

Abstract
Clinical studies have shown that integrase strand transfer inhibitors (INSTIs) can be used effectively against HIV-1 infection. To date, no resistance substitution has been found in INSTI-naive patients treated with the new integrase inhibitor dolutegravir (DTG). In a recent selection study with DTG, using a virus bearing the H51Y substitution in integrase, the emergence of an R to K substitution at position 262 (R262K) was observed. We characterized this double mutant with respect to integrase strand transfer activity and susceptibility to DTG both biochemically and in tissue culture. We showed that the addition of R262K to H51Y decreased recombinant integrase strand transfer activity but improved integrase DNA-binding affinity, compared to wild-type or H51Y-containing enzymes. The defect in strand transfer activity did not translate into a decrease in HIV-1 infectivity. The combination of H51Y and R262K substitutions slightly decreased susceptibility to DTG (fold change = 1.87) in cell-based resistance assays. Although viral replication was not affected and enzyme efficiency was impaired by the addition of R262K to H51Y, there was an overall increase in the level of biochemical drug resistance against DTG. Our findings suggest that the R at position 262 plays an important role in DNA binding.
INTRODUCTION
New antiretroviral (ARV) drug classes against HIV are constantly being developed, such as the integrase strand transfer inhibitors (INSTIs) that target the HIV-1 integrase (IN) enzyme (1–3). INSTIs that are currently available clinically, such as raltegravir (RAL), elvitegravir, and dolutegravir (DTG) (4), act by inhibiting the strand transfer step of viral integration, which occurs after nuclear entry and covalently links the 3′-hydroxyl groups of viral DNA to host DNA (5). Among INSTIs, DTG possesses the greatest genetic barrier against the emergence of resistance substitutions and, until now, no major resistance substitution has been observed in treatment-naive patients who have failed treatment with this drug (6, 7).
Our group identified an R263K variant during tissue culture selection experiments with DTG (8). This preclinical observation has since been confirmed for several patients who were part of a cohort of highly treatment-experienced, INSTI-naive patients who failed DTG-based therapy (9).
In order to further characterize DTG resistance substitutions, we identified a new R262K substitution that emerged under DTG pressure from a virus containing an H51Y substitution, which had been previously identified as a DTG secondary substitution (8, 10, 11). Primary human cord blood mononuclear cells (CBMCs) were infected with viruses bearing the primary resistance substitution H51Y and were grown in the presence of DTG in order to mimic drug pressure and potential viral escape. At week 25, a new substitution at position 262 of integrase was observed (11). Although R262E, in association with other substitutions, had previously been detected in a patient with HIV-1 subtype B treated with RAL (12), this is the first report of a substitution at position 262 in the context of DTG resistance. Furthermore, R262 has been shown to play a role in DNA binding (13). Therefore, we characterized R262K in association with H51Y in regard to strand transfer activity, infectivity, and resistance against DTG in cell-based assays. Our results showed that the combination of H51Y and R262K substitutions conferred decreased susceptibility to DTG but not RAL, while decreasing integrase strand transfer activity.
MATERIALS AND METHODS
Antiviral compounds and cell lines.
DTG and RAL were obtained from GlaxoSmithKline/ViiV Healthcare and Merck Inc., respectively. The TZM-bl cell line was obtained from John C. Kappes, Xiaoyun Wu, and Tranzyme, Inc., through the NIH AIDS Reagent Program, and the 293T cell line was obtained from the American Type Culture Collection (ATCC CRL-11268). Both cell lines were maintained in Dulbecco's minimal essential medium (DMEM) supplemented with 10% fetal bovine serum, 1% (vol/vol) penicillin-streptomycin, and 1% l-glutamine. All cells were incubated at 37°C in a humidified 5% CO2 atmosphere.
Plasmids, protein expression, and protein purification.
The creation of pET-15b expression plasmids coding for soluble wild-type (WT) and H51Y-mutated HIV subtype B IN (INB) was described previously (14). The R262K substitution was introduced to create the pET-15bINB(R262K) and pET-15bINB(H51Y/R262K) plasmids by using a Q5 site-directed mutagenesis kit (New England BioLabs), according to the manufacturer's instructions. The primers used were INB(R262K)-F (5′-TAGTGACATAAAAGTAGTGCCAAAAAGAAAAGCAAAGATCATCAC-3′) and INB(R262K)-R (5′-CCTGATGATCTTTGCTTTTCTTTTTGGCACTACTTTTATGTCACT-3′). Following mutagenesis, plasmids were treated for 4 h at 37°C with DpnI and transformed into Escherichia coli strain XL10-Gold ultracompetent cells (Stratagene), according to the manufacturer's instructions. Plasmids were recovered from bacteria using a QIAprep miniprep kit (Qiagen), according to the manufacturer's instructions. Plasmids were quantified using a NanoDrop spectrophotometer and were subjected to DNA sequencing.
