

Abstract
BC-3781, a pleuromutilin antimicrobial agent, is being developed for the treatment of patients with acute bacterial skin and skin structure infections (ABSSSI) and community-acquired bacterial pneumonia. Data from a phase 2 study of patients with ABSSSI were used to refine a previous population pharmacokinetic (PK) model and explore potential predictors of PK variability. The previously derived population PK model based on data from three phase 1 studies was applied to sparse sampling data from a phase 2 ABSSSI study and modified as necessary. Covariate analyses were conducted to identify descriptors (e.g., body size, renal function, age) associated with interindividual variability in PK. All population PK analyses were conducted by using Monte Carlo parametric expectation maximization implemented in S-ADAPT 1.5.6. The population PK data set contained 1,167 concentrations from 129 patients; 95% of the patients had 5 or more PK samples (median, 11). The previous population PK model (three-compartment model with first-order elimination and nonlinear protein binding) provided an acceptable and unbiased fit to the data from the 129 patients. Population PK parameters were estimated with acceptable precision; individual clearance values were particularly well estimated (median individual precision of 9.15%). Graphical covariate evaluations showed no relationships between PK and age or renal function but modest relationships between body size and clearance and volume of distribution, which were not statistically significant when included in the population PK model. This population PK model will be useful for subsequent PK-pharmacodynamic analyses and simulations conducted to support phase 3 dose selection. (This study has been registered at ClinicalTrials.gov under registration no. NCT01119105.)
INTRODUCTION
BC-3781 is a pleuromutilin antimicrobial agent that demonstrates very good in vitro microbiological activity against a wide range of bacterial pathogens, including the most common skin pathogens, such as Staphylococcus aureus (including methicillin-resistant strains), Streptococcus pyogenes, and Streptococcus agalactiae (1). BC-3781 is also active against respiratory pathogens such as Streptococcus pneumoniae, Haemophilus influenzae, Legionella pneumophila, Moraxella catarrhalis, Chlamydophila pneumoniae, and Mycoplasma pneumoniae (2). Given the in vitro spectrum of microbiological activity, BC-3781 is initially being developed for the treatment of acute bacterial skin and skin structure infections (ABSSSI). The routes of administration that are being developed include the oral and intravenous (i.v.) routes, which will potentially allow an i.v.-to-oral switch (i.e., step-down therapy).
Data from three phase 1 clinical studies, a single-dose escalation study (EudraCT number 2009-010318-31), a single- and multiple-dose study (EudraCT number 2009-014053-33), and a third study examining the effects of gender and age on the pharmacokinetics (PK) of BC-3781 (IND number 106594), have been used to construct a population PK model describing the disposition of BC-3781 in healthy subjects (3). The previous population PK analysis data set contained 1,677 PK samples collected from 66 subjects. The most robust fit to the data was obtained by using a three-compartment model with zero-order infusion and first-order (linear) elimination. The nonlinearity of protein binding observed in an in vitro protein binding (equilibrium dialysis) study in which the mean (standard deviation [SD]) percentage unbound increased from 12.1 (0.3)% at 1 μg/ml to 17.1 (1.7)% at 3 μg/ml to 27.3 (6.1)% at 10 μg/ml (the highest concentration tested) was incorporated into the model, as it provided a statistically significant improvement of the fit (compared to the model without nonlinear protein binding). Overall, this model fit the individual plasma concentration data with a high degree of precision and minimal bias. This phase 1 population PK model was used, in combination with nonclinical PK-pharmacodynamic (PK-PD) targets, to conduct Monte Carlo simulations to aid in dose selection and to define the optimal, sparse PK sampling scheme employed in a phase 2 study of patients with ABSSSI (NAB-BC-3781-2001, study 2001).
This report describes the population PK analysis conducted for BC-3781 by using data from study 2001. The primary aim of this analysis was to describe individual PK exposures in patients with ABSSSI. A covariate analysis was also undertaken to identify those patient characteristics associated with the interindividual variability of BC-3781 PK.
