We determined the MICs of VAN (Vancocin CP; Baxter, Brazil), SAM (Unasyn; Pfizer, Italy), and TZP (Tazocin; Wyeth Lederle, Italy) by broth microdilution following CLSI methods (17). Although the studied strains were beta-lactamase negative, ampicillin (AMP) and piperacillin (PIP) were used as combinations with beta-lactamase inhibitors.
Inoculum preparation.
Strains were stored at −70°C and recovered by two successive streaks on brain heart infusion (BHI) agar (Becton Dickinson) with 5% sheep blood, followed by incubation for 24 h at 37°C under ambient air (aerobic groups) or by placing the plates within a sealed anaerobiosis bag (GazPak EZ; Becton Dickinson) in a 10% CO2 incubator (anaerobic groups). The GazPak EZ system provides an atmosphere with <1% oxygen and >13% CO2 within 24 h. We named this step “phase 0.”
Before in vivo experiments, we standardized the in vitro growth kinetics under aerobic and anaerobic incubation conditions in broth to define the time and optical density at 580 nm (OD580) (Spectro 22H; Labomed Inc., Culver City, CA) required to reach ∼8 log10 CFU/ml in the log phase (data not shown). For all strains, 3 to 5 colonies from phase 0 were suspended in 10 ml of broth, serially diluted 4 times (1:10), and incubated overnight at 37°C, keeping the atmosphere the same as in the previous step (phase 1). The most diluted tube with complete turbidity was selected, diluted further (1:100) in fresh broth, and incubated using the same atmosphere as that from phase 1, until it reached an OD580 corresponding to ∼8.0 log10 CFU/ml (phase 2). The broths used in phases 1 and 2 were BHI (Becton Dickinson) for the aerobic groups and thioglycolate USP (a reducing medium from Oxoid, United Kingdom) for the anaerobic groups. Bacteria from phase 2 were diluted in the corresponding medium to the desired final inoculum size, which varied from 5 to 8 log10 CFU/ml according to the experiment. In some in vivo experiments (described below), the inoculum from phase 2 was supplemented with mucin just before inoculation by mixing autoclaved 10% (wt/vol) porcine stomach mucin (Sigma-Aldrich) with the culture broth (BHI or thioglycolate) in equal volumes, obtaining a final mucin concentration of 5% (18).
Animal model.
The University of Antioquia Animal Experimentation Ethics Committee reviewed and approved the experimental protocol. Six-week-old murine-pathogen-free female mice of the strain Udea:ICR(CD-2), weighing 23 to 27 g, were rendered neutropenic (<100 neutrophils/mm3) by intraperitoneal injection of two doses of cyclophosphamide (Endoxan; Baxter, Germany), given 4 days (150 mg/kg of body weight) and 1 day (100 mg/kg) before infection (19). Sixteen hours later, isoflurane (Isorane; Abbott, Argentina)-anesthetized animals were infected intramuscularly in each thigh with 0.1 ml of the selected inoculum. During the standardization of the model, mice were separated into four experimental groups (n ≥ 10 animals/group) according to the inoculum conditions used, as follows: aerobiosis (nonsupplemented broth), aerobiosis plus mucin (incubated under an aerobic atmosphere in broth supplemented with 5% porcine mucin), anaerobiosis (nonsupplemented broth), and anaerobiosis plus mucin (incubated under an anaerobic atmosphere in broth supplemented with 5% mucin). A minimum of two animals per group were picked from their cages at random and sacrificed immediately after inoculation (0 h), at five or more time points (1, 2, 4, 6, 8, 10, 12, 14, and/or 18 h), and by the end of the model (26 h after infection). Thighs were aseptically removed, homogenized (PRO200 tissue homogenizer; ProScientific), diluted, and automatically plated (DS+ spiral plater; Interscience, France). Bacterial counting (Scan 500 automatic colony counter; Interscience, France) was done after 20 h of incubation at 37°C in ambient air. Earlier experiments showed that there was no significant difference in bacterial counts for thigh samples plated on BHI agar incubated under aerobiosis or anaerobiosis conditions (data not shown). All experiments were performed at least twice. If not specified otherwise, each symbol in all figures represents the geometric mean of results for both thighs from one mouse.
To control for the potential effect of the broth supplement on the animals, a group of 10 neutropenic mice were injected in the thighs with sterile 5% mucin and examined daily for signs of local or systemic toxicity for 10 days.