The aforementioned substitutions were inserted into the pNL4-3 WT vector, which was obtained through the NIH AIDS Reagent Program, with the same primers as described above, following the same protocol except that DpnI-digested plasmids were transformed into NEB 5-alpha competent E. coli cells (New England BioLabs). Luria-Bertani (LB) broth (Multicell) prepared with Milli-Q water and supplemented with 100 μg/ml ampicillin was used for all bacterial growth. Expression and purification of N-terminally His-tagged IN recombinant proteins were performed as described previously (15). E. coli BL21(DE3) Gold cells (Stratagene) were used to express proteins. Recombinant protein concentrations were measured using a calculated extinction coefficient of 50,420 M−1 cm−1 (16). Recombinant protein aliquots were stable at −80°C without degradation or loss of activity.
Integrase strand transfer activities.
Strand transfer activities of the recombinant WT, H51Y-containing, R262K-containing, and H51Y/R262K-containing subtype B integrase proteins were measured using a microtiter plate assay, as described previously (8, 15). Briefly, functional DNA long terminal repeat (LTR) duplexes were covalently bound to Costar DNA-Bind 96-well plates (Corning) at 4°C for 48 h. The plates were then blocked with 0.5% bovine serum albumin (BSA) for 18 h at 4°C. Before use, the plates were washed with phosphate-buffered saline (PBS) (pH 7.4) and with assay buffer (50 mM morpholinopropanesulfonic acid [MOPS] [pH 6.8], 50 μg/ml BSA, 50 mM NaCl, 0.15% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate [CHAPS], 30 mM MnCl2). Purified integrase proteins were then added, and the plates were incubated for 30 min at 25°C. The biotinylated target DNA duplex (sense, 5′-TGACCAAGGGCTAATTCACT-3′-biotin; antisense, 5′-AGTGAATTAGCCCTTGGTCA-3′-biotin) was incubated for 1 h at 37°C in order for the strand transfer reaction to occur. Plates were then washed twice with wash buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 0.05% Tween 20, 2 mg/ml BSA). A solution containing europium-labeled streptavidin (PerkinElmer) diluted to 0.025 μg/ml in wash buffer in the presence of 50 μM diethylenetriaminepentaacetic acid (DTPA) (Sigma) was then added to the plates, and the plates were incubated for 30 min in the dark. The plates were then washed, and Wallac enhancement solution was added (PerkinElmer). Time-resolved fluorescence (TRF) was measured with a FLUOstar Optima multilabel plate reader (BMG Labtech).
Integrase DNA-binding activities.
The DNA-binding activities of the WT, H51Y, R262K, and H51Y/R262K integrase subtype B proteins were measured using a fluorescence-based, microtiter plate, DNA-binding assay, as described previously (17). Briefly, Corning 96-well black flat-bottom polystyrene high-binding microplates (Corning catalog no. 3925) were used to measure the DNA-binding activities of the recombinant proteins. The plates were coated for 18 h at 4°C with different concentrations of the recombinant integrase proteins diluted in PBS (pH 7.4). The plates were then washed with PBS (pH 7.4) to remove unbound proteins and were blocked with 5% BSA at 25°C for 2 h. After blocking, the coated plates were washed twice with ice-cold PBS and once with binding buffer (20 mM MOPS [pH 7.2], 20 mM NaCl, 7.5 mM MgCl2 or MnCl2, 5 mM dithiothreitol [DTT]). Fluorescently labeled RhoR-LTR duplex (25 nM) (LTR-D1, 5′-CTTTTAGTCAGTGTGGAAAATCTCTAGCAGT-3′; LTR-D2, 5′-RhoR-XN/ACTGCTAGAGATTTTCCACACTGACTAAAAG-3′) was then added to the plates, and the plates were incubated for 1 h at 25°C in the dark. After incubation, the plates were washed 3 times with ice-cold PBS. Finally, PBS was added to each well, and the fluorescence signals were measured with a FLUOstar Optima multilabel plate reader.
Assay of 3′ processing.
The 3′ processing activity of the purified recombinant integrase proteins was determined as described previously (18). Briefly, 3′-biotinylated LTR duplex was covalently linked at various concentrations to Costar DNA-Bind plates, under conditions similar to those described for strand transfer. To initiate 3′ processing, purified integrase proteins (400 nM) in reaction buffer (50 nM MOPS [pH 6.8], 50 mg/ml BSA, 50 mM NaCl, 20 mM MnCl2, 0.015% CHAPS, 5 mM DTT) were incubated on the plates for 2 h at 37°C. Negative-control wells had only reaction buffer added. After 2 h, the plates were quickly washed three times with wash buffer to remove all traces of unbound material. All subsequent steps of the assay were as described above for the strand transfer assay.
Generation of replication-competent virus particles for studies of HIV infectivity and drug resistance.
