

Abstract
In a previous report (T. J. Dougherty, A. Nayar, J. V. Newman, S. Hopkins, G. G. Stone, M. Johnstone, A. B. Shapiro, M. Cronin, F. Reck, and D. E. Ehmann, Antimicrob Agents Chemother 58:2657–2664, 2014), a novel bacterial type II topoisomerase inhibitor, NBTI 5463, with activity against Gram-negative pathogens was described. First-step resistance mutations in Pseudomonas aeruginosa arose exclusively in the nfxB gene, a regulator of the MexCD-OprJ efflux pump system. The present report describes further resistance studies with NBTI 5463 in both Pseudomonas aeruginosa and Escherichia coli. Second-step mutations in P. aeruginosa arose at aspartate 82 of the gyrase A subunit and led to 4- to 8-fold increases in the MIC over those seen in the parental strain with a first-step nfxB efflux mutation. A third-step mutant showed additional GyrA changes, with no changes in topoisomerase IV. Despite repeated efforts, resistance mutations could not be selected in E. coli. Genetic introduction of the Asp82 mutations observed in P. aeruginosa did not significantly increase the NBTI MIC in E. coli. However, with the aspartate 82 mutation present, it was possible to select second-step mutations in topoisomerase IV that did lead to MIC increases of 16- and 128-fold. As with the gyrase aspartate 82 mutation, the mutations in topoisomerase IV did not by themselves raise the NBTI MIC in E. coli. Only the presence of mutations in both targets of E. coli led to an increase in NBTI MIC values. This represents a demonstration of the value of balanced dual-target activity in mitigating resistance development.
INTRODUCTION
Gram-negative infections are currently a focus of increased concern in the medical community. The options for treatment of these pathogens have narrowed as a myriad of resistance mechanisms have emerged to challenge virtually every class of existing antibiotic (1–4). At the same time, attempts to identify fundamentally new targets and effective novel compounds to control multidrug-resistant (MDR) Gram-negative infections have largely fallen short of their goals. Although many novel compounds have been found in high-throughput screening (HTS) campaigns, it has proven extremely difficult to advance these to leads that can progress to the clinical testing phase. While some promising compounds, such as ceftazidime-avibactam, ceftolozane-tazobactam, plazomicin, and eravacycline, are in the late stages of clinical development (5–7), ongoing effective control of infections caused by MDR Gram-negative pathogens will demand additional new therapeutic compounds.
Given the failure of several novel target efforts to develop compounds that are effective in patients (8, 9), one strategy that has emerged is to identify new compounds that engage existing validated antimicrobial targets. There have been several reports of novel, nonquinolone compounds that interact with bacterial type II topoisomerases gyrase and topoisomerase IV (TopoIV); however, these are almost exclusively active primarily against Gram-positive pathogens, although one class of tricyclic compounds has broader activity (10–17). We have recently reported on a chemical class of topoisomerase inhibitors with activity against several Gram-negative multidrug resistant pathogens; these compounds have been termed novel bacterial type II topoisomerase inhibitors (NBTI) (11, 18). As previously reported, these compounds inhibit the two topoisomerase targets in the bacterial cell, gyrase and topoisomerase IV, in a manner distinct from that of the fluoroquinolone class, with inhibition not resulting in a cleavage complex (19, 20). The NBTI compounds were also found to maintain MIC activity against strains with topoisomerase mutations that impair fluoroquinolone target inhibition (18). In the initial report, we identified first-step resistance mutations in Pseudomonas aeruginosa exclusively in the nfxB regulator gene controlling the expression of the MexCD, OprJ efflux pump system (21). Subsequently, additional resistance development studies were performed to obtain a more detailed understanding of the NBTI compound resistance mechanisms.
In the present report, we describe target-based resistance mutations in the type II topoisomerases of both P. aeruginosa and Escherichia coli that affect susceptibility to NBTI 5463. These mutations were mapped in the target genes and found to be located on sites in the target enzymes that are distinct from the amino acid changes observed in fluoroquinolone resistance mutations. An interesting finding reported herein is that in the case of E. coli, single-target mutations in either gyrase or topoisomerase IV did not result in a significant increase in the MIC to the NBTI compound. Only after the introduction of mutations in both target enzymes was an increase in the MIC observed.
MATERIALS AND METHODS
Strains and media.
Experiments to generate resistant strains for characterization were performed with P. aeruginosa PAO1 and E. coli W3110. Experiments for resistant mutant selection and characterization were performed in LB broth and on LB agar. Susceptibility testing was performed in cation-adjusted Mueller-Hinton broth according to Clinical and Laboratory Standards Institute (CLSI) guidelines (22). A complete list of the strains employe d in this study is presented in Table 1.
TABLE 1.