To elucidate further the contribution of mucin to the development of the model, groups of immunocompetent mice were infected with E. faecalis ATCC 29212 prepared under anaerobiosis or anaerobiosis-plus-mucin conditions. To reduce the number of animals used in this case, the mice were sacrificed only 0, 1, 2, 14, and 26 h after infection.
Impact of culture conditions on systemic dissemination.
To measure the impact of inoculum preparation on systemic dissemination, we sacrificed at least 7 neutropenic mice from two independent experiments to quantify bacterial dissemination to the blood and spleen 26 h after infection with E. faecium ATCC 19434 prepared under conditions of aerobiosis, aerobiosis plus mucin, or anaerobiosis plus mucin. Each spleen was homogenized and diluted to 1 ml in sterile saline before plating; blood (0.1 ml) was plated directly on the agar. The limits of detection were 1 log10 CFU per ml of blood and 2.3 log10 CFU per gram of spleen.
Impact of inoculum conditions on antimicrobial pharmacodynamics.
Neutropenic mice were infected with E. faecium ATCC 19434 (VAN and SAM) or E. faecalis ATCC 29212 (TZP) prepared under conditions of aerobiosis, aerobiosis plus mucin, or anaerobiosis plus mucin. Subcutaneous treatment (0.2 ml) started at 2 h postinfection, allocating 2 mice per dose and encompassing at least 6 doses from no effect to maximal effect. The doses ranged from 2.34 to 600, 18.75 to 2,400, and 18.75 to 4,800 mg/kg/day for VAN, SAM, and TZP, respectively, in all cases divided every 3 h (q3h). At the end of treatment, the thighs of each mouse were dissected aseptically, homogenized independently, serially diluted, plated, and incubated overnight at 37°C. The limit of detection was 100 CFU/thigh, which in this model was equivalent to 2 log10 CFU/g. In addition, we included six untreated control mice per experiment; these received 0.2-ml subcutaneous injections of sterile saline q3h and were sacrificed and processed in the same way as the experimental groups at 3 time points: just after inoculation (−2 h) and at the onset (0 h) and end (24 h) of therapy.
Statistical analysis.
Bacterial counts were stored in an Excel database (Microsoft, Seattle, WA) for subsequent analysis. In vivo growth (G) was established by use of an empirical equation and as a parameter derived from nonlinear regression (NLR). In the former case, the net growth (ΔG26−2) was defined simply by subtraction of the mean number of bacteria observed 2 h after infection from that reached at 26 h (19, 20). In the latter case, in vivo growth-time curves were fitted using an NLR analysis described by a modified Gompertz equation (see Table S1 in the supplemental material) (21). The magnitudes of the parameters derived from this equation were compared by curve-fitting analysis (CFA; Prism 6.04) (22, 23). A successful in vivo model was defined as one that attained the following four criteria: (i) net growth (ΔG26−2) of ≥1.5 log10 CFU/g, (ii) significant parameters derived from Gompertz's equation that fulfilled the homoscedasticity and normality assumptions, (iii) a difference between the maximal and minimal predicted bacterial counts of ≥2 log10 CFU/g (see the Auctus column in Table S1), and (iv) a standard error of the estimate (Sy|x) of ≤0.5 log10. Dissemination data for the blood and spleen at 26 h postinfection were compared by analysis of variance (ANOVA) for normally distributed data (see Fig. S1 in the supplemental material); otherwise, the Kruskal-Wallis (KW) test was used.
For PD data, the net antibacterial effect (dependent variable) of each product's dose (independent variable) was calculated by subtracting the number of CFU/g for untreated controls at 24 h from the number of CFU/g remaining in treated mice. Hill's equation was fitted to these data to describe the sigmoid dose-response relationship and to estimate the primary PD (PDP) parameters maximum effect (Emax), 50% effective dose (ED50), and Hill's slope (N), as we have described elsewhere (23, 24). A parameter was considered invalid if the nonlinear regression failed any of the presumptions of normality, homoscedasticity, or nonmulticolinearity. Additionally, the secondary parameters bacteriostatic dose (BD) and maximal kill dose (MKD) were calculated (23). For PK analysis, we used data from our previous studies of infected ICR mice treated with VAN, piperacillin, and ampicillin (24–26), fitted with Pmetrics by using the nonparametric adaptive grid (NPAG) algorithm, to obtain the population PK parameters (27). For PK/PD analysis, the area under the concentration-time curve for the free, unbound fraction of the drug (fAUC) between 0 h and 24 h (VAN) and the time in a 24-h period that the drug concentration for the free, unbound fraction exceeded the MIC under steady-state pharmacokinetic conditions (fT>MIC) (AMP and PIP) were calculated using ADAPT 5 (28, 29). For these calculations, we used protein binding levels in mice of 29%, 20%, and 3% for VAN, PIP, and AMP, respectively (24, 30, 31). PD parameters were compared by curve-fitting analysis (23).