293T cells were transfected with 12.5 μg of plasmid using Lipofectamine 2000 (Life Technologies), according to the manufacturer's instructions. Fresh medium was added after 6 h of incubation, and the cells were incubated for an additional 48 h. Supernatants were then harvested and passed through a 0.45-μm filter in order to remove remaining cellular debris. Virus particles were divided into aliquots and stored at −80°C. The concentrations of virus particles were determined by measuring reverse transcriptase (RT) activity, as described previously (19). HIV infectivity was measured using noncompetitive short-term infectivity assays using TZM-bl cells, as described previously (8). HIV susceptibility to DTG and RAL was determined using short-term resistance assays with TZM-bl cells, as described previously (8).
Homology modeling.
A WT homology model of the HIV-1 integrase was constructed based on the available crystal structures of the prototype foamy virus (PFV) (20, 21). The amino acid sequence of WT subtype B was submitted to the I-TASSER three-dimensional protein prediction server (22, 23). The published PFV target capture complex crystal structure (Protein Data Bank [PDB] identification no. 4E7K) was used as a lead template to generate a WT subtype B dimeric model (20, 21). The ProtMod server was used to remove any sampling errors that might have been introduced by multiple-threading alignments and iterative template fragment assembly simulations of I-TASSER (22, 23); the WT subtype B model generated by I-TASSER served as a template while WT, H51Y, and R262K/H51Y amino acid sequences were input as queries to generate models using the program Modeler (24, 25). Ramachandran diagram analysis (26, 27) was used to verify that all homology models possessed >90% of residues in the favored and allowed orientations, reflecting the stability of the protein structure. DNA interaction hints were obtained by overlaying the HIV-1 homology models with the PFV crystal structure (PDB identification no. 4E7K) (20, 21). The Research Collaboratory for Structural Bioinformatics PDB Protein Comparison Tool was used to assess model quality based on root mean square deviation (RMSD) values (28). Rotamer orientations for mutated residues and key residues in the active site were carefully examined, and the best backbone-dependent rotamers were selected (29, 30). The molecular visualization program PyMOL, version 1.3 (http://pymol.org), was used for structural visualization and image processing.
Statistical analysis.
All experiments consisted of at least 2 sets of experiments performed in triplicate, to yield 6 independent values for each data point, unless indicated otherwise. For each experiment, strand transfer values measured in the absence of drug were arbitrarily set as 100%. When various concentrations of target DNA were employed, strand transfer results were fitted to the Michaelis-Menten equation with the use of GraphPad Prism 6.0 software, to generate values for maximal enzyme activity (Vmax) and Km. Enzyme efficiency (Vmax/Km) was determined as described previously (14). The strand transfer activity of the wild-type and mutant enzymes in the presence of INSTIs was determined by using a competitive inhibition model and by constraining the results with Km values based on the delta target DNA results. The Km values for the wild-type subtype B integrase (INBWT), INBH51Y, INBR262K, and INBH51Y/R262K enzymes were 2.75, 7.12, 5.12, and 3.41, respectively. Using replication capacity experiments in TZM-bl cells in the presence of DTG or RAL, we determined 50% effective concentrations (EC50s) for the WT and mutant viruses, using the sigmoidal dose-response function of GraphPad Prism 6.0. The null hypothesis for equally variant data sets was evaluated using the Student t test. Significant differences were defined as those with P values of <0.05, and the underlying distributions were two-tailed for all tests. The open source statistical package OpenEpi (http://www.openepi.com/Menu/OE_Menu.htm) was used for all tests.
RESULTS
The addition of R262K to H51Y decreases integrase strand transfer activities.
First, we characterized the effects of R262K, alone or in combination with H51Y, on integrase strand transfer activities of purified recombinant subtype B integrase (INB) enzymes (Fig. 1). Using various concentrations of the different recombinant enzymes, we showed that INBR262K and INBH51Y/R262K possessed lower strand transfer activities than did INBWT or INBH51Y (Fig. 1A), while integrase activity in the presence of H51Y was not significantly different from WT values. Similar findings were obtained when the amount of target DNA in the strand transfer reaction was varied (Fig. 1B). Enzyme kinetic results showed that differences in strand transfer activities between INBWT and INBH51Y were not significant (Fig. 1B and C). Further investigation revealed that the Km value of INBR262K was higher than the WT value (Km values of 6.977 and 5.825 nM, respectively) (Fig. 1C), but the combination of H51Y and R262K was restorative (Km = 5.087 nM), implying that the two substitutions in tandem restored enzyme affinity for the substrate. However, the combination of H51Y and R262K also resulted in a marked decrease in maximal DNA binding (Vmax), compared to the WT value (Vmax values of 63.09 and 107.6 relative fluorescence units [RFU]/h, respectively). Figure 1D shows that enzyme efficiency (Vmax/Km) values for INBR262K and INBH51Y/R262K were lower than values for INBWT and INBH51Y.
FIG 1.

Effects of the addition of R262K to H51Y on integrase strand transfer activity. (A) Relative strand transfer activities with various recombinant protein concentrations. Columns represent the means and standard errors of three independent experiments. (B) Relative strand transfer activities in the presence of various target DNA concentrations. Points represent the means and standard errors of seven independent experiments. (C) Km values. Columns represent the means and standard errors of seven independent observations. (D) Recombinant enzyme efficiencies, as determined by dividing Vmax by Km. Columns represent the means and standard errors of seven independent experiments.