MATERIALS AND METHODS
PK data collection and assay.
Data were obtained from study 2001, a phase 2 clinical study of patients with ABSSSI. Patients were randomized to receive one of two doses of BC-3781 (100 mg i.v. twice daily [q12h] or 150 mg i.v. q12h) or vancomycin (1 g i.v. q12h, adjusted individually according to institution guidelines) for 5 to 14 days. BC-3781 infusions were to be given over 2 h; actual infusion durations were used in the creation of the population PK data set. Two hundred ten patients were randomized, with approximately 70 patients per treatment group. Only those patients randomized to receive BC-3781 were included in the population PK analysis.
Subject demographic and disease characteristics collected prior to the administration of the study drug were used to characterize the analysis population and to evaluate their ability to explain a portion of the interindividual variability of selected PK parameters. Demographic information included age, weight, body surface area (BSA), body mass index (BMI), sex, and race. The only laboratory information included in the analysis was serum creatinine (to calculate creatinine clearance [CLCR]) and serum albumin. BSA was calculated by using the method of Gehan and George (4). The BMI was calculated as height in centimeters divided by weight in kilograms squared. CLCR was calculated from the baseline serum creatinine level, age, and body weight by using the Cockcroft and Gault equation (5) and then normalized to BSA.
The PK sampling strategy used was selected by using optimal sampling theory based on the phase 1 population PK model for BC-3781 (3). Plasma PK samples were to be collected as follows: on day 1, at 2, 3 to 5, and 8 to 12 h after the start of the infusion; on day 5 and the final treatment day, before and 2, 4 to 6, and 10 to 12 h after the start of the infusion.
The determination of BC-3781 in 1,423 human plasma samples was performed by means of liquid chromatography with tandem mass spectrometric detection. The method was validated according to the FDA's Guidance for Industry on Bioanalytical Method Validation (6) and covered a calibration range of 1.00 ng/ml, the lower limit of quantitation (LLOQ), to 1,000 ng/liter, the upper limit of quantitation (ULOQ). Samples exceeding the ULOQ were diluted with blank plasma and reassayed. The predefined acceptance criteria regarding accuracy (85.0 to 115%; 80.0 to 120% at the LLOQ) and precision (15.0%; 20.0% at the LLOQ) were met during prestudy validation. Batch acceptance criteria, as detailed in the FDA Guidance for Industry on Bioanalytical Method Validation (6), were met for all of the batches reported.
General data handling.
The actual dates and times of dose administration and PK sample collection were used in the construction of the population PK data set. An outlier was defined as an aberrant observation that substantially deviated from the rest of the observations within an individual. Outliers were excluded from this analysis, given the potential for these observations to negatively impact the convergence and/or parameter estimates (7). Suspected outlier observations were tested and, if justified, excluded. Concentrations in the data set that were below the limit of quantitation (BLQ) were flagged. The population analysis program then applied the Beal M3 method (8), such that the algorithm considered this BLQ value as a normally distributed, random value somewhere between negative infinity and the limit of quantification.
Population PK analysis methods.
Candidate PK models were fitted to the BC-3781 plasma concentration-time data by using Monte Carlo parametric expectation maximization as implemented in the open-source software program S-ADAPT (9, 10, 11). S-ADAPT analyses were performed on a Windows operating system and compiled using Intel FORTRAN 9.1. The model selection criteria used to discriminate between candidate PK models included (i) evaluation of individual and population mean parameter estimates and their precision (standard error of the mean [SEM]), (ii) graphical examination of standard goodness-of-fit plots and plots of the observed versus individual predicted concentrations, (iii) reduction in both residual variability and interindividual variability, and (iv) comparison of objective function for nested models or the Akaike information criterion (12) for either nested or nonnested models.