RESULTS
Impacts of the atmosphere of incubation, mucin supplementation, and immune status on in vivo growth.
Figure 1 illustrates that anaerobiosis-plus-mucin conditions were required with E. faecalis 29212 (∼5 log10 CFU/g) to achieve a successful in vivo model (as defined in Materials and Methods) in neutropenic (Fig. 1a) and immunocompetent (Fig. 1c) mice. The net growth under aerobiosis-plus-mucin conditions was enhanced in neutropenic mice (Fig. 1b) (mean ΔG26−2 = 2.07 log10 CFU/g), but the regression failed the assumption of homoscedasticity (no fit to Gompertz's equation). In immunocompetent mice (Fig. 1c), anaerobiosis without mucin led to exponential decay of the bacterial count (below the limit of detection), with complete clearance of infection after 14 h. Due to the low growth, high error, and high similarity to aerobiosis encountered with the anaerobiosis condition (see Table S1 in the supplemental material), it was not tested anymore.
FIG 1.
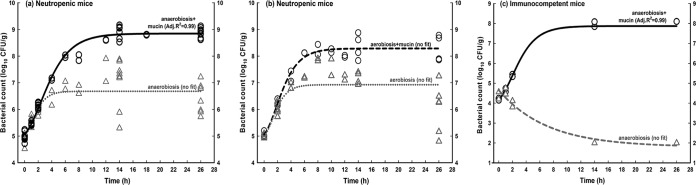
Impacts of atmosphere of incubation, mucin supplementation, and immune status on the in vivo growth of E. faecalis ATCC 29212. In all panels, solid lines represent significant and valid regressions using Gompertz's equation, and dotted lines indicate that the regression did not fit or failed the presumptions of normality or homoscedasticity (see Table S1 in the supplemental material). (a) Growth in neutropenic mice infected with inocula prepared under conditions of anaerobiosis (dotted gray line) or anaerobiosis plus mucin (solid black line). (b) Growth in neutropenic mice infected with inocula prepared under conditions of aerobiosis (dotted gray line) or aerobiosis plus mucin (black dotted line). (c) In vivo growth curves for immunocompetent animals infected with inocula prepared under conditions of anaerobiosis plus mucin (solid black line) or anaerobiosis (gray interrupted line). In panel c, data points at 26 h (black) and 14 and 26 h (gray) overlap.
The comparison of immunocompetent and neutropenic mice for anaerobiosis plus mucin (inoculum of ∼4 log10 CFU per thigh) indicated that neutropenia enhanced the ΔG26−2 by 1.26 log10 CFU/g. The results with other strains are summarized in Table S1 in the supplemental material.
The animals injected with 5% sterile mucin had swelled thighs without signs of systemic illness during the first day, after which the inflammation resolved completely. On day 10 after mucin injection, all mice were alive and healthy.
Impact of mucin on systemic dissemination of E. faecium ATCC 19434.
Taking into account the similar levels of in vivo growth reached in mice inoculated with E. faecium 19434 under anaerobiosis-plus-mucin versus aerobiosis-plus-mucin conditions (see Table S1 in the supplemental material), we compared the levels of dissemination to distant organs 26 h after infection. Higher bacterial burdens in the spleen (P < 0.001 by ANOVA) and blood (P = 0.0002 by KW test) were observed for anaerobiosis plus mucin (see Fig. S1 for details).
Antimicrobial pharmacodynamics.
The MICs of VAN and SAM against E. faecium 19434 were 1 and 2 mg/liter, respectively; against E. faecalis 29212, the MIC of TZP was 2 mg/liter. The PK parameters of VAN, AMP, and PIP on infected neutropenic mice are shown in Table 1, and the PD parameters of VAN, SAM, and TZP obtained using different inoculum conditions are shown in Table 2. In all cases, mucin supplementation led to high in vivo growth (ΔG26−2 ≥ 1.85 log10 CFU/g) and yielded significant PD parameters (adjusted R2 ≥ 0.94). In contrast, the aerobiosis group displayed minimal net growth (ΔG26−2 ≤ 0.41 log10 CFU/g) and did not fit Hill's equation with VAN (adjusted R2 = 0.25) and TZP (adjusted R2 = 0.31), showing erratic PD profiles (Fig. 2a and b). For SAM (adjusted R2 = 0.84), aerobiosis (Fig. 2c) displayed invalid parameters with a high multicolinearity (variance inflation factor [VIF] of >166 for the bacteriostatic dose).