Strain details
| Strain | Genotype |
|---|---|
| P. aeruginosa PA01 | |
| AZ301 | 4-bp deletion from bp 490 to 493 in nfxB |
| AZ391 | D82E in GyrA, 4-bp deletion from bp 490 to 493 in nfxB |
| AZ392 | D82E in GyrA, 4-bp deletion from bp 490 to 493 in nfxB |
| AZ393 | D82E in GyrA, 4-bp deletion from bp 490 to 493 in nfxB |
| AZ394 | D82N in GyrA, 4-bp deletion from bp 490 to 493 in nfxB |
| AZ395 | D82N in GyrA, 4-bp deletion from bp 490 to 493 in nfxB |
| AZ396 | D82G in GyrA, 4-bp deletion from bp 490 to 493 in nfxB |
| AZ397 | D82E in GyrA, 4-bp deletion from bp 490 to 493 in nfxB |
| AZ398 | D82N in GyrA, 4-bp deletion from bp 490 to 493 in nfxB |
| AZ789 | D82E and D87Y in GyrA, 4-bp deletion from bp 490 to 493 in nfxB |
| AZ790 | D82E and D87Y in GyrA, 4-bp deletion from bp 490 to 493 in nfxB |
| AZ791 | D82E and D87Y in GyrA, 4-bp deletion from bp 490 to 493 in nfxB |
| AZ792 | D82E and D87Y in GyrA, 4-bp deletion from bp 490 to 493 in nfxB |
| AZ799 | AZ392 with pMMB67 |
| AZ800 | AZ392 with pMMB67::nfxB |
| AZ801 | AZ394 with pMMB67 |
| AZ802 | AZ394 with pMMB67::nfxB |
| AZ803 | AZ396 with pMMB67 |
| AZ804 | AZ396 with pMMB67::nfxB |
| E. coli W3110 | |
| AZ480 | D82E in GyrA, yfaL is replaced by kan |
| AZ481 | D82G in GyrA, yfaL is replaced by kan |
| AZ484 | D82E in GyrA, K277N and K278L in ParC, yfaL is replaced by kan |
| AZ486 | D82E in GyrA, P439Q in ParE, yfaL is replaced by kan |
| AZ489 | D82G in GyrA, D79G in ParC, yfaL is replaced by kan |
| AZ530 | D82E in GyrA, K277N and K278L in ParC, markerless deletion of yfaL |
| AZ674 | D82E in GyrA, K277N and K278L in ParC, markerless deletion of yfaL, pKD46 (23) is present |
| AZ687 | D82E in GyrA, K277N and K278L in ParC, markerless deletion of yfaL, ygiU is replaced by kan |
| AZ699 | K277N and K278L in ParC, markerless deletion of yfaL, ygiU is replaced by kan |
| AZ704 | D82E in GyrA, D79G in ParC, markerless deletion of yfaL |
| AZ711 | D82E in GyrA, D79G in ParC, markerless deletion of yfaL, pKD46 is present |
| AZ717 | D82E in GyrA, D79G in ParC, markerless deletion of yfaL, ygiU is replaced by kan |
| AZ727 | D79G in ParC, markerless deletion of yfaL, ygiU is replaced by kan |
Resistant mutant selection.
P. aeruginosa PAO1 (AZ301) and E. coli W3110 were both grown overnight in LB broth with shaking at 37°C. The P. aeruginosa PAO1 strain designated AZ301 had a 4-bp deletion in the nfxB coding region (18), leading to an MIC increase against the NBTI compound over that of the baseline PAO1 parent strain. For selection, 2 ml of cells from an overnight culture were centrifuged, resuspended in 0.5 ml of LB, and plated on 1×, 2×, 4×, and 8× MIC of NBTI 5463 on large-diameter (150-mm) plates. In addition, 10-fold serial dilutions of the liquid cultures were plated on antibiotic-free LB agar to determine the total number of bacteria. Plates were examined for potentially resistant colonies after 24 and 48 h of incubation at 37°C.
Gene amplification and sequencing.
The genes for the gyrase and TopoIV subunits GyrA, GyrB, ParC, and ParE were amplified from P. aeruginosa and E. coli genomic DNA using previously described methods (18). DNA primers employed are listed in Table S1 in the supplemental material. Dideoxy DNA sequencing was performed using an Applied Biosystems 3100 series genetic analyzer.
Introduction of Asp82Glu and Asp82Gly in GyrA of E. coli.
Wild-type gyrA was cloned into pET-46 EK/LIC (EMD Millipore, Billerica, MA) to obtain pJT596. pAN125 and pAN126 were obtained by introducing the missense mutations Asp82Glu and Asp82Gly into pJT596 by site-directed mutagenesis using the QuikChange kit from Agilent Technologies (Santa Clara, CA). The first 500 bp of the gyrA gene were amplified from pAN125 and pAN126. The PCR products were transformed into E. coli BW25113 (23, 24). Two different isolates with the Asp82Glu or Asp82Gly mutation encoded in gyrA were selected at a concentration of 1× MIC of NBTI5463 in the BW25113 background and sequenced to confirm the presence of the mutations. Since the change in MIC of these mutants to NBTI5463 was found to be minimal, to readily move these gyrA mutations to a clean genetic background in W3110 via P1 phage transduction, strains with a kanamycin resistance gene in adjacent nonessential genes were constructed.
Construction of the kanamycin-resistant strains for P1 phage cotransduction.
P1 cotransduction relies on proximity of the genes. Hence, the yfaL gene, located adjacent to the gyrA gene (Fig. 1), was replaced by a kanamycin-resistant (Kanr) gene (23) in strains containing the Asp82Glu and Asp82Gly missense mutations encoded in gyrA. P1 cotransduction was performed to move the tightly linked Kanr gene and Asp82 mutations in gyrA into W3110 to obtain AZ480 and AZ481 (25, 26) by selecting for kanamycin resistance. The proximity of the Kanr marker to the gyrA gene yielded greater than 95% cotransduction of the two genes with the P1 transducing phage. The cotransduction of the gyrA mutation with kanamycin resistance was confirmed by sequencing.
FIG 1.
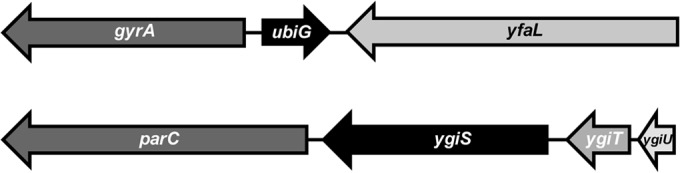
Closely linked genes are highly cotransduced by P1 phage generalized transduction. The kanamycin resistance genes were placed in yfaL and ygiU. (Top) Gene context for E. coli gyrA and yfaL, spanning approximately 6 kb. (Bottom) Gene context for E. coli parC and ygiU, spanning roughly 4 kb. P1 cotransduction of these closely linked genes exceeds 95%.
Mobilizing parC mutations in W3110.
Strains AZ484 and AZ489 were used to mobilize parC mutations in E. coli W3110. The first step was to delete the Kanr gene from yfaL using PCP20 (23) for flippase-mediated excision from AZ484 and AZ489. Subsequently, the open reading frame (ORF) of ygiU (selected due to its proximity to the parC gene) (Fig. 1) was replaced by the Kanr gene for cotransduction of parC mutations along with kanamycin resistance into the W3110 background. Thus, strains AZ687 and AZ717 were created, containing the Kanr gene in ygiU in addition to the gyrA and parC mutations. Two separate P1 lysates were prepared from AZ687 and AZ717 to transduce W3110 (24). Thus, strains with only ParC mutations were obtained: AZ699 (Lys277Asn and Lys278Leu encoded in parC) and AZ727 (Asp79Gly encoded in parC). Strain genotypes are listed in Table 1.