Given the previous population PK analysis for BC-3781 (3), model development focused on three-compartment models with zero-order input and first-order elimination. In addition to a model incorporating saturable protein binding, a model was attempted in which total drug concentrations were used. Interindividual variability was estimated for each structural population PK model parameter, where possible, by using an exponential-error model. Residual variability represents a composite of assay variability, intrasubject variability, model misspecification, errors in the timing of dose administration or PK sample collection, subject noncompliance, and other unexplained errors and was initially described by using an additive plus proportional coefficient of variation (CV) error model. Other models for residual variability were to be explored as necessary.
Covariate model development.
The structural population PK model for BC-3781 was utilized to assess the ability of the selected subject covariates to explain a portion of the interindividual variability in the total clearance and primary volume of distribution parameters. Bayesian estimates of the clearance and volume of distribution parameters were obtained, and plots of the individual parameters versus each of the subject covariates were examined for observable trends. Potential functional forms of the relationship between the PK parameter and the covariate were assessed.
A forward-stepwise approach was used to construct the covariate models in S-ADAPT. Each parameter-covariate pair showing a visually apparent relationship was added, one at a time, to the model. That covariate for which the addition resulted in the largest significant improvement in the objective function (α = 0.05) was retained, provided that (i) the model-fitting procedure minimized successfully, (ii) there was a decrease in interindividual variability for the parameter on which the covariate was added; and (iii) there was not a substantial compensatory increase in interindividual variability for the other PK parameters.
The resulting model was used for the next step. The process was then repeated until none of the remaining covariates provided an improvement in the objective function when added to the model. This full multivariate model was then subjected to a backward elimination procedure in which the criteria for retention in the model were made more stringent (α = 0.001 or an increase in the value of the objective function of >10.83 units after the removal of that covariate).
The reduced multivariable model, including all significant covariates, was then evaluated for any remaining biases with respect to the structural and residual variability models. The model was also checked for possible simplifications such as power functions that could be reduced to linear functions (e.g., when power is approximately equal to 1) or significant categorical covariates that could be redefined by using fewer groups (e.g., race categories of Caucasian, black, and Hispanic changed to Caucasian and other).
Calculation of PK exposure estimates.
PK exposure estimates were calculated for each patient by simulating a concentration-time profile by using the final population PK model. In this simulation, the dosing history and post hoc PK parameter estimates for each patient were used to generate a PK profile (both free- and total-drug levels) from 0 to 24 h postdose after the first dose of BC-3781. The area under the concentration-time curve from 0 to 24 h (AUC0–24) was calculated by integrating the PK profile over time. Cmax (maximum concentration of drug in serum) values were obtained by simulating the concentration immediately following the end of the infusion.
RESULTS
Analysis data.
There were 141 patients with ABSSSI randomized to receive BC-3781 in study 2001; of these, the 10 patients enrolled from site 20 were excluded because of consistent, irreconcilable issues with sample documentation. Of the remaining 131 patients, 1 was excluded for having only one PK sample (patient 014-003) and a second was excluded because of an inability to reconcile obvious issues with sample timing, which resulted in an inability to fit the model to the patient's aberrant concentration-time profile. These two patients accounted for 9 of the 40 outlier samples excluded from the analysis; the remaining 31 samples were removed for various reasons. The final analysis data set contained 129 patients and 1,167 samples. Sixty-four patients had received 100 mg q12h, and 65 had received 150 mg q12h. All of the subjects had at least two PK samples, and the majority had six or more. Samples were spread throughout the postdose intervals in a manner consistent with the intended sampling schedule.
The demographic characteristics were relatively consistent between the patients in the two dose groups, as shown in Table 1. The analysis population was predominantly male (66.7%) and Caucasian (75.2%) and had normal renal function (mean CLCR of 118 ml/min/1.73 m2). The mean age was 41.6 years, and the ages ranged from 18 to 73 years. The mean weight was 92.3 kg, and the weights ranged from 43.8 to 161 kg.