The addition of R262K to H51Y alters integrase LTR DNA-binding activities.
Previous studies have shown that R262 in IN is important for DNA binding (13). Therefore, we investigated the DNA-binding capacity of the H51Y, R262K, and H51Y/R262K mutants as well as the WT enzyme, as described previously (17), in the presence of either MgCl2 or MnCl2. In the presence of Mg2+ ions, the inclusion of the R262K substitution decreased integrase DNA-binding efficiency (Table 1). In contrast, in the presence of Mn2+ ions, all of the mutant IN preparations appeared to have higher DNA-binding activities than did WT enzyme (Table 1), although differences between INBH51Y and INBWT were not significant (Table 1). In contrast, INBR262K and INBH51Y/R262K bound DNA more efficiently than did the WT and H51Y enzymes. Overall, the addition of R262K to H51Y altered the efficiency of integrase strand transfer and improved DNA-binding activities.
TABLE 1.
DNA-binding parameters for purified integrase proteinsa
| Genotype | Mn2+ |
Mg2+ |
||||
|---|---|---|---|---|---|---|
| Vmax (mean ± SD) (RFU/h) | Km (mean ± SD) (nM) | Enzyme efficiency (mean ± SD) (RFU/h/nM) | Vmax (mean ± SD) (RFU/h) | Km (mean ± SD) (nM) | Enzyme efficiency (mean ± SD) (RFU/h/nM) | |
| WT | 124.3 ± 9.2 | 473.7 ± 87.6 | 0.26 ± 0.11 | 129.3 ± 5.6 | 1293 ± 144 | 0.1 ± 0.04 |
| H51Y | 146 ± 11.6 | 454.7 ± 116.6 | 0.32 ± 0.10 | 209.9 ± 10.8 | 2964 ± 300.6 | 0.07 ± 0.04 |
| R262K | 148.2 ± 11.6 | 309.5 ± 72.9 | 0.48 ± 0.16 | 208.4 ± 21.6 | 3961 ± 720.6 | 0.05 ± 0.03 |
| H51Y/R262K | 119.7 ± 4.0 | 126.9 ± 19.1 | 0.94 ± 0.21b | 207.6 ± 24.1 | 4256 ± 904.6 | 0.05 ± 0.03 |
DNA-binding activities were determined with various concentrations of recombinant integrase enzymes in the presence of either MnCl2 or MgCl2. Vmax and Km values were calculated using GraphPad Prism software. Recombinant enzyme efficiency was determined by dividing Vmax by Km. All studies with Mn2+ were performed using three replicate samples, and all studies with Mg2+ were performed in duplicate. SD, standard deviation.
P < 0.05.
The addition of R262K to H51Y does not affect 3′ processing.
In order to verify the observations from the DNA-binding experiments, we tested the ability of the different mutant enzymes to perform 3′ processing, as described previously (10). The R262K and H51Y/R262K enzymes had similar 3′ processing activities, compared to the WT enzyme. Although the H51Y substitution appeared to result in decreased 3′ processing (Fig. 2A), further investigation failed to reveal significant differences in overall enzyme efficiencies, compared to the WT enzyme (Fig. 2B). Thus, the addition of R262K to H51Y did not affect 3′ processing activity.
FIG 2.
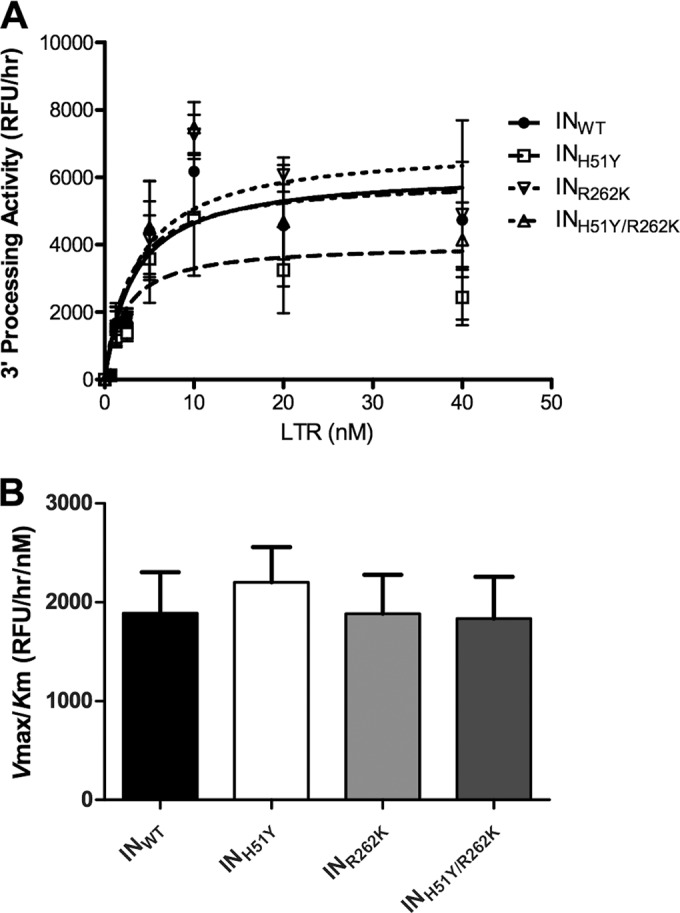
Effects of the addition of R262K to H51Y on integrase 3′ processing activity. (A) 3′ processing activity with various concentrations of viral LTR. Points represent the means and standard errors of two independent experiments. (B) Recombinant enzyme efficiency, as determined by the ratio of Vmax to Km. Columns represent the means and standard errors of two independent experiments.