Mutant topoisomerase production.
The gene sequences encoding E. coli ParC (1 to 752) Asp79Gly and E. coli GyrA (1 to 875) Asp82Gly were synthesized with an N-terminal 6×His tag and a tobacco etch virus (TEV) protease cleavage recognition sequence (GenScript, Piscataway, NJ). The genes were cloned into a modified version of the pET-28b expression plasmid. The plasmids were designated pJT984 and pJT985, respectively. Purification of mutant proteins followed methods similar to those used for the wild-type proteins (13, 27), employing a column chromatography sequence of immobilized metal affinity chromatography, followed by size exclusion chromatography. The His6 and TEV purification tags were not removed, and protein purities were verified by SDS-PAGE and liquid chromatograph-mass spectrometry (LC-MS).
Topoisomerase activity assays.
Measurement of catalytic activity and compound inhibition of the E. coli wild-type and mutant gyrase and TopoIV enzymes was performed using assays that detect the formation of phosphate from DNA-dependent ATP hydrolysis (13, 28).
RESULTS
Resistance in P. aeruginosa.
We previously reported that the first-step resistance mutations identified in P. aeruginosa with the NBTI compounds were all found to be located in the nfxB regulator for the MexCD-OprJ efflux system (18). Using one of these strains, which contained a 4-bp deletion (490 to 493) in the 564-bp nfxB sequence, second-step resistance mutants were isolated by plating dilutions of cells (ca. 5 × 109 CFU) on multiple plates containing increasing concentrations of NBTI 5463. Table 2 indicates the resistance rates, which were low. The resistant strains exhibited 8- to 16-fold increases in the MIC to NBTI 5463 compared to that of the parental first-step mutant strain (Table 3). Genomic DNA was isolated from several of the independently isolated resistant mutants, and the four topoisomerase genes (gyrA, gyrB, parC, and parE) were sequenced. Mutations were found exclusively in the gyrA gene (Table 3) and represented single base pair changes that affected the codon for aspartate 82 to generate glutamate, glycine, or asparagine. The asparagine substitution was the only change to significantly reduce fluoroquinolone susceptibility as well.
TABLE 2.
Resistance rates of second-step P. aeruginosa mutants derived from first-step nfxB mutant
| Concn, × MIC (8 μg/ml) | Mutation rate |
|---|---|
| 1× | >5.4 × 10−9 |
| 2× | 1.1 × 10−9 |
| 4× | 5.0 × 10−10 |
| 8× | <5.4 × 10−10 |
TABLE 3.
MICs and sequence data for gyrase A second-step P. aeruginosa mutants
| Straina | GyrA mutation | MIC (μg/ml) |
||
|---|---|---|---|---|
| NBTI 5463 | Ciprofloxacin | Levofloxacin | ||
| PAO1 | 0.5 | 0.13 | 0.5 | |
| AZ301 (nfxB Δ490–493) | 16 | 1 | 2 | |
| AZ391 | D82E | 256 | 1 | 2 |
| AZ392 | D82E | 256 | 1 | 2 |
| AZ393 | D82E | 256 | 1 | 2 |
| AZ394 | D82N | 256 | 8 | 16 |
| AZ395 | D82N | 256 | 8 | 16 |
| AZ396 | D82G | 128 | 2 | 4 |
| AZ397 | D82E | 128 | 1 | 2 |
| AZ398 | D82N | 256 | 8 | 16 |
AZ391, -392, -393, and -394 were isolated at 2× MIC (32 μg/ml). AZ395, -396, -397, and -398 were isolated at 4× MIC (64 μg/ml).
In order to test the effect of the GyrA mutations in the absence of the background NfxB expression, isolates with each of the three GyrA D82 mutations were transformed with the plasmid pMMB67 containing an intact nfxB gene from P. aeruginosa PAO1, as well as an empty vector control for each strain. Table S2 in the supplemental material contains the results, which illustrate that as expected, the GyrA D82 mutations by themselves raise the MIC level to NBTI, but except for D82N (which has a very modest effect), there is no impact of GyrA D82 mutations on fluoroquinolone resistance.
Although the MIC values of the second-step mutants were well beyond any clinically relevant level (128 to 256 μg/ml), attempts were made to select higher-level, next-step mutants. Beginning with a strain that had both a 4-bp NfxB deletion and an Asp82Glu GyrA change (AZ397), a few resistant mutants were obtained on LB plates with high levels of NBTI 5463. All had MIC values of >1,024 μg/ml. Sequencing of the four topoisomerase genes of four independent resistant isolates identified an identical additional change in GyrA of aspartate 87 to tyrosine in all four independent isolates (Table 4) evaluated. The strains also had increased ciprofloxacin and levofloxacin MIC values. High-level quinolone resistance has been previously reported with changes at the GyrA aspartate 87 locus (29).
TABLE 4.
P. aeruginosa third-step high-level-resistance isolates
| Straina | MIC (μg/ml)b |
Mutationc |
|||
|---|---|---|---|---|---|
| NBTI 5463 | Ciproflox. | Levoflox. | GyrA | NfxB | |
| AZ397 | 128 | 1 | 2 | D82E | (490–493) 4-bp deletion |
| AZ789 | >1,024 | 8 | 8 | D82E, D87Y | (490–493) 4-bp deletion |
| AZ790 | >1,024 | 8 | 8 | D82E, D87Y | (490–493) 4-bp deletion |
| AZ791 | >1,024 | 8 | 8 | D82E, D87Y | (490–493) 4-bp deletion |
| AZ792 | >1,024 | 8 | 8 | D82E, D87Y | (490–493) 4-bp deletion |
AZ789, -790, -791, and -792 were isolated at 8× MIC (1,024 μg/ml).