TABLE 1.
Subject demographic characteristics of the PK analysis population
| Variable | 100-mg group (n = 64) |
150-mg group (n = 65) |
All patients (n = 129) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| No. (%) | Mean no. (CV [%]) | Median (min-max) | No. (%) | Mean no. (CV [%]) | Median (min-max) | No. (%) | Mean no. (CV [%]) | Median (min-max) | |
| Age (yr) | 41.3 (12.7) | 41.0 (18.0–73.0) | 42.0 (13.4) | 42.0 (20.0–73.0) | 41.6 (13.0) | 41.0 (18.0–73.0) | |||
| Wt (kg) | 89.0 (20.6) | 85.1 (43.8–146) | 95.6 (23.4) | 89.1 (46.4–161) | 92.3 (22.2) | 87.5 (43.8–161) | |||
| Ht (cm) | 173 (10.5) | 174 (152–191) | 173 (9.70) | 173 (150–196) | 173 (10.0) | 173 (150–196) | |||
| BSA (m2) | 2.07 (0.258) | 2.04 (1.51–2.71) | 2.15 (0.286) | 2.10 (1.41–2.81) | 2.11 (0.274) | 2.05 (1.41–2.81) | |||
| BMI (kg/m2) | 30.0 (7.53) | 29.2 (12.1–53.7) | 32.1 (7.82) | 31.4 (19.4–55.5) | 31.1 (7.73) | 30.3 (12.1–55.5) | |||
| CLCR (ml/min/1.73 m2) | 122 (33.3) | 118 (49.6–207) | 114 (32.5) | 113 (35.6–181) | 118 (33.0) | 117 (35.6–207) | |||
| Serum albumin level (g/liter) | 42.1 (4.62) | 42.0 (28.0–52.0) | 41.6 (4.47) | 42.0 (29.0–50.0) | 41.9 (4.54) | 42.0 (28.0–52.0) | |||
| No. (%) of males | 43 (67.2) | 43 (66.2) | 86 (66.7) | ||||||
| No. (%) of females | 21 (32.8) | 22 (33.9) | 43 (33.3) | ||||||
| No. (%) of Caucasians | 52 (81.3) | 45 (69.2) | 97 (75.2) | ||||||
| No. (%) of blacks | 7 (10.9) | 13 (20.0) | 20 (15.5) | ||||||
| No. (%) of Asians | 0 | 1 (1.54) | 1 (0.775) | ||||||
| No. (%) of Native American/Alaskan Natives | 2 (3.13) | 4 (6.15) | 6 (4.65) | ||||||
| No. (%) of Native Hawaiians/Pacific Islanders | 2 (3.13) | 2 (3.08) | 4 (3.10) | ||||||
| No. (%) of mixed race | 1 (1.56) | 0 | 1 (0.775) | ||||||
Semilog scatterplots of plasma BC-3781 concentrations versus time since the start of the last dose, stratified by dosage regimen, are provided in Fig. 1. In general, concentrations appeared to increase proportionally with increasing doses despite some overlap in the distribution of concentrations between doses. Smoothers through the data from each visit suggested that the data were consistent with a three-compartment PK model, similar to that which was observed for the phase 1 data (3).
FIG 1.

Semilog scatterplots of plasma drug concentrations versus time since the start of the last dose, stratified by study visit and separated by dose.
Structural model development.
Initial structural model development involved the fitting of two population PK models to the data from study 2001, i.e., the final model from the previous analysis, which included nonlinear protein binding, and a simplified model in which total drug concentrations were modeled. It is important to note that it was necessary to fix the parameters that defined the nonlinear protein binding process to those values obtained from the in vitro protein binding study. Although it resulted in improved fits to the data, allowing these parameters to be estimated by the total drug PK profiles resulted in free-fraction estimates well above those observed in vitro (25 to 30% free as opposed to the 12 to 15% observed in vitro at clinically relevant concentrations [data on file, Nabriva Therapeutics]). Although it is theoretically possible that the dynamics of the in vivo protein binding results in significantly lower plasma protein binding than that observed from the static in vitro equilibrium dialysis, the real-time free-drug concentrations cannot be measured in vivo and thus there are no data to support the larger free-drug fraction.