The addition of R262K to H51Y does not impair viral infectivity.
Next, we characterized the effects of these substitutions on HIV-1 infectivity. Since the secondary R262K substitution has never been observed alone (11), we focused on three viruses, i.e., WT, H51Y, and H51Y/R262K. TZM-bl cells were infected with various amounts of pNL4.3WT, pNL4.3H51Y, and pNL4.3H51Y/R262K viruses. In agreement with our biochemical assays, none of the H51Y, R262K, or H51Y/R262K substitutions affected HIV-1 infectivity in tissue culture (Fig. 3).
FIG 3.

Effects of the addition of R262K to H51Y on viral infectivity. The effects of the WT, H51Y, and H51Y/R262K substitutions on HIV infectivity in TZM-bl cells were measured with various amounts of virus. Points represent the means and standard errors of two independent experiments.
The addition of R262K to H51Y decreases HIV-1 susceptibility to DTG.
To further characterize the R262K secondary substitution in the presence of H51Y, we performed resistance assays using TZM-bl cells and various concentrations of DTG or RAL (Fig. 4), which generated EC50s for each virus as well as fold change (FC) values (Table 2). Although the primary H51Y substitution did not confer resistance against DTG or RAL (FC values of 1.25 and 0.98, respectively), consistent with previous results (11), the addition of R262K did result in low-level but significant biochemical resistance to DTG but not to RAL (FC values of 1.9 and 0.92, respectively).
FIG 4.
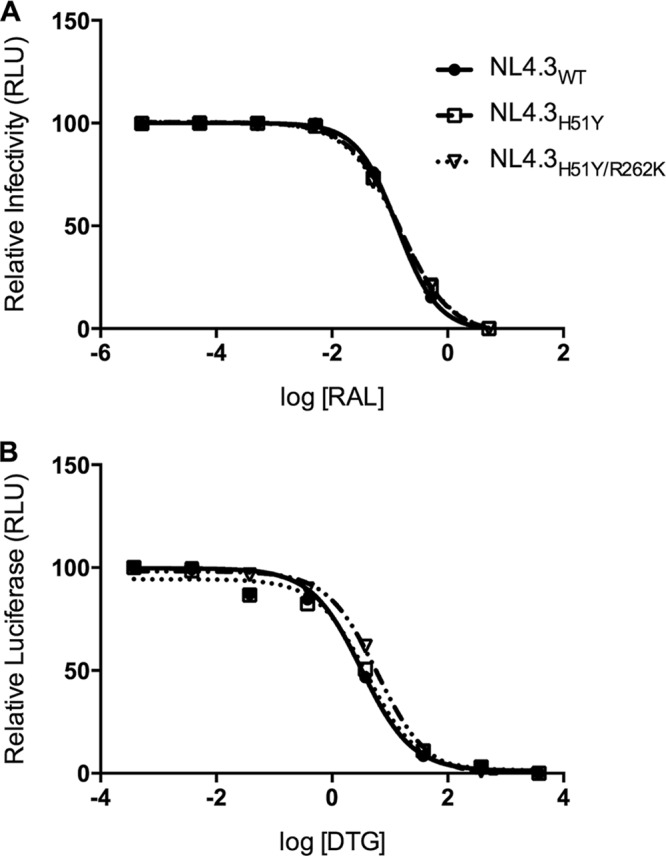
Effects of the addition of R262K to H51Y on susceptibility to INSTIs. (A) Resistance curves for WT, H51Y, and H51Y/R262K viruses with various concentrations of RAL. Points represent the means and standard errors of two independent experiments. (B) Resistance curves for WT, H51Y, and H51Y/R262K viruses with various concentrations of DTG. Points represent the means and standard errors of two independent experiments.
TABLE 2.
Effects of H51Y and R262K substitutions on EC50s for DTG and RAL in cell-based assaysa
| Genotype | DTG |
RAL |
||
|---|---|---|---|---|
| EC50 (mean ± SD) (nM) | FC | EC50 (mean ± SD) (nM) | FC | |
| WT | 3.23 ± 1.06 | 1.00 | 0.16 ± 1.25 | 1.00 |
| H51Y | 4.02 ± 1.25 | 1.24 | 0.15 ± 1.16 | 0.98 |
| H51Y/R262K | 6.05 ± 1.09 | 1.87b | 0.14 ± 1.35 | 0.92 |
The effects of the primary INSTI resistance substitutions H51Y and R262K on the EC50 values for DTG and RAL were determined in cell culture assays. DTG EC50 values are representative of three independent experiments. RAL EC50 values are representative of two independent experiments. SD, standard deviation.