TABLE 1.
Pharmacokinetic parameters in infected mice, derived from a two-compartment model for vancomycin and ampicillin or a one-compartment model for piperacillina
| Parameter | Value |
||
|---|---|---|---|
| Vancomycin | Ampicillin | Piperacillin | |
| kel (h−1) | 1.67 | 20.4 | 3.16 |
| Vc (liters) | 0.01 | 0.01 | 0.04 |
| kcp (h−1) | 3.16 | 78.2 | NA |
| kpc (h−1) | 62.8 | 15.0 | NA |
| ka (h−1) | 2.52 | 6.45 | 7.45 |
Using data from references 24 to 26. Abbreviations: kel, elimination rate constant; Vc, volume of the central compartment; kcp, transfer rate constant from the central to peripheral compartment; kpc, transfer rate constant from the peripheral to central compartment; ka, absorption rate constant; NA, not applicable.
TABLE 2.
Pharmacodynamics of VAN, TZP, and SAM against enterococci under different inoculum conditionsa
| Parameter | Value |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| VAN vs E. faecium 19434 |
TZP vs E. faecalis 29212 |
SAM vs E. faecium 19434 |
|||||||
| Aerobiosis | Aerobiosis + mucin | Anaerobiosis + mucin | Aerobiosis | Aerobiosis + mucin | Anaerobiosis + mucin | Aerobiosis | Aerobiosis + mucin | Anaerobiosis + mucin | |
| Emax (log10 CFU/g) | IVP | 3.34 ± 0.07 | 3.05 ± 0.07 | IVP | 4.37 ± 0.12 | 4.86 ± 0.12 | 3.75 ± 0.26 | 6.36 ± 0.37 | 6.80 ± 0.23 |
| ED50 (mg/kg/day) | IVP | 18.5 ± 0.77 | 26.0 ± 1.62 | IVP | 215 ± 9.94 | 425 ± 19.0 | 122 ± 19.3 | 259 ± 30.9 | 218 ± 10.9 |
| N | IVP | 3.03 ± 0.36 | 5.27 ± 0.91 | IVP | 4.53 ± 0.57 | 3.64 ± 0.41 | 2.18 ± 0.71 | 2.39 ± 0.58 | 3.53 ± 0.46 |
| BD | IVP | 44.3 ± 2.29 | 52.8 ± 2.91 | IVP | 23.5 ± 0.50 | 32.0 ± 0.40 | IVP | 31.3 ± 2.23 | 28.3 ± 0.89 |
| MKD | IVP | IVP | 83.6 ± 9.82 | IVP | 34.2 ± 1.77 | 41.1 ± 1.29 | IVP | IVP | 45.7 ± 2.16 |
Abbreviations: Emax, maximum effect; ED50, 50% effective dose; N, Hill's slope; BD, bacteriostatic dose in terms of fAUC/MIC (VAN) or fT>MIC (SAM and TZP); MKD, the dose (exposure) required for maximal killing of 0.8 log (VAN), 2.3 log (TZP), and 4 log (SAM) bacteria in terms of fAUC/MIC (VAN) or fT>MIC (SAM and TZP). All values are presented as means ± standard errors. IVP, invalid parameter because it was not possible to fit Hill's equation to data or the nonlinear regression failed any of the presumptions of normality, homoscedasticity, and nonmulticolinearity (variance inflation factor of <10).
FIG 2.
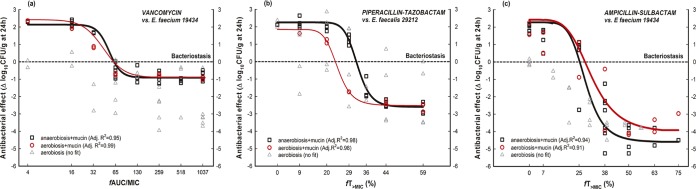
Pharmacodynamics of vancomycin (a), piperacillin-tazobactam (b), and ampicillin-sulbactam (c) in the following groups: aerobiosis (gray triangles and lines), aerobiosis plus mucin (red circles and lines), and anaerobiosis plus mucin (black squares and lines). For the aerobiosis group (gray triangles), no antibiotic fit Hill's equation in terms of fAUC/MIC (VAN) or fT>MIC (SAM and TZP) against E. faecium ATCC 19434 or E. faecalis ATCC 29212.