Ultimately, the model incorporating nonlinear protein binding was deemed most appropriate for the following reasons. (i) The minimum value of the objective function was lower for the nonlinear protein binding model. Despite the addition of two PK parameters, the drop in the objective function (8 units) represents a statistically significant improvement in fit (P < 0.05). (ii) In general, the precision of the population PK parameters was higher. (iii) The overall goodness of fit was improved, both on an individual basis and on a population basis. (iv) This model allowed the prediction of free-drug concentrations, which can be important for the appropriate conduct of PK-PD target attainment analyses when translating preclinical PK-PD targets between species.
Given that robust estimates of the population PK parameters were obtained by using the phase 2 PK data alone, it was not necessary to pool the phase 2 data with those from the phase 1 studies.
Covariate model development.
The covariate screening plots shown in Fig. 2 revealed that no strong relationships between patient descriptors and primary PK parameters (total clearance of free drug [CLtu], volume of the central compartment for free drug [Vcu], and steady-state volume of distribution of free drug [Vssu]) were evident. The only relationships of potential significance were those between CLtu and body size (BSA and weight). Several models were attempted in S-ADAPT to test their statistical significance. Although the addition of these relationships resulted in a slight lowering of the interindividual variability of CLtu and Vcu, they did so by reducing the overall goodness of fit (exhibited by the increase in the objective function in each case). Since none of these models met the statistical criteria for inclusion of covariate relationships and the improvements in the interindividual variability of CLtu were modest (10 to 15%), no covariate relationships were added to the population PK model.
FIG 2.

Covariate screening plots. Abbreviations: AGEY, age in years; WTKG, weight in kilograms; HTCM, height in centimeters; ALB, serum albumin concentration in grams per deciliter; BSA, body surface area in square meters; BMI, body mass index in kilograms per square meter; CLCR, creatinine clearance in milliliters per minute per 1.73 m2; CLtu, total clearance of free drug; Vcu, volume of the central compartment for free drug; Vssu, steady-state volume of distribution of free drug.
Final population PK model.
The final population PK model for the data from study 2001 was a three-compartment model with zero-order infusion and first-order (linear) elimination. Saturable (i.e., nonlinear) protein binding was implemented in the population PK model by modeling free-drug concentrations and converting fitted free-drug concentrations back to total-drug values by using the principles of mass balance and a nonlinear relationship between free- and total-drug concentrations.
The population PK parameter estimates and associated standard errors for the model are provided in Table 2. Note that all of the parameters are scaled to free-drug concentrations; for example, the total clearance estimate (CLtu) of 120 liters/h represents the clearance of the free drug and is therefore significantly higher than the CLt estimate from the model that did not contain saturable protein binding (∼18 liters/h). In general, the magnitude of the interindividual variability was relatively low, with the exception of Vcu, which was quite variable (CV of 139%), likely because of the fact that the PK sampling scheme was designed to maximize the ability to estimate clearance while sacrificing the ability to estimate volume parameters. The goodness-of-fit plots for this model are provided in Fig. 3. Excellent fits to the data were obtained, as evidenced by the overall coefficient of determination of 0.892 based on observed versus individual fitted concentrations (upper left panel). In general, the other plots show consistent scatter about zero, indicating that there were no significant biases in the fit of the data.
TABLE 2.