Ciproflox., ciprofloxacin; Levoflox., levofloxacin.
Mutations were not observed in gyrB, parC, or parE in any of these strains.
We attempted to define the target potency of NBTI 5463 toward purified gyrase and TopoIV isolated from P. aeruginosa, but despite multiple attempts in protein production, we were unable to obtain either enzyme with satisfactory specific activity to allow for biochemical assays.
Resistance to NBTI in E. coli.
As with P. aeruginosa, dense cultures (2 × 109 cells) of E. coli were spread on large-diameter LB agar plates containing multiples of the compound MIC (2×, 4×, 8×, 16×). This procedure was repeated multiple times, but no colonies grew on plates at 2× MIC or above. Five colonies were isolated during several attempts with 1× MIC (0.5 μg/ml) of NBTI 5463. However, when retested, they did not exhibit elevated MIC values compared with those for the parent strain. Genomic DNA was also isolated from these 1× MIC strains, and the topoisomerase genes were sequenced. None of these isolates had any mutations in the four type II topoisomerase subunits.
Because repeated efforts did not isolate resistant mutants of E. coli, the Wanner λ red recombineering system in plasmid pKD46 was employed to introduce changes at aspartate 82 of GyrA, analogous to the changes observed in resistant P. aeruginosa described above. The Asp82Glu and Asp82Gly mutations were successfully introduced into E. coli, but repeated attempts to introduce the Asp82Asn mutation were unsuccessful. Surprisingly, the introduction of these changes, which had a significant impact on NBTI resistance in P. aeruginosa, caused minimal change in susceptibility to either the NBTI compound or fluoroquinolones in E. coli (Table 5).
TABLE 5.
MICs of E. coli strains with topoisomerase target mutationsa
| Strainb | MIC (μg/ml) |
Mutation(s) |
||||
|---|---|---|---|---|---|---|
| NBTI 5463 | Ciproflox. | Levoflox. | GyrA | ParC | ParE | |
| W3110 | 0.5 | 0.03 | 0.06 | |||
| AZ480 | 1 | 0.015 | 0.03 | D82E | ||
| AZ481 | 0.5 | 0.06 | 0.125 | D82G | ||
| AZ484 | 8 | 0.03 | 0.03 | D82E | K277N, K278L | |
| AZ486 | 32 | 0.03 | 0.06 | D82E | P439Q | |
| AZ489 | 64 | 0.125 | 0.25 | D82G | D79G | |
| AZ699 | 0.5 | 0.03 | 0.03 | K277N, K278L | ||
| AZ727 | 0.5 | 0.03 | 0.03 | D79G | ||
For all strains in the table, all four topoisomerase genes (gyrA, gyrB, parC, and parE) were sequenced.
AZ484 was isolated at 2× MIC (2 μg/ml), AZ486 was isolated at 4× MIC (4 μg/ml), and AZ489 was isolated at 16× MIC (8 μg/ml).
In order to explore further the effect of GyrA mutations on resistance to NBTI 5463 in E. coli, the GyrA Asp82Glu and Asp82Gly mutant strains were used for a second round of selection on LB plates against NBTI 5463. In this case, it was possible to select mutants at frequencies in the order of 10−8 to 10−9 with significantly decreased susceptibility (8 to 128-fold MIC increase) to NBTI 5463 (Table 5). Initially, these second-step mutants grew slowly on plates with NBTI 5463, taking 36 to 48 h to form small colonies, but immediately upon subsequent passage on LB plates with NBTI at the selection concentration grew at a normal rate. The initial slow growth may reflect a physiological shift or acquisition of additional compensatory mutations outside of the two topoisomerases. These second-step mutants had mutations in the ParC or ParE subunit of topoisomerase IV. The decreased susceptibility was confined to the NBTI compound, with no significant impact on the two fluoroquinolones tested.
The above results raised the question of whether the TopoIV mutations were solely responsible for the reduced susceptibility to NBTI 5463 or if it was also necessary to have the gyrA mutations in the strains' genetic background. To address this question, strains containing only the TopoIV mutations were constructed. Again, the DNA recombineering system was employed to first remove the Kanr marker from yfaL and to subsequently place a Kanr marker in the nonessential gene ygiU, adjacent to parC in the ASN484 and -489 strains. These were subsequently transduced via P1 phage into the parental E. coli W3110 background with selection for kanamycin resistance. The cotransduction of the TopoIV mutation was confirmed by DNA sequencing. As seen in Table 5, the presence of TopoIV subunit mutations alone was not sufficient to affect the susceptibility to NBTI 5463 or to the two fluoroquinolones tested.
With the observation that strains containing mutations in a single topoisomerase were not resistant to NBTI 5463, we next assessed whether the target-based mutations altered enzyme inhibition by NBTI 5463. Mutant E. coli GyrA and ParC proteins containing Asp82Gly and Asp79Gly, respectively, were expressed and purified. The mutant GyrA and ParC proteins were reconstituted with their parental GyrB or ParE partners, and enzyme activity was measured. Both reconstituted mutant topoisomerases exhibited specific activities comparable to those of their wild-type counterparts, and therefore, DNA-dependent ATPase assays were performed to measure 50% inhibitory concentrations (IC50s) for NBTI 5463 (Table 6). While the IC50s for the wild-type enzymes were within 2-fold of each other, the aspartate-to-glycine mutations in both enzymes resulted in large increases in the IC50. The NBTI 5463 MIC values of E. coli strains containing only one of the mutant type II topoisomerases, with the other being wild type, were the same as the MIC of the wild-type strain (Table 6).
TABLE 6.
Inhibition of gyrase and topoisomerase IV by NBTI 5463
| E. coli enzyme | GyrA/ParC status | IC50 (nM) of ATPasea | MIC (μg/ml) for E. colib |
|---|---|---|---|
| Gyrase | GyrA wild type | 5 ± 1 | 0.5 |
| GyrA D82G | >200 | 0.5 | |
| TopoIV | ParC wild type | 2.6 ± 0.1 | 0.5 |
| ParC D79G | >200 | 0.5 |
IC50s (mean ± SD) for inhibition of the indicated enzyme by NBTI 5463.