P < 0.05.
In silico studies of WT and mutant HIV-1 integrases.
To gain insight into the impact of the H51Y and R262K substitutions on HIV-1 IN, we performed structural modeling of the enzyme in the presence of long terminal repeat (LTR) DNA, target DNA, and divalent Mn2+ ions. Homology models of IN for the WT, H51Y, and R262K/H51Y enzymes were created using the Modeler server (24, 25), with the published crystal structure of the prototype foamy virus (PFV) integrase (PDB identification no. 4E7K) as a lead template (21). The modeled HIV-1 IN showed good global agreement in the N-terminal, catalytic core, and C-terminal domains (Fig. 5A). Rotamer interrogation of the key catalytic residues (D64, D116, and E152) showed that they had orientations similar to those seen in the PFV structure (data not shown). In the models generated, the positions of the catalytic residues were not altered and possessed the proper orientations needed to coordinate Mn2+ ions, which were superimposed from the PFV structure (Fig. 5B and C). The positions of H51Y and R262K were unchanged from H51 and R262 (data not shown), and there were no major global shifts in the secondary structures (Fig. 5A). These results are in agreement with the absence of major changes in infectivity when the H51Y and R262K substitutions are present.
FIG 5.

Homology model of HIV-1 integrase subtype B. A homology model was generated based on the crystal structure of the target capture complex of PFV (PDB identification no. 4E7K). Secondary structure overlays and catalytic triad orientations and positions were examined for WT and mutant HIV-1 integrases. (A) Global overlay of the INWT, INH51Y, and INR262K/H51Y models. (B and C) Close-up overlays, showing the relative positions of the catalytic triad of D64, D116, and E152 in INWT (B and C), INH51Y (B), and INR262K/H51Y (C) coordinating Mn2+ ions. Mn2+ ion positions were simulated by inserting their coordinates from the PFV structure (PDB identification no. 4E7K) in the active site, and the ions are represented as small black spheres. Coordinates of the viral LTR and target DNA were inserted in the active site of the integrase homology models by overlap of the PFV crystal structure (PDB identification no. 4E7K). All images were processed using PyMOL software. Diagrams and carbon atom coloration differentiate the INWT (white), INH51Y (black), and INR262K/H51Y (gray) homology models.
DISCUSSION
The HIV-1 IN protein contains three functional domains essential for the integration of viral DNA into the host genome (31, 32). R262 is part of the C-terminal domain, which is known to bind to DNA in a nonspecific manner (31, 33, 34), and R262 is known to be important for DNA binding (13). Substitutions at position 262 from R to E or A negatively affected DNA binding (13), in agreement with our DNA-binding assays in the presence of Mg2+ ions (Table 1). In contrast, a change to R262K increased viral DNA binding in the presence of Mn2+ ions (Table 1). A possible explanation for these observations is that mutating R (a positively charged residue) to either E (negatively charged) or A (nonpolar) changes both the length and the charge, resulting in a loss of electrostatic interactions or repulsion between similar charges in the microenvironment. However, an R262K change maintains the overall positive charge of the side chain while causing it to be slightly shortened. This may be why the emergence of R262K is favored over that of R262A or R262E under DTG pressure (11).
R262K was first shown to be present in cord blood mononuclear cells (CBMCs) infected with HIV containing the H51Y substitution (11). Although the H51Y substitution was previously shown to be nondetrimental to viral fitness (10), as confirmed here, this study extends these findings by showing that the addition of R262K to H51Y decreased enzyme strand transfer activity (Fig. 1) without affecting 3′ processing activity (Fig. 2) or viral infectivity (Fig. 3). A possible interpretation of these results is that the addition of R262K to H51Y may improve DNA-binding efficiency while decreasing strand transfer activity, resulting in a balanced overall integration process. R262K in association with H51Y also conferred low-level in vitro resistance against DTG in short-term cell-based assays (Table 2), an observation that helps to explain the selection of this substitution in viruses containing the H51Y substitution (11). H51Y alone does not affect viral infectivity (Fig. 3) or resistance to RAL or DTG (Fig. 4). Although R262K decreases integrase affinity for target DNA (Fig. 1), this is apparently balanced by improved integrase binding to viral DNA (Table 1), resulting in viruses that possess close to WT infectivity (Fig. 3). This is consistent with the notion that INSTIs partly inhibit integration by competing with the processed viral LTR DNA ends that bind to the integrase enzyme during the integration process. Therefore, the growth of viruses with greater LTR binding is likely to be favored in the presence of INSTIs.