Final population PK model parameter estimates and standard errors
| Parametera | Final estimated mean value (% SEM) |
|
|---|---|---|
| Population mean | Magnitude of interindividual variability (CV %) | |
| Vcu (liters) | 66.1 (18.6) | 139 (29.4) |
| Vp1u (liters) | 582 (7.51) | 48.0 (17.3) |
| CLd1u (liters/h) | 60.8 (11.0) | 35.1 (30.4) |
| Vp2u (liters) | 211 (10.2) | 64.5 (19.0) |
| CLd2u (liters/h) | 199 (10.1) | 39.3 (25.8) |
| CLtu (liters/h) | 120 (4.04) | 17.8 (14.4) |
| Bmax | 12.7 | |
| KD | 2.07 | |
| SDin | 0.0005 | |
| SDsl | 0.217 (2.62) | |
Volumes and clearances are scaled to free-drug concentrations. The minimum value of the objective function is −1,888. Vp1u, volume of distribution of the first peripheral compartment; Vp2u, volume of distribution of the second peripheral compartment; CLd2u, second distributional clearance; Bmax, parameter describing the maximum extent of protein binding; SDin, intercept (additive) term for residual variability model for plasma concentrations; SDsl, slope (proportional) term for residual variability model.
FIG 3.

Goodness-of-fit plots for the final population PK model. Obs., observed. r2, overall coefficient of determination.
The precision of the mean PK parameters was universally high, supported by percent SEM values that were all below 20%. The precision of the interindividual variability estimates was also excellent (maximum of 30% for the first distributional clearance [CLd1u]). The intraindividual variability, which is approximated by the slope (proportional) term for the residual variability model (SDsl), was approximately 22%.
To assess the accuracy of drug exposure estimates, the precision of the individual estimates of CLtu was examined. Given that the mathematical algorithms used to fit the population PK model result in a distribution of not only the mean population PK parameters but also distributions of likely PK parameters for the individual patients, individual percent CV estimates were reflective of the precision of the individual clearance estimates and therefore the predicted AUC estimates. As shown in Fig. 4, the individual percent CV estimates for CLtu were all relatively low (median of 9.15, maximum of 25%), indicating that the precision of the individual estimates of clearance and, by inference, the AUC was high.
FIG 4.
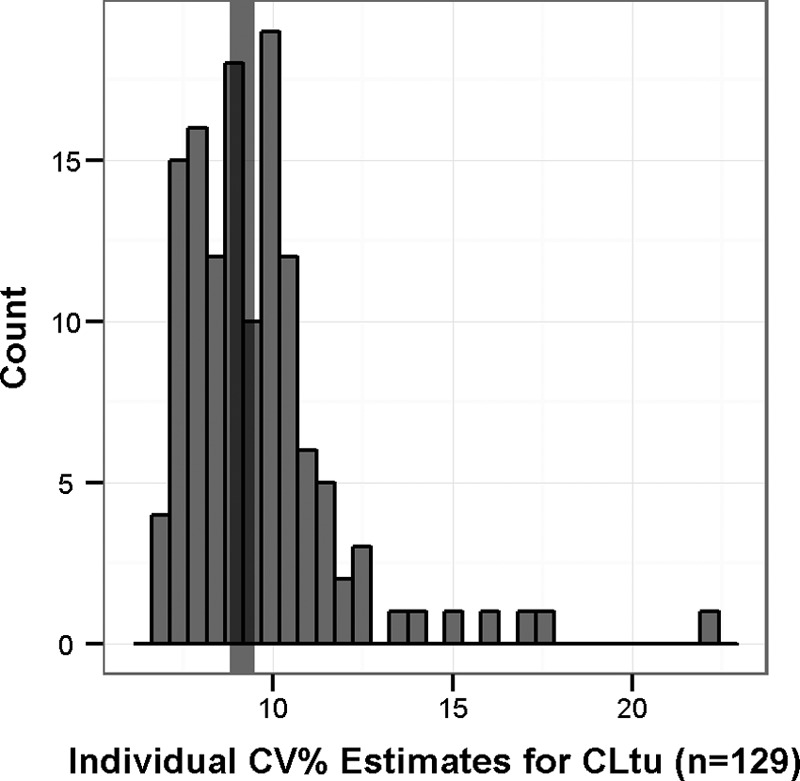
Precision of the individual clearance estimates from the final population PK model. The vertical band represents the median value (9.15%).