Regarding the pharmacodynamics of VAN against E. faecium 19434 (Fig. 2a), there were statistically significant differences in the primary parameters (Emax, ED50, and N) between the aerobiosis-plus-mucin and anaerobiosis-plus-mucin groups (P < 0.0001 by global CFA). Moreover, it was not possible to estimate a valid MKD under aerobiosis-plus-mucin conditions due to high multicolinearity (VIF = 30).
Similar to the case for VAN, the PD of TZP against E. faecalis 29212 (Fig. 2b) changed depending on the group tested, and there were significant differences (P < 0.0001 by global CFA) in PD parameters (Table 2) in comparing aerobiosis plus mucin and anaerobiosis plus mucin. Specifically, the magnitude of the exposure required for stasis was significantly lower for aerobiosis plus mucin than for anaerobiosis plus mucin (BD = 23.5% versus 32.0% in terms of fT>MIC).
Finally, SAM was highly bactericidal against E. faecium 19434 (Fig. 2c), and anaerobiosis plus mucin was necessary to calculate the exposure required (in terms of fT>MIC) to kill 4 log bacteria (MKD) (Table 2). In contrast, aerobiosis plus mucin yielded an invalid parameter.
DISCUSSION
At least two problems characterize the available animal models of enterococcal infection: minimal bacterial growth and a high level of data dispersion (8–13). Here we solved these problems by using anaerobiosis plus mucin to attain an optimized thigh infection model suitable for PD studies.
Previously, Safdar et al. evaluated the PD of daptomycin against Staphylococcus aureus, Streptococcus pneumoniae, and E. faecium cultured under aerobic conditions without mucin supplementation (13). They concluded that more studies with strains that grew better were needed because low growth (ΔG26−2 < 0.4 log10 CFU/g) led to overestimation of the drug's efficacy against E. faecium (AUC/MIC for stasis was 0.94 to 1.67) compared to that with S. aureus (388 to 537) or S. pneumoniae (75 to 237) (13). Other authors have obtained similarly biased results (9).
Our data demonstrate that only anaerobiosis-plus-mucin conditions yielded high tissue growth and the excellent fit with significant parameters required to define the exposure-response relationship. In contrast, the use of aerobiosis plus mucin yielded a questionably low magnitude of exposure required for stasis with TZP compared with the values reported for other beta-lactams (∼30% for BD) (32), and in the case of VAN and SAM, it was not possible to estimate the maximum killing dose.
We have several hypotheses that may explain the importance of anaerobiosis plus mucin for attaining a successful in vivo model and that deserve to be tested in the future. Previous studies have found differential gene expression in enterococci according to the incubation atmosphere (33). Day et al. showed that there are three different phenotypic profiles of cytolysin expression in E. faecalis, based on the atmosphere of incubation (34). We found that E. faecalis ATCC 29212 corresponds to Day et al.'s phenotype 2 (data not shown), meaning that beta-hemolysis appears only under an anaerobic atmosphere. In particular, this toxin in enterococci has been characterized as a heat-labile, oxygen-stable molecule (35), suggesting that it is available to induce damage in vivo even after the atmosphere switches. Regarding mucin, it is a heavily glycosylated cysteine-rich protein that covers the epithelium of the gastrointestinal tract and is a potential natural source of amino acids and sugars for enterococci (36–39).
There has been at least one previous attempt to improve the enterococcal thigh infection model. Eguchi et al. supplemented the inoculum with carrageenan, a tissue irritant used in models of inflammatory pain, enhancing net growth (ΔG26−2 = 1.41 log) in neutropenic mice but also increasing the standard deviation (SD = 1.04 log10 CFU/g at 26 h) (40). We think that our model with anaerobiosis plus mucin is better because it enhanced the net growth of susceptible and resistant strains (ΔG26−2 > 1.6 log) (see Table S1 in the supplemental material) without increasing the dispersion of the data (coefficient of variation of <40%, versus 74% for the work of Eguchi et al.). This is a major point, because the power of any statistical method for quantitative variables and the estimation of the sample size are critically dependent on the standard deviation of the data. As a novelty of our approach, mucin enhanced growth even in immunocompetent mice, probably by a protective “coating effect” on bacteria (41).