As stated above, the addition of R262K to H51Y resulted in decreased susceptibility to DTG (FC = 1.87) (Table 2), a finding consistent with the emergence of R262K under DTG selection pressure. However, the low levels of resistance that result may preclude the clinical occurrence of the H51Y/R262K double mutant, consistent with the fact that no DTG resistance substitution has yet been reported among treatment-naive patients receiving this drug clinically.
ACKNOWLEDGMENTS
This project was supported by the Canadian Institutes for Health Research. S.H. is the recipient of a doctoral studentship from the Fonds de la Recherche du Québec en Santé. K.A. is the recipient of a fellowship from the Canadian Institutes for Health Research.
We declare no competing financial interests.
V.C. designed and performed experiments, analyzed data, and wrote the manuscript, T.M. designed and performed experiments, analyzed data, and revised the manuscript, K.A. performed experiments and analyzed data, S.H. performed the homology modeling analyses, and M.A.W. supervised the project and revised the manuscript. All the authors read and approved the final manuscript.
REFERENCES
- 1.Quashie PK, Mesplede T, Wainberg MA. 2013. HIV drug resistance and the advent of integrase inhibitors. Curr Infect Dis Rep 15:85–100. doi: 10.1007/s11908-012-0305-1. [DOI] [PubMed] [Google Scholar]
- 2.Wainberg MA, Mesplede T, Quashie PK. 2012. The development of novel HIV integrase inhibitors and the problem of drug resistance. Curr Opin Virol 2:656–662. doi: 10.1016/j.coviro.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 3.Hightower KE, Wang R, Deanda F, Johns BA, Weaver K, Shen Y, Tomberlin GH, Carter HL III, Broderick T, Sigethy S, Seki T, Kobayashi M, Underwood MR. 2011. Dolutegravir (S/GSK1349572) exhibits significantly slower dissociation than raltegravir and elvitegravir from wild-type and integrase inhibitor-resistant HIV-1 integrase-DNA complexes. Antimicrob Agents Chemother 55:4552–4559. doi: 10.1128/AAC.00157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballantyne AD, Perry CM. 2013. Dolutegravir: first global approval. Drugs 73:1627–1637. doi: 10.1007/s40265-013-0121-4. [DOI] [PubMed] [Google Scholar]
- 5.Craigie R. 2001. HIV integrase, a brief overview from chemistry to therapeutics. J Biol Chem 276:23213–23216. doi: 10.1074/jbc.R100027200. [DOI] [PubMed] [Google Scholar]
- 6.Wainberg MA, Mesplede T, Raffi F. 2013. What if HIV were unable to develop resistance against a new therapeutic agent? BMC Med 11:249. doi: 10.1186/1741-7015-11-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mesplede T, Quashie PK, Wainberg MA. 2012. Resistance to HIV integrase inhibitors. Curr Opin HIV AIDS 7:401–408. doi: 10.1097/COH.0b013e328356db89. [DOI] [PubMed] [Google Scholar]
- 8.Quashie PK, Mesplede T, Han YS, Oliveira M, Singhroy DN, Fujiwara T, Underwood MR, Wainberg MA. 2012. Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. J Virol 86:2696–2705. doi: 10.1128/JVI.06591-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cahn P, Pozniak AL, Mingrone H, Shuldyakov A, Brites C, Andrade-Villanueva JF, Richmond G, Buendia CB, Fourie J, Ramgopal M, Hagins D, Felizarta F, Madruga J, Reuter T, Newman T, Small CB, Lombaard J, Grinsztejn B, Dorey D, Underwood M, Griffith S, Min S. 2013. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet 382:700–708. doi: 10.1016/S0140-6736(13)61221-0. [DOI] [PubMed] [Google Scholar]
- 10.Mesplede T, Quashie PK, Osman N, Han Y, Singhroy DN, Lie Y, Petropoulos CJ, Huang W, Wainberg MA. 2013. Viral fitness cost prevents HIV-1 from evading dolutegravir drug pressure. Retrovirology 10:22. doi: 10.1186/1742-4690-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oliveira M, Mesplede T, Quashie PK, Moisi D, Wainberg MA. 2014. Resistance mutations against dolutegravir in HIV integrase impair the emergence of resistance against reverse transcriptase inhibitors. AIDS 28:813–819. doi: 10.1097/QAD.0000000000000199. [DOI] [PubMed] [Google Scholar]
- 12.Canducci F, Sampaolo M, Marinozzi MC, Boeri E, Spagnuolo V, Galli A, Castagna A, Lazzarin A, Clementi M, Gianotti N. 2009. Dynamic patterns of human immunodeficiency virus type 1 integrase gene evolution in patients failing raltegravir-based salvage therapies. AIDS 23:455–460. doi: 10.1097/QAD.0b013e328323da60. [DOI] [PubMed] [Google Scholar]