Exposure estimates and secondary PK parameters.
Summary statistics for the fitted PK exposure estimates by dose group are provided in Table 3. While the medians differ as expected between groups, there was a modest degree of overlap in AUC0–24 values between the 100- and 150-mg dose levels.
TABLE 3.
Summary statistics for BC-3781 PK exposure estimates and half-life
| Parameter and dose (mg) | Mean (SD) | Median (min, max) |
|---|---|---|
| Day 1 Cmax (μg/ml) | ||
| 100 | 1.57 (0.974) | 1.46 (0.515, 8.11) |
| 150 | 1.90 (0.705) | 1.88 (0.633, 4.38) |
| Day 1 AUC0–24 (μg · h/ml) | ||
| 100 | 10.9 (5.97) | 9.87 (3.86, 48.3) |
| 150 | 14.1 (5.76) | 12.8 (4.29, 35.1) |
| Half-life (h) | ||
| 100 | 11.0 (5.18) | 9.74 (0.838, 23.5) |
| 150 | 13.2 (5.79) | 12.4 (3.38, 23.5) |
DISCUSSION
The objectives of the analysis described herein were 3-fold. The first objective was to apply the population PK model for BC-3781 developed from phase 1 data to the PK data from a phase 2 study, thereby assessing the utility of the phase 1 model vis-à-vis phase 2 patients undergoing sparse sampling. The plan allowed for modifications of this model as necessary to obtain an adequate fit to the data. The second objective was to identify any patient descriptors associated with the interindividual variability in BC-3781 population PK parameters. The third, and most important, objective was to obtain accurate estimates of PK exposure in the phase 2 patients in order to facilitate the conduct of PK-PD analyses (13).
The previously developed PK model for BC-3781, which was a three-compartment model with zero-order infusion and first-order (linear) elimination, was based upon intensive PK sampling data from three phase 1 studies of subjects who received a wide range of single and multiple doses (3). The model had been used to define an optimal sampling strategy to enable robust clearance estimates within the logistical framework of this phase 2 study. The phase 1 population PK model provided for a robust description of the PK of BC-3781, as evidenced by the lack of changes in the structure of the model that were required to obtain an adequate fit of the BC-3781 concentrations from the phase 2 study. Additionally, the assumption of nonlinear protein binding was tested in this analysis and the model fits indicated that this assumption should be retained for the phase 2 data. This added complexity in the model also has the advantage of allowing for more accurate estimates of free-drug exposures, which are useful for translational exercises such PK-PD target attainment analyses to support dose selection or in vitro susceptibility breakpoint selection.
The covariate exploration analysis revealed that no strong relationships between patient descriptors and PK parameters were evident. This was first apparent visually via the covariate screening plots and confirmed statistically by implementing several covariate models in S-ADAPT. It is important to note that this does not imply that there are no relationships between patient descriptors and BC-3781 PK, only that no relationships were evident in these data. Future population PK analyses using larger, more diverse patient populations may provide further insight into those patient characteristics that help explain the interindividual variability in BC-3781 PK.
Given that the primary goal of this analysis was to obtain accurate estimates of drug exposure, the precision of the individual estimates of CLtu, which is the only PK parameter of importance when estimating the AUC (in a linear-elimination model such as this), was examined. Consistent with the overall goodness of fit of the model, individual estimates of the percent CV of CLtu were all relatively low (median of 9.15%, maximum of 25%). This finding indicates that (i) the optimal sampling strategy resulted in highly informative concentration-time profiles and (ii) the precision of the individual clearance and AUC estimates was high.