MIC versus strains W3110 for unaltered enzymes and strains AZ481 and AZ727 for the respective altered enzymes.
DISCUSSION
The NBTI series of compounds, including NBTI 5463, was developed with the goal of improved Gram-negative pathogen coverage and minimizing the potential for hERG cardiac channel inhibition (30). Earlier compounds with chemical similarities to the NBTI series were focused primarily on Gram-positive antibacterial activities (10–16).
We previously reported on the properties of the Gram-negative-series compound NBTI 5463, including both in vitro and animal infection model studies (18). In the original report, the preliminary resistance studies found that in P. aeruginosa, a number of different mutations that affected the nfxB regulator of the MexCD, OprJ efflux pump system arose upon selection. In the present study, we extended the study of NBTI 5463 resistance in P. aeruginosa by employing one of the nfxB mutants to select second-step mutations. These next-level mutants arose at a low level, and employing increasing concentrations of compound led to a decrease in the selection rate (Table 2). Sequencing of several mutants revealed that the resistance mutations were all found exclusively in the GyrA subunit, and the changes observed were isolated to a single locus, aspartate 82 of GyrA (Table 3). This residue is predicted to form a key compound binding interaction with the target, and mutations at the equivalent aspartate 83 in S. aureus have been reported to confer resistance to other NBTIs but not to alter fluoroquinolone susceptibility (10, 11). In P. aeruginosa, the aspartate 82 mutations all resulted in high-level NBTI resistance; however, fluoroquinolone susceptibility was unchanged in the Asp82Gly and Asp82Glu mutants but reduced 8-fold in the Asp82Asn mutant. As a possible explanation, given the proximity of aspartate 82 to threonine 83, which is known to influence fluoroquinolone susceptibility (29), it is conceivable that the Asp82Asn substitution alters the positioning of Thr83 enough to affect the water-metal ion bridge necessary for fluoroquinolone binding.
Employing these second-step gyrase mutants, a few isolates with extremely high-level resistance could also be selected (MIC > 1,024 μg/ml). In the clones isolated and tested, these mutations in response to NBTI 5463 were all found in this case to be in a second locus in GyrA, Asp87, that is associated with quinolone resistance, and indeed these isolates were cross resistant to the two fluoroquinolones tested. In P. aeruginosa, high-level fluoroquinolone resistance is associated with mutations in both gyrase and TopoIV, and mutations in gyrase appear to precede mutations in TopoIV (29, 31). The observed pattern for NBTI 5463, where in a GyrA mutant background, resistant mutations appeared again in GyrA, is an unusual finding. The appearance of the third-step mutant in a known fluoroquinolone resistance locus raises a concern about preexisting resistance to NBTI 5463. However, in a panel of 108 P. aeruginosa strains, NBTI 5463 displayed a MIC90 of 8 μg/ml for the 57 fluoroquinolone-resistant isolates tested, versus a MIC90 of 4 μg/ml for the 51 fluoroquinolone-susceptible strains (30). The isolates were not genotyped in that study, and future studies with genetically characterized strains that are susceptible and resistant to NBTI 5463 and fluoroquinolones are warranted.
A surprising finding was the repeated failure to raise any resistant mutants of E. coli against NBTI 5463. After several attempts, recombineering techniques were employed to introduce into E. coli the GyrA aspartate 82 mutations seen in P. aeruginosa by cotransduction with a selectable marker (Kanr) in an adjacent gene. Neither the Asp82Gly nor Asp82Glu changes in E. coli GyrA led to an increase in the MIC against the NBTI compound or the two fluoroquinolones tested. However, employing these strains with gyrase mutations selected isolates on NBTI 5463 plates that did have increased MIC values for the NBTI, and mutations in several locations in parC, or in one case parE, were found in the resistant mutants. One of the parC mutants (AZ489) contained an Asp79Gly mutation that is analogous to the GyrA Asp82Gly mutation in its parent strain. As with gyrase, aspartate 79 in ParC is adjacent to a common fluoroquinolone resistance mutation location, serine 80 (32). It is noteworthy that the single Asp79Gly mutant constructed by transduction had no change in fluoroquinolone susceptibility.
One possibility was that parC mutations in the double mutant were solely responsible for the decreased susceptibility to NBTI 5463. To test this, two of the NBTI-resistant mutants (AZ484 and AZ489) were used to produce two strains with parC mutations only. The resulting strains were found to be susceptible to both NBTI 5463 and fluoroquinolones, similar to the GyrA-only mutants. We conclude that NBTI 5463 resistance requires mutations in two topoisomerase target genes in E. coli, since mutations in either gene alone did not affect susceptibility to NBTI 5463. This explains the initial failure to select mutants on plates with compound, since it would require the very unlikely creation of two simultaneous mutations in the gyrase and topoisomerase IV targets to select a resistant isolate.
The parental and mutant forms of the enzymes were compared for levels of enzyme inhibition. Two pieces of information emerged from this study (Table 6). One, the IC50s for inhibition of gyrase and topoisomerase IV ATPase activities were very closely matched (within 2-fold) for the parent forms. Thus, the NBTI inhibitor is virtually equipotent in its E. coli target inhibition profile. Second, both mutant forms of the topoisomerase enzymes were resistant to NBTI 5463 inhibition. The maintained susceptibility seen in the single-target mutants demonstrates that NBTI 5463 is a dual-target inhibitor in E. coli. As a result of this property, the compound may encounter an E. coli strain with either a gyrA or parC single-target resistance mutation, but the equipotent nature of NBTI 5463 inhibition does not impact the MIC to permit this resistant mutant to survive and emerge.
