

Abstract
The apicoplast is an essential plastid organelle found in Plasmodium parasites which contains several clinically validated antimalarial-drug targets. A chemical rescue screen identified MMV-08138 from the “Malaria Box” library of growth-inhibitory antimalarial compounds as having specific activity against the apicoplast. MMV-08138 inhibition of blood-stage Plasmodium falciparum growth is stereospecific and potent, with the most active diastereomer demonstrating a 50% effective concentration (EC50) of 110 nM. Whole-genome sequencing of 3 drug-resistant parasite populations from two independent selections revealed E688Q and L244I mutations in P. falciparum IspD, an enzyme in the MEP (methyl-d-erythritol-4-phosphate) isoprenoid precursor biosynthesis pathway in the apicoplast. The active diastereomer of MMV-08138 directly inhibited PfIspD activity in vitro with a 50% inhibitory concentration (IC50) of 7.0 nM. MMV-08138 is the first PfIspD inhibitor to be identified and, together with heterologously expressed PfIspD, provides the foundation for further development of this promising antimalarial drug candidate lead. Furthermore, this report validates the use of the apicoplast chemical rescue screen coupled with target elucidation as a discovery tool to identify specific apicoplast-targeting compounds with new mechanisms of action.
INTRODUCTION
Despite encouraging progress over the past decade, malaria caused by Plasmodium parasites continues to pose an enormous disease burden (1). New antimalarials with novel mechanisms of action are needed to circumvent existing or emerging drug resistance (2). The apicoplast is a plastid organelle unique to Plasmodium spp. (and other pathogenic Apicomplexa parasites) and is a key target for development of new antimalarials. Due to its prokaryotic origin and evolution as a secondary plastid, it contains pathways that have no counterpart in the human host (3, 4). The apicoplast in Plasmodium is essential for both intraerythrocytic and intrahepatic development in the human host (5, 6).
Despite efforts to develop inhibitors of apicoplast function, to date, there have been no primary agents for treatment of acute malaria whose mechanism of action targets this unusual plastid organelle. Antibiotics that inhibit prokaryotic transcription and translation, such as doxycycline and clindamycin, block expression of the apicoplast genome and are active against Plasmodium parasites (5). Unfortunately, these drugs show a “delayed death” phenotype, in which growth inhibition occurs only after 2 replication cycles (96 h). The slow kinetics limit the use of doxycycline and clindamycin to chemoprophylaxis or as partner drugs in combination therapies with faster-acting compounds. Fosmidomycin, which inhibits the enzyme DoxR/IspC for MEP (methyl-d-erythritol-4-phosphate) isoprenoid precursor biosynthesis in the apicoplast, has immediate onset but shows high recrudescence rates clinically when used as monotherapy (7, 8). The efficacy of fosmidomycin-based combination therapy is currently being evaluated, with mixed results (9–12). Development of new apicoplast inhibitors as antimalarials has been challenging due to gaps in our knowledge of apicoplast biology and specific pathways and proteins to target.
As an alternative to target-specific approaches, several large-scale “chemical genetics” screens have been carried out to identify compounds with antimalarial activity, defined by growth inhibition of Plasmodium falciparum blood cultures (13–15). This approach (i) directly measures a disease-relevant phenotype while (ii) interrogating all cellular pathways in (iii) an unbiased manner to identify the most drug-sensitive nodes, even if the target proteins had not been previously obvious or even characterized (16). Forward chemical genetics is particularly useful in Plasmodium, where the lack of practical methods for large-scale mutant analysis to deconvolute new pathways or of conditional knockouts to investigate essential functions prohibits the genetic approaches that have been so powerful in the past in other model systems (17). However, major drawbacks to this approach are that, once a compound is identified as growth inhibitory, its target protein or pathway may be difficult to decipher and may be one of multiple targets if the compound is nonspecific (16).
Our previous work demonstrated that the essential function of the apicoplast in P. falciparum blood-stage parasites is the production of isoprenoid precursors, isopentenyl pyrophosphate (IPP) and its isomer dimethylallyl pyrophosphate (DMAPP), by the prokaryotic MEP pathway (18). The remaining plastid pathways are required to house this critical biosynthetic activity and to supply it with cofactors and substrates. We demonstrated this by generating P. falciparum parasites that lacked apicoplasts but could be chemically rescued by addition of IPP to the growth media. IPP chemical rescue represents an exciting opportunity to carry out a simple pathway-specific screen to identify small molecules that target the apicoplast. Compounds whose antimalarial growth inhibition is eliminated by the addition of IPP would be revealed to target essential pathways for apicoplast function. IPP has already been shown to rescue growth inhibition by fosmidomycin and antibiotics (18). This chemical rescue screen retains all the benefits of an unbiased, phenotypic screen but overcomes the main drawbacks by (i) ensuring specificity and (ii) providing important insight into the biological target and mechanism of action.
In principle, a chemical rescue screen, followed by target elucidation, enables discovery of apicoplast inhibitors with new mechanisms of action. However, this strategy has yet to be proven as a discovery tool. Recently, the inhibitor MMV-08138 was identified by an IPP chemical rescue screen as having specific activity against the apicoplast (19, 20). Unfortunately, the target of the inhibitor was unknown and therefore the mechanism of apicoplast dysfunction was unclear. Here, we provide evidence for the mechanism of action of MMV-08138 via targeting of IspD, an enzyme in the key isoprenoid precursor biosynthesis pathway in the apicoplast.
MATERIALS AND METHODS
Chemicals.