To our knowledge, this is the first in vivo report of the magnitudes of the PD index fAUC/MIC of VAN against E. faecium necessary for stasis (52.8 ± 2.91) and maximal bacterial killing (83.6 ± 9.82). In fact, the only previous data regarding the in vivo PD of VAN came from a retrospective clinical study, which found that an fAUC/MIC of 87 (adjusted by 30% protein binding) was necessary for a satisfactory clinical outcome of Gram-positive infections (42). The similarity of these magnitudes between mice and humans is remarkable and constitutes a case of predictive validation of our optimized model (5, 43). Although there are no reports of PD indices of TZP and SAM against enterococci, the fT>MIC magnitudes found here were similar to those reported for other penicillins (32, 44). In conclusion, we have validated an animal model, optimized by preparing the inoculum under anaerobic conditions and supplementing it with mucin (5), that is useful for accurately assessing antimicrobial PD against enterococci.
Supplementary Material
ACKNOWLEDGMENTS
This project was funded by the Colombian Government Department of Science, Technology and Innovation (Colciencias) (grant 111540820499) and by the University of Antioquia (Estrategia de Sostenibilidad 2013–2014).
We do not have conflicts of interest.
We thank Jefferson Perez and Alexis Santamaria for animal care support and for participation during some of the experimental procedures.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02352-13.
REFERENCES
- 1.Gilmore MS, Coburn PS, Nallapareddy SR, Murray BE. 2002. Enterococcal virulence, p 301–354. In Gilmore MS. (ed), The enterococci: pathogenesis, molecular biology, and antibiotic resistance. ASM Press, Washington, DC. [Google Scholar]
- 2.Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK, National Healthcare Safety Network Team, Participating National Healthcare Safety Network Facilities . 2008. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect Control Hosp Epidemiol 29:996–1011. doi: 10.1086/591861. [DOI] [PubMed] [Google Scholar]
- 3.Huycke MM. 2002. Physiology of enterococci, p 133–175. In Gilmore MS. (ed), The enterococci: pathogenesis, molecular biology, and antibiotic resistance. ASM Press, Washington, DC. [Google Scholar]
- 4.Houlihan HH, Stokes DP, Rybak MJ. 2000. Pharmacodynamics of vancomycin and ampicillin alone and in combination with gentamicin once daily or thrice daily against Enterococcus faecalis in an in vitro infection model. J Antimicrob Chemother 46:79–86. doi: 10.1093/jac/46.1.79. [DOI] [PubMed] [Google Scholar]
- 5.Zuluaga AF, Rodriguez CA, Agudelo M, Vesga O. 2014. About the validation of animal models to study the pharmacodynamics of generic antimicrobials. Clin Infect Dis 59:459–461. doi: 10.1093/cid/ciu306. [DOI] [PubMed] [Google Scholar]
- 6.Fantin B, Leclercq R, Arthur M, Duval J, Carbon C. 1991. Influence of low-level resistance to vancomycin on efficacy of teicoplanin and vancomycin for treatment of experimental endocarditis due to Enterococcus faecium. Antimicrob Agents Chemother 35:1570–1575. doi: 10.1128/AAC.35.8.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sande MA. 1981. Evaluation of antimicrobial agents in the rabbit model of endocarditis. Rev Infect Dis 3(Suppl):S240–S249. [PubMed] [Google Scholar]
- 8.Arias CA, Singh KV, Panesso D, Murray BE. 2007. Evaluation of ceftobiprole medocaril against Enterococcus faecalis in a mouse peritonitis model. J Antimicrob Chemother 60:594–598. doi: 10.1093/jac/dkm237. [DOI] [PubMed] [Google Scholar]
- 9.Dandekar PK, Tessier PR, Williams P, Nightingale CH, Nicolau DP. 2003. Pharmacodynamic profile of daptomycin against Enterococcus species and methicillin-resistant Staphylococcus aureus in a murine thigh infection model. J Antimicrob Chemother 52:405–411. doi: 10.1093/jac/dkg337. [DOI] [PubMed] [Google Scholar]