The concept of dual targeting as an advantage for topoisomerase inhibitors has been recognized from the fluoroquinolone precedent (33–35). In Gram-positive pathogens, fluoroquinolones in general display greater potency toward TopoIV than gyrase, and as a result of this asymmetric target profile, earlier generations of quinolones readily selected for first-step ParC mutants. Recognizing that balanced, dual targeting should overcome this liability, subsequent evolution of the scaffold resulted in compounds with lower resistance rates that indeed possessed balanced S. aureus and S. pneumoniae gyrase-TopoIV profiles (36–39). In P. aeruginosa and E. coli, however, fluoroquinolones consistently select for gyrase first-step mutants and display greater target potency toward gyrase (29, 38).
For the NBTI class, knowledge is building on the target preferences in Gram-positive and Gram-negative pathogens and consequent ability to select for first-step target mutants. In one study, for example, NBTI analogs displayed equipotent target affinity for Streptococcus pneumoniae gyrase and TopoIV but an asymmetrical profile toward the Staphylococcus aureus enzymes, with greater affinity for S. aureus gyrase (40). Another group concluded that an asymmetrical target profile in S. aureus led to unacceptably high resistance frequencies (3 × 10−6 at 4× MIC) (41). This group directed compound optimization toward a balanced target profile in S. aureus and demonstrated that an analog with equipotent target affinities had a reduced spontaneous resistance mutation frequency (5 × 10−8 at 4× MIC).
Our work is the first to shed light on NBTI target preference and resistance potential in Gram-negative pathogens. P. aeruginosa and E. coli appear to follow different paths to resistance development. In our studies, changes in first-step mutants of P. aeruginosa appeared exclusively in an efflux pump regulator gene (42), and subsequent mutations appeared exclusively in two loci in gyrase only. The ability to select resistant strains carrying a mutation in gyrase suggests two possibilities: either NBTI 5463 has an unbalanced target profile in P. aeruginosa such that its TopoIV target affinity is significantly weaker than that for gyrase, or NBTI does inhibit topoisomerase IV potently in the mutant gyrase background but relies solely upon gyrase inhibition for antibacterial activity, which translates to an increased MIC in the resistant mutants with GyrA mutations. With further investigation, NBTI 5463 may become a valuable tool compound for understanding P. aeruginosa topoisomerase inhibitor pharmacology.
The situation for NBTI 5463 in E. coli is clearly different from that in P. aeruginosa. In E. coli, the compound displayed exquisitely balanced target affinity such that inhibition of only one of the two targets translated to the same MIC value as if both targets were inhibited. This represents, to our knowledge, the first demonstration of such balanced, dual targeting of topoisomerases in a Gram-negative pathogen. As a result, in E. coli, NBTI 5463 exemplifies the ideal of requiring two simultaneous mutations for resistance development. Even though NBTI 5463 did not possess a preclinical safety profile to justify its progression (30), we believe that future NBTI molecules that realize the potential of this series to be successful Gram-negative antibacterial agents will be developed.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kathy MacCormack and Keith Ferguson for support with DNA sequencing. Richard Alm is acknowledged for providing the E. coli wild-type gyrA clone.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04077-14.
REFERENCES
- 1.Livermore DM. 2009. Has the era of untreatable infections arrived? J Antimicrob Chemother 64(Suppl 1):i29–i36. doi: 10.1093/jac/dkp255. [DOI] [PubMed] [Google Scholar]
- 2.Kunz AN, Brook I. 2010. Emerging resistant Gram-negative aerobic bacilli in hospital-acquired infections. Chemotherapy 56:492–500. doi: 10.1159/000321018. [DOI] [PubMed] [Google Scholar]
- 3.Tamma PD, Cosgrove SE, Maragakis LL. 2012. Combination therapy for treatment of infections with Gram-negative bacteria. Clin Microbiol Rev 25:450–470. doi: 10.1128/CMR.05041-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu ZQ, Flavin MT, Flavin J. 2014. Combating multidrug-resistant Gram-negative bacterial infections. Expert Opin Investig Drugs 23:163–182. doi: 10.1517/13543784.2014.848853. [DOI] [PubMed] [Google Scholar]
- 5.Lagacé-Wiens P, Walkty A, Karlowsky JA. 2014. Ceftazidime-avibactam: an evidence-based review of its pharmacology and potential use in the treatment of Gram-negative bacterial infections. Core Evid 9:13–25. doi: 10.2147/CE.S40698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhanel GG, Lawson CD, Zelenitsky S, Findlay B, Schweizer F, Adam H, Walkty A, Rubinstein E, Gin AS, Hoban DJ, Lynch JP, Karlowsky JA. 2012. Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin and amikacin. Expert Rev Anti Infect Ther 10:459–473. doi: 10.1586/eri.12.25. [DOI] [PubMed] [Google Scholar]