- 13.Lutzke RA, Plasterk RH. 1998. Structure-based mutational analysis of the C-terminal DNA-binding domain of human immunodeficiency virus type 1 integrase: critical residues for protein oligomerization and DNA binding. J Virol 72:4841–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quashie PK, Mesplede T, Han YS, Veres T, Osman N, Hassounah S, Sloan RD, Xu HT, Wainberg MA. 2013. Biochemical analysis of the role of G118R-linked dolutegravir drug resistance substitutions in HIV-1 integrase. Antimicrob Agents Chemother 57:6223–6235. doi: 10.1128/AAC.01835-13 (Erratum, 58:633, 2014, doi:.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bar-Magen T, Donahue DA, McDonough EI, Kuhl BD, Faltenbacher VH, Xu H, Michaud V, Sloan RD, Wainberg MA. 2010. HIV-1 subtype B and C integrase enzymes exhibit differential patterns of resistance to integrase inhibitors in biochemical assays. AIDS 24:2171–2179. doi: 10.1097/QAD.0b013e32833cf265. [DOI] [PubMed] [Google Scholar]
- 16.Walker JM. 2005. The proteomics protocols handbook. Humana Press, Totowa, NJ. [Google Scholar]
- 17.Han YS, Xiao WL, Quashie PK, Mesplede T, Xu H, Deprez E, Delelis O, Pu JX, Sun HD, Wainberg MA. 2013. Development of a fluorescence-based HIV-1 integrase DNA binding assay for identification of novel HIV-1 integrase inhibitors. Antiviral Res 98:441–448. doi: 10.1016/j.antiviral.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 18.Han YS, Quashie P, Mesplede T, Xu HT, Mekhssian K, Fenwick C, Wainberg MA. 2012. A high-throughput assay for HIV-1 integrase 3′-processing activity using time-resolved fluorescence. J Virol Methods 184:34–40. doi: 10.1016/j.jviromet.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Xu HT, Asahchop EL, Oliveira M, Quashie PK, Quan Y, Brenner BG, Wainberg MA. 2011. Compensation by the E138K mutation in HIV-1 reverse transcriptase for deficits in viral replication capacity and enzyme processivity associated with the M184I/V mutations. J Virol 85:11300–11308. doi: 10.1128/JVI.05584-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. 2010. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464:232–236. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hare S, Maertens GN, Cherepanov P. 2012. 3′-processing and strand transfer catalysed by retroviral integrase in crystallo. EMBO J 31:3020–3028. doi: 10.1038/emboj.2012.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Webb B, Sali A. 2014. Protein structure modeling with MODELLER. Methods Mol Biol 1137:1–15. doi: 10.1007/978-1-4939-0366-5_1. [DOI] [PubMed] [Google Scholar]
- 25.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. 2007. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci Chapter 2:Unit 2.9. doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- 26.Sheik SS, Sundararajan P, Hussain AS, Sekar K. 2002. Ramachandran plot on the web. Bioinformatics 18:1548–1549. doi: 10.1093/bioinformatics/18.11.1548. [DOI] [PubMed] [Google Scholar]
- 27.Kolaskar AS, Sawant S. 1996. Prediction of conformational states of amino acids using a Ramachandran plot. Int J Pept Protein Res 47:110–116. [DOI] [PubMed] [Google Scholar]
- 28.Prlic A, Bliven S, Rose PW, Bluhm WF, Bizon C, Godzik A, Bourne PE. 2010. Pre-calculated protein structure alignments at the RCSB PDB website. Bioinformatics 26:2983–2985. doi: 10.1093/bioinformatics/btq572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhuyan MS, Gao X. 2011. A protein-dependent side-chain rotamer library. BMC Bioinformatics 12(Suppl 14):S10. doi: 10.1186/1471-2105-12-S14-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zembower DE, Liao S, Flavin MT, Xu ZQ, Stup TL, Buckheit RW Jr, Khilevich A, Mar AA, Sheinkman AK. 1997. Structural analogues of the calanolide anti-HIV agents: modification of the trans-10,11-dimethyldihydropyran-12-ol ring (ring C). J Med Chem 40:1005–1017. [DOI] [PubMed] [Google Scholar]
- 31.Vink C, Oude Groeneger AM, Plasterk RH. 1993. Identification of the catalytic and DNA-binding region of the human immunodeficiency virus type I integrase protein. Nucleic Acids Res 21:1419–1425. doi: 10.1093/nar/21.6.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bushman FD, Engelman A, Palmer I, Wingfield P, Craigie R. 1993. Domains of the integrase protein of human immunodeficiency virus type 1 responsible for polynucleotidyl transfer and zinc binding. Proc Natl Acad Sci U S A 90:3428–3432. doi: 10.1073/pnas.90.8.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woerner AM, Marcus-Sekura CJ. 1993. Characterization of a DNA binding domain in the C-terminus of HIV-1 integrase by deletion mutagenesis. Nucleic Acids Res 21:3507–3511. doi: 10.1093/nar/21.15.3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engelman A, Hickman AB, Craigie R. 1994. The core and carboxyl-terminal domains of the integrase protein of human immunodeficiency virus type 1 each contribute to nonspecific DNA binding. J Virol 68:5911–5917. [DOI] [PMC free article] [PubMed] [Google Scholar]