- 10.Griffith DC, Rodriguez D, Corcoran E, Dudley MN. 2008. Pharmacodynamics of RWJ-54428 against Staphylococcus aureus, Streptococcus pneumoniae, and Enterococcus faecalis in a neutropenic mouse thigh infection model. Antimicrob Agents Chemother 52:244–247. doi: 10.1128/AAC.00776-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lefort A, Lafaurie M, Massias L, Petegnief Y, Saleh-Mghir A, Muller-Serieys C, Le Guludec D, Fantin B. 2003. Activity and diffusion of tigecycline (GAR-936) in experimental enterococcal endocarditis. Antimicrob Agents Chemother 47:216–222. doi: 10.1128/AAC.47.1.216-222.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vouillamoz J, Moreillon P, Giddey M, Entenza JM. 2006. Efficacy of daptomycin in the treatment of experimental endocarditis due to susceptible and multidrug-resistant enterococci. J Antimicrob Chemother 58:1208–1214. doi: 10.1093/jac/dkl406. [DOI] [PubMed] [Google Scholar]
- 13.Safdar N, Andes D, Craig WA. 2004. In vivo pharmacodynamic activity of daptomycin. Antimicrob Agents Chemother 48:63–68. doi: 10.1128/AAC.48.1.63-68.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santamaria A, Rodriguez CA, Agudelo M, Vesga O, Zuluaga AF. 2010. Abstr 3rd Int ASM Conf Enterococci, Portland, Oregon, p 33–34. [Google Scholar]
- 15.Zuluaga AF, Agudelo M, Perez JA, Gonzalez JM, Rodriguez CA, Vesga O. 2011. Successful models of thigh infection in neutropenic mice by vancomycin-susceptible (VSE) and -resistant enterococci (VRE), abstr B-056. Abstr 51st Intersci Conf Antimicrob Agents Chemother, Chicago, IL ASM Press, Washington, DC. [Google Scholar]
- 16.Zuluaga AF, Rodriguez CA, Perez JA, Gonzalez JM, Vesga O. 2013. Redefining the pharmacodynamics of three antienterococcal drugs in the optimized neutropenic mouse thigh infection model, abstr A-018. Abstr 53rd Intersci Conf Antimicrob Agents Chemother, Denver, CO ASM Press, Washington, DC. [Google Scholar]
- 17.CLSI. 2013. Performance standards for antimicrobial susceptibility testing: twenty-third informational supplement. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 18.Lorian V. 2005. Antibiotics in laboratory medicine, 5th ed, p 670 Lippincott William & Wilkins, Philadelphia, PA. [Google Scholar]
- 19.Zuluaga AF, Salazar BE, Rodriguez CA, Zapata AX, Agudelo M, Vesga O. 2006. Neutropenia induced in outbred mice by a simplified low-dose cyclophosphamide regimen: characterization and applicability to diverse experimental models of infectious diseases. BMC Infect Dis 6:55. doi: 10.1186/1471-2334-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gudmunsson S, Erlensdóttir H. 1999. Murine thigh infection model, p 137–144. In Zak O, Sande MA (ed), Handbook of animal models of infection. Academic Press, San Diego, CA. [Google Scholar]
- 21.Zwietering MH, Jongenburger I, Rombouts FM, van't Riet K. 1990. Modeling of the bacterial growth curve. Appl Environ Microbiol 56:1875–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glantz SA. 2005. Primer of biostatistics, 6th ed. McGraw-Hill Medical Publishing, New York, NY. [Google Scholar]
- 23.Zuluaga AF, Agudelo M, Cardeno JJ, Rodriguez CA, Vesga O. 2010. Determination of therapeutic equivalence of generic products of gentamicin in the neutropenic mouse thigh infection model. PLoS One 5:e10744. doi: 10.1371/journal.pone.0010744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vesga O, Agudelo M, Salazar BE, Rodriguez CA, Zuluaga AF. 2010. Generic vancomycin products fail in vivo despite being pharmaceutical equivalents of the innovator. Antimicrob Agents Chemother 54:3271–3279. doi: 10.1128/AAC.01044-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez CA, Agudelo M, Gonzalez M, Zuluaga AF, Vesga O. 2013. Generic piperacillin-tazobactam (TZP) enriches resistant subpopulations of Escherichia coli after exposure in the neutropenic mouse thigh infection model (NMTIM), abstr A-479. Abstr 53rd Intersci Conf Antimicrob Agents Chemother, Denver, CO ASM Press, Washington, DC. [Google Scholar]
- 26.Zuluaga AF, Salazar BE, Loaiza SA, Agudelo M, Vesga O. 2004. Therapeutic equivalence with the original compound of 8 generic products of ampicillin determined in the neutropenic murine thigh infection model, abstr E-2033. Abstr 44th Intersci Conf Antimicrob Agents Chemother American Society for Microbiology, Washington, DC. [Google Scholar]
- 27.Neely MN, van Guilder MG, Yamada WM, Schumitzky A, Jelliffe RW. 2012. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit 34:467–476. doi: 10.1097/FTD.0b013e31825c4ba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Argenio DZ, Schumitzky A, Wang X. 2009. ADAPT 5 user's guide: pharmacokinetic/pharmacodynamic systems analysis software. Biomedical Simulations Resource, Los Angeles, CA. [Google Scholar]