- 7.Sutcliffe JA, O'Brien W, Fyfe C, Grossman TH. 2013. Antibacterial activity of eravacycline (TP-434), a novel fluorocycline, against hospital and community pathogens. Antimicrob Agents Chemother 57:5548–5558. doi: 10.1128/AAC.01288-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hernandez V, Crépin T, Palencia A, Cusack S, Akama T, Baker SJ, Bu W, Feng L, Freund YR, Liu L, Meewan M, Mohan M, Mao W, Rock FL, Sexton H, Sheoran A, Zhang Y, Zhang YK, Zhou Y, Nieman JA, Anugula MR, Keramane el M, Savariraj K, Reddy DS, Sharma R, Subedi R, Singh R, O'Leary A, Simon NL, De Marsh PL, Mushtaq S, Warner M, Livermore DM, Alley MR, Plattner JJ. 2013. Discovery of a novel class of boron-based antibacterials with activity against Gram-negative bacteria. Antimicrob Agents Chemother 57:1394–1403. doi: 10.1128/AAC.02058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gwynn MN, Portnoy A, Rittenhouse SF, Payne DJ. 2010. Challenges of antibacterial discovery revisited. Ann N Y Acad Sci 1213:5–19. doi: 10.1111/j.1749-6632.2010.05828.x. [DOI] [PubMed] [Google Scholar]
- 10.Black MT, Stachyra T, Platel D, Girard AM, Claudon M, Bruneau JM, Miossec C. 2008. Mechanism of action of the antibiotic NXL101, a novel nonfluoroquinolone inhibitor of bacterial type II topoisomerases. Antimicrob Agents Chemother 52:3339–3349. doi: 10.1128/AAC.00496-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bax BD, Chan PF, Eggleston DS, Fosberry A, Gentry DR, Gorrec F, Giordano I, Hann MM, Hennessy A, Hibbs M, Huang J, Jones E, Jones J, Brown KK, Lewis CJ, May EW, Saunders MR, Singh O, Spitzfaden CE, Shen C, Shillings A, Theobald AJ, Wohlkonig A, Pearson ND, Gwynn MN. 2010. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466:935–940. doi: 10.1038/nature09197. [DOI] [PubMed] [Google Scholar]
- 12.Singh SB, Kaelin DE, Wu J, Miesel L, Tan CM, Meinke PT, Olsen D, Lagrutta A, Bradley P, Lu J, Patel S, Rickert K, Smith R, Soisson S, Wei C, Fukuda H, Kishii R, Takei M, Fukuda Y. 2014. Oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad spectrum antibacterial agents. ACS Med Chem Lett 5:609–614. doi: 10.1021/ml500069w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reck F, Alm R, Brassil P, Newman J, Dejonge B, Eyermann CJ, Breault G, Breen J, Comita-Prevoir J, Cronin M, Davis H, Ehmann D, Galullo V, Geng B, Grebe T, Morningstar M, Walker P, Hayter B, Fisher S. 2011. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II: broad-spectrum antibacterial agents with reduced hERG activity. J Med Chem 54:7834–7847. doi: 10.1021/jm2008826. [DOI] [PubMed] [Google Scholar]
- 14.Reck F, Alm RA, Brassil P, Newman JV, Ciaccio P, McNulty J, Barthlow H, Goteti K, Breen J, Comita-Prevoir J, Cronin M, Ehmann DE, Geng B, Godfrey AA, Fisher SL. 2012. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II with reduced pKa: antibacterial agents with an improved safety profile. J Med Chem 55:6916–6933. doi: 10.1021/jm300690s. [DOI] [PubMed] [Google Scholar]
- 15.Miles TJ, Axten JM, Barfoot C, Brooks G, Brown P, Chen D, Dabbs S, Davies DT, Downie DL, Eyrisch S, Gallagher T, Giordano I, Gwynn MN, Hennessy A, Hoover J, Huang J, Jones G, Markwell R, Miller WH, Minthorn EA, Rittenhouse S, Seefeld M, Pearson N. 2011. Novel amino-piperidines as potent antibacterials targeting bacterial type IIA topoisomerases. Bioorg Med Chem Lett 21:7489–7495. doi: 10.1016/j.bmcl.2011.09.117. [DOI] [PubMed] [Google Scholar]
- 16.Miles TJ, Hennessy AJ, Bax B, Brooks G, Brown BS, Brown P, Cailleau N, Chen D, Dabbs S, Davies DT, Esken JM, Giordano I, Hoover JL, Huang J, Jones GE, Sukmar SK, Spitzfaden C, Markwell RE, Minthorn EA, Rittenhouse S, Gwynn MN, Pearson ND. 2013. Novel hydroxyl tricyclics (e.g., GSK966587) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg Med Chem Lett 23:5437–5441. doi: 10.1016/j.bmcl.2013.07.013. [DOI] [PubMed] [Google Scholar]
- 17.Tari LW, Li X, Trzoss M, Bensen DC, Chen Z, Lam T, Zhang J, Lee SJ, Hough G, Phillipson D, Akers-Rodriguez S, Cunningham ML, Kwan BP, Nelson KJ, Castellano A, Locke JB, Brown-Driver V, Murphy TM, Ong VS, Pillar CM, Shinabarger DL, Nix J, Lightstone FC, Wong SE, Nguyen TB, Shaw KJ, Finn J. 2013. Tricyclic GyrB/ParE (TriBE) inhibitors: a new class of broad-spectrum dual-targeting antibacterial agents. PLoS One 8(12):e84409. doi: 10.1371/journal.pone.0084409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dougherty TJ, Nayar A, Newman JV, Hopkins S, Stone GG, Johnstone M, Shapiro AB, Cronin M, Reck F, Ehmann DE. 2014. NBTI 5463 is a novel bacterial type II topoisomerase inhibitor with activity against Gram-negative bacteria and in vivo efficacy. Antimicrob Agents Chemother 58:2657–2664. doi: 10.1128/AAC.02778-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laponogov I, Sohi MK, Veselkov DA, Pan XS, Sawhney R, Thompson AW, McAuley KE, Fisher LM, Sanderson MR. 2009. Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nat Struct Mol Biol 16:667–669. doi: 10.1038/nsmb.1604. [DOI] [PubMed] [Google Scholar]
- 20.Laponogov I, Pan XS, Veselkov DA, McAuley KE, Fisher LM, Sanderson MR. 2010. Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS One 5(6):e11338. doi: 10.1371/journal.pone.0011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purssell A, Poole K. 2013. Functional characterization of the NfxB repressor of the mexCD-oprJ multidrug efflux operon of Pseudomonas aeruginosa. Microbiology 159:2058–2073. doi: 10.1099/mic.0.069286-0. [DOI] [PubMed] [Google Scholar]
- 22.Clinical and Laboratory Standards Institute. 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Approved standard M07-A8, 8th ed. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 23.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy KC. 1998. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J Bacteriol 180:2063–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singer M, Baker TA, Schnitzler G, Deischel SM, Goel M, Dove W, Jaacks KJ, Grossman AD, Erickson JW, Gross CA. 1989. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol Rev 53:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomason LC, Costantino N, Court DL. 2007. E. coli genome manipulation by P1 transduction. Curr Protoc Mol Biol Chapter 1:Unit 1.17.1. doi: 10.1002/0471142727.mb0117s79. [DOI] [PubMed] [Google Scholar]