In summary, population PK methods allowed for a robust description of the plasma PK of BC-3781 after i.v. administration to patients in this phase 2 study. The population PK model was deemed “fit for purpose,” given the desire to define the free- and total-drug exposures experienced by those patients randomized to receive BC-3781. Precise estimates of drug clearance, and therefore the AUC, were obtained which can then be used to conduct clinical PK-PD analyses of efficacy and safety.
ACKNOWLEDGMENT
This analysis was funded by Nabriva Therapeutics AG, Vienna, Austria.
REFERENCES
- 1.Sader HS, Biedenbach DJ, Paukner S, Ivezic-Schoenfeld Z, Jones RN. 2012. Antimicrobial activity of the investigational pleuromutilin compound BC-3781 tested against Gram-positive organisms commonly associated with acute bacterial skin and skin structure infections. Antimicrob Agents Chemother 56:1619–1623. doi: 10.1128/AAC.05789-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sader HS, Paukner S, Ivezic-Schoenfeld Z, Biedenbach DJ, Schmitz FJ, Jones RN. 2012. Antimicrobial activity of the novel pleuromutilin antibiotic BC-3781 against organisms responsible for community-acquired respiratory tract infections (CARTIs). J Antimicrob Chemother 67:1170–1175. doi: 10.1093/jac/dks001. [DOI] [PubMed] [Google Scholar]
- 3.Rubino CM, Forrest A, Bhavnani SM, Prince WT, Ivezic-Schoenfeld Z, Wicha WW, Ambrose PG. 2010. Population pharmacokinetics of BC-3781 using phase 1 data, abstr A1-018. Abstr. 50th Intersci Conf Antimicrob Agents Chemother. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gehan EA, George SL. 1970. Estimation of human body surface area from height and weight. Cancer Chemother Rep 54:225–235. [PubMed] [Google Scholar]
- 5.Cockcroft DW, Gault MH. 1976. Prediction of creatinine clearance from serum creatinine. Nephron 16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 6.Food and Drug Administration. 2001. Guidance for industry: bioanalytical method validation. Department of Health and Human Services, Washington, DC. [Google Scholar]
- 7.Anonymous. 1999. Guidance for industry on population pharmacokinetics; availability. Food and Drug Administration, HHS. Notice Fed Regist 64:6663–6664. [PubMed] [Google Scholar]
- 8.Beal SL. 2001. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn 28:481–504. doi: 10.1023/A:1012299115260. [DOI] [PubMed] [Google Scholar]
- 9.Bauer RJ. 2006. S-ADAPT/MCPEM. User's guide: software for pharmacokinetic, pharmacodynamic, and population data analysis. Robert J. Bauer, Berkeley, CA: http://bmsr.usc.edu/downloads/s-adapt. [Google Scholar]
- 10.D'Argenio DZ, Schumitzky A. 1979. A program package for simulation and parameter estimation in pharmacokinetic systems. Comput Programs Biomed 9:115–134. doi: 10.1016/0010-468X(79)90025-4. [DOI] [PubMed] [Google Scholar]
- 11.D'Argenio DZ, Schumitzky A. 2003. ADAPT II user's guide: pharmacokinetic/pharmacodynamic systems analysis software. BMSR Biomedical Simulations Resource, Los Angeles, CA. [Google Scholar]
- 12.Akaike H. 1979. Bayesian extension of the minimum AIC procedure of autoregressive model fitting. Biometrika 66:237–242. doi: 10.1093/biomet/66.2.237. [DOI] [Google Scholar]
- 13.Bhavnani SM, Hammel JP, Rubino CM, Bulik CC, Reynolds DK, Ivezic-Schoenfeld Z, Wicha WW, Novak R, Prince WT, Ambrose PG. 2011. Pharmacokinetic-pharmacodynamic analysis for efficacy of BC-3781 using new clinical trial endpoints for patients with acute bacterial skin and skin structure infection, abstr A2-025. Abstr 51st Intersci Conf Antimicrob Agents Chemother. [Google Scholar]