- 29.Louie A, Grasso C, Bahniuk N, Van Scoy B, Brown DL, Kulawy R, Drusano GL. 2010. The combination of meropenem and levofloxacin is synergistic with respect to both Pseudomonas aeruginosa kill rate and resistance suppression. Antimicrob Agents Chemother 54:2646–2654. doi: 10.1128/AAC.00065-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Ogtrop ML, Mattie H, Sekh BR, van Strijen E, van Furth R. 1992. Comparison of the antibacterial efficacies of ampicillin and ciprofloxacin against experimental infections with Listeria monocytogenes in hydrocortisone-treated mice. Antimicrob Agents Chemother 36:2375–2380. doi: 10.1128/AAC.36.11.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bulik CC, Tessier PR, Keel RA, Sutherland CA, Nicolau DP. 2012. In vivo comparison of CXA-101 (FR264205) with and without tazobactam versus piperacillin-tazobactam using human simulated exposures against phenotypically diverse Gram-negative organisms. Antimicrob Agents Chemother 56:544–549. doi: 10.1128/AAC.01752-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Craig WA. 2003. Basic pharmacodynamics of antibacterials with clinical applications to the use of beta-lactams, glycopeptides, and linezolid. Infect Dis Clin North Am 17:479–501. doi: 10.1016/S0891-5520(03)00065-5. [DOI] [PubMed] [Google Scholar]
- 33.Shepard BD, Gilmore MS. 1999. Identification of aerobically and anaerobically induced genes in Enterococcus faecalis by random arbitrarily primed PCR. Appl Environ Microbiol 65:1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Day AM, Cove JH, Phillips-Jones MK. 2003. Cytolysin gene expression in Enterococcus faecalis is regulated in response to aerobiosis conditions. Mol Genet Genomics 269:31–39. doi: 10.1007/s00438-003-0819-1. [DOI] [PubMed] [Google Scholar]
- 35.Van Tyne D, Martin MJ, Gilmore MS. 2013. Structure, function, and biology of the Enterococcus faecalis cytolysin. Toxins 5:895–911. doi: 10.3390/toxins5050895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pultz NJ, Hoskins LC, Donskey CJ. 2006. Vancomycin-resistant enterococci may obtain nutritional support by scavenging carbohydrate fragments generated during mucin degradation by the anaerobic microbiota of the colon. Microb Drug Resist 12:63–67. doi: 10.1089/mdr.2006.12.63. [DOI] [PubMed] [Google Scholar]
- 37.Corfield AP, Wagner SA, Clamp JR, Kriaris MS, Hoskins LC. 1992. Mucin degradation in the human colon: production of sialidase, sialate O-acetylesterase, N-acetylneuraminate lyase, arylesterase, and glycosulfatase activities by strains of fecal bacteria. Infect Immun 60:3971–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monti E, Bonten E, D'Azzo A, Bresciani R, Venerando B, Borsani G, Schauer R, Tettamanti G. 2010. Sialidases in vertebrates: a family of enzymes tailored for several cell functions. Adv Carbohydr Chem Biochem 64:403–479. doi: 10.1016/S0065-2318(10)64007-3. [DOI] [PubMed] [Google Scholar]
- 39.Monti E, Preti A, Venerando B, Borsani G. 2002. Recent development in mammalian sialidase molecular biology. Neurochem Res 27:649–663. doi: 10.1023/A:1020276000901. [DOI] [PubMed] [Google Scholar]
- 40.Eguchi K, Kanazawa K, Eriguchi Y, Ueda Y. 2009. Pharmacodynamics of SMP-601 (PTZ601) against vancomycin-resistant Enterococcus faecium and methicillin-resistant Staphylococcus aureus in neutropenic murine thigh infection models. Antimicrob Agents Chemother 53:3391–3398. doi: 10.1128/AAC.00972-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olitzki L. 1948. Mucin as a resistance-lowering substance. Bacteriol Rev 12:149–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hyatt JM, McKinnon PS, Zimmer GS, Schentag JJ. 1995. The importance of pharmacokinetic/pharmacodynamic surrogate markers to outcome. Focus on antibacterial agents. Clin Pharmacokinet 28:143–160. [DOI] [PubMed] [Google Scholar]
- 43.Varga OE, Hansen AK, Sandoe P, Olsson IA. 2010. Validating animal models for preclinical research: a scientific and ethical discussion. Altern Lab Anim 38:245–248. [DOI] [PubMed] [Google Scholar]
- 44.Craig WA. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis 26:1–10. doi: 10.1086/516284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.