- 27.Shapiro A, Jahic H, Prasad S, Ehmann DE, Thresher J, Gao N, Hajec L. 2010. A homogeneous, high-throughput fluorescence anisotropy-based DNA supercoiling assay. J Biomol Screen 15:1088–1098. doi: 10.1177/1087057110378624. [DOI] [PubMed] [Google Scholar]
- 28.Shapiro AB, Andrews B. 2012. Allosteric inhibition of the DNA-dependent ATPase activity of Escherichia coli gyrase by a representative of a novel class of inhibitors. Biochem Pharmacol 84:900–904. doi: 10.1016/j.bcp.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Akasaka T, Tanaka M, Yamaguchi A, Sato K. 2001. Type II topoisomerase mutations in fluoroquinolone-resistant clinical strains of Pseudomonas aeruginosa isolated in 1998 and 1999: role of target enzyme in mechanism of fluoroquinolone resistance. Antimicrob Agents Chemother 45:2263–2268. doi: 10.1128/AAC.45.8.2263-2268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reck F, Ehmann DE, Dougherty TJ, Newman JV, Hopkins S, Stone G, Agrawal N, Ciaccio P, McNulty J, Barthlow H, O'Donnell J, Goteti K, Breen J, Comita-Prevoir J, Cornebise M, Cronin M, Eyermann CJ, Geng B, Carr GR, Pandarinathan L, Tang X, Cottone A, Zhao L, Bezdenejnih-Snyder N. 2014. Optimization of physicochemical properties and safety profile of novel inhibitors of bacterial topoisomerase type II (NBTIs) with activity against Pseudomonas aeruginosa. Bioorg Med Chem 22:5392–5409. doi: 10.1016/j.bmc.2014.07.040. [DOI] [PubMed] [Google Scholar]
- 31.Bruchmann S, Dötsch A, Nouri B, Chaberny IS, Häussler S. 2013. Quantitative contributions to target alteration and decreased drug accumulation to Pseudomonas aeruginosa fluoroquinolone resistance. Antimicrob Agents Chemother 57:1361–1368. doi: 10.1128/AAC.01581-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgan-Linnell SK, Becnel Boyd L, Steffen D, Zechiedrich L. 2009. Mechanisms accounting for fluoroquinolone resistance in Escherichia coli clinical isolates. Antimicrob Agents Chemother 53:235–241. doi: 10.1128/AAC.00665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ng EY, Trucksis M, Hooper DC. 1996. Quinolone resistance mutations in topoisomerase IV: relationship to the flqA locus and genetic evidence that topoisomerase IV is the primary target and DNA gyrase is the secondary target of fluoroquinolones in Staphylococcus aureus. Antimicrob Agents Chemother 40:1881–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan XS, Ambler J, Mehtar S, Fisher LM. 1996. Involvement of topoisomerase IV and DNA gyrase as ciprofloxacin targets in Streptococcus pneumoniae. Antimicrob Agents Chemother 40:2321–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malik M, Hoatam G, Chavda K, Kerns RJ, Drlica K. 2010. Novel approach for comparing the abilities of quinolones to restrict the emergence of resistant mutants during quinolone exposure. Antimicrob Agents Chemother 54:149–156. doi: 10.1128/AAC.01035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ince D, Zhang X, Silver LC, Hooper DC. 2002. Dual targeting of DNA gyrase and topoisomerase IV: target interactions of garenoxacin (BMS-284756, T-3811ME), a new desfluoroquinolone. Antimicrob Agents Chemother 46:3370–3380. doi: 10.1128/AAC.46.11.3370-3380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strahilevitz J, Hooper DC. 2005. Dual targeting of topoisomerase IV and gyrase to reduce mutant selection: direct testing of the paradigm by using WCK-1734, a new fluoroquinolone, and ciprofloxacin. Antimicrob Agents Chemother 49:1949–1956. doi: 10.1128/AAC.49.5.1949-1956.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cambau E, Matrat S, Pan XS, Roth Dit Bettoni R, Corbel C, Aubry A, Lascols C, Driot JY, Fisher LM. 2009. Target specificity of the new fluoroquinolone besifloxacin in Streptococcus pneumoniae, Staphylococcus aureus and Escherichia coli. J Antimicrob Chemother 63:443–450. doi: 10.1093/jac/dkn528. [DOI] [PubMed] [Google Scholar]
- 39.Nilius AM, Shen LL, Hensey-Rudloff D, Almer LS, Beyer JM, Balli DJ, Cai Y, Flamm RK. 2003. In vitro antibacterial potency and spectrum of ABT-492, a new fluoroquinolone. Antimicrob Agents Chemother 47:3260–3269. doi: 10.1128/AAC.47.10.3260-3269.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitton-Fry MJ, Brickner SJ, Hamel JC, Brennan L, Casavant JM, Chen M, Chen T, Ding X, Driscoll J, Hardink J, Hoang T, Hua E, Huband MD, Maloney M, Marfat A, McCurdy SP, McLeod D, Plotkin M, Reilly U, Robinson S, Schafer J, Shepard RM, Smith JF, Stone GG, Subramanyam C, Yoon K, Yuan W, Zaniewski RP, Zook C. 2013. Novel quinoline derivatives as inhibitors of bacterial DNA gyrase and topoisomerase IV. Bioorg Med Chem Lett 23:2955–2961. doi: 10.1016/j.bmcl.2013.03.047. [DOI] [PubMed] [Google Scholar]
- 41.Surivet JP, Zumbrunn C, Rueedi G, Hubschwerlen C, Bur D, Bruyère T, Locher H, Ritz D, Keck W, Seiler P, Kohl C, Gauvin JC, Mirre A, Kaegi V, Dos Santos M, Gaertner M, Delers J, Enderlin-Paput M, Boehme M. 2013. Design, synthesis, and characterization of novel tetrahydropyran-based bacterial topoisomerase inhibitors with potent anti-Gram-positive activity. J Med Chem 56:7396–7415. doi: 10.1021/jm400963y. [DOI] [PubMed] [Google Scholar]
- 42.Monti MR, Morero NR, Miguel V, Argaraña CE. 2013. NfxB as a novel target for analysis of mutation spectra in Pseudomonas aeruginosa. PLoS One 8(6):e66236. doi: 10.1371/journal.pone.0066236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.