Racemic MMV-08138 was purchased from Sigma. Diastereomers of MMV-08138 were purchased as custom syntheses from NuChem Therapeutics; enantiopurity was verified by high-pressure liquid chromatography with a UV detector (HPLC-UV) on a ChiralceOID-RH column.
P. falciparum cultures.
P. falciparum W2 (MRA-157), D10 (MRA-201), and D10 ACPL-green fluorescent protein (GFP) (MRA-568) were obtained from MR4. Parasites were grown in human erythrocytes (2% hematocrit [Hct]) in RPMI 1640 media supplemented with 0.25% Albumax II (Gibco Life Technologies), 2 g/liter sodium bicarbonate, 0.1 mM hypoxanthine, 25 mM HEPES (pH 7.4), and 50 μg/liter gentamicin, at 37°C, 5% O2, and 6% CO2. For D10 ACPL-GFP, the medium was also supplemented with100 nM pyrimethamine (Sigma). For passage of drug-treated, IPP-rescued parasites, the medium was supplemented with 5 μM drug and 200 μM IPP (Isoprenoids LC). For comparisons of the growth rates determined under different treatment conditions, cultures were carried simultaneously and handled identically with respect to changes of media and addition of blood cells.
Growth inhibition assays.
P. falciparum W2 cultures (125 μl) were grown in 96-well plates containing serial dilutions of drugs in triplicate. Medium was supplemented with 200 μM IPP as indicated. Growth was initiated with ring-stage parasites at 1% parasitemia (P) and 0.5% hematocrit. Plates were incubated for 72 h. Growth was terminated by fixation with 1% formaldehyde, and parasitized cells were stained with 50 nM YOYO-1 (Invitrogen). Parasitemia was determined by flow cytometry. Data were analyzed by the use of BD C6 Accuri C-Sampler software and EC50 curves plotted by GraphPad Prism. For reporting EC50s, fresh drug stocks made from dry powder were used to minimize loss of efficacy of the compound during storage in solution.
Selection of drug-resistant mutants. (i) Selection 1.
P. falciparum W2 cultures were grown as described above in 6-well culture plates. Each well contained 10 ml of culture media at 2% hematocrit (Hct) and a starting parasitemia (percent P) of 5%. On day 0, cultures were treated with 9.3 μM MMV-08138 (Sigma; racemic mix), a concentration equal to 12 times the EC50. The parasites were maintained under drug pressure with a daily medium change, and cultures were split 1:2 and supplemented with fresh erythrocytes every 7 days. Parasite growth was monitored 2 times a week until ring-stage parasites were observed.
(ii) Selection 2.
A two-step resistance selection was carried out as follows. A culture of W2 parasites (500 ml, 2% Hct, 15% P, ∼1.5 × 1010 parasites) was treated with an amount of the 1R,3S diastereomer of MMV-08138 corresponding to the IC75 (600 nM). After 7 days of daily changes of media, no parasites were visible on a Giemsa-stained smear. The culture was maintained with changes of media every 3 days and split 1:2 with fresh erythrocytes. Resistant parasites were observed emerging on day 43 of the treatment, and on day 49 a split of the resistant line was seeded (50 ml, 2% Hct, 7.6% P, ∼8 × 108 parasites) in a standard T-150 flask with treatment increased to a 6 μM concentration, or 30 times the EC50, of the 1R,3S diastereomer of MMV-08138. After 7 days of daily changes of media, no parasites were visible on a GIEMSA-stained smear, and the culture was maintained thereafter with changes of media every 3 days. On day 75, a resistant line emerged in the stepped-up split with visible growth defects that disappeared when the treatment was reduced to a 2 μM concentration of the 1R,3S diastereomer of MMV-08138. Concentrations were adjusted according to EC50 measurements performed on drug stocks used in the selection and may have differed slightly from the reported EC50 on freshly made drug stocks.
gDNA isolation.
In order to isolate genomic DNA (gDNA) from drug-resistant parasites, parasitized cultures underwent two consecutive cycles of synchronization (per 5% sorbitol treatment). Once highly synchronous (>90% ring stage) and at 10% parasitemia, parasites were extracted by lysing red blood cells (RBCs) with 0.1% saponin (Sigma) for 5 min. Intact parasites were washed twice with phosphate-buffered saline (PBS) and resuspended in buffer (150 mM NaCl, 10 mM EDTA, 50 mM Tris, pH 7.5). Subsequently, resuspended parasites were lysed by overnight incubation at 37°C by addition of 0.1% l-loril sarkosil (Teknova) and 200 μg/ml proteinase K (NEB). Following parasite lysis, nucleic acids were extracted with phenol-chloroform-isoamyl alcohol (Ambion) (25:24:1; pH 7.88 to 7.92) using phase-lock tubes (5 Prime). RNA was digested with 100 μg/ml RNase A (Qiagen) treatment for 1 h at 37°C. Subsequently, gDNA was extracted twice more as described above and once with 100% chloroform. Ethanol precipitation was carried out using conventional methods, and purified gDNA was stored at −20°C.
Whole-genome sequencing.
Illumina-compatible 131-nucleotide (nt) paired-end libraries were made from 25 ng of purified gDNA using a Nextera DNA Sample Prep kit (Epicentre) per the supplier's instructions (however, the biotinylated PCR [bPCR] step was decreased to 6 cycles from 9 cycles with a modified extension step to 60°C for 6 min). Additionally, Illumina-compatible adaptors were included at this PCR step in place of Nextera Adapter 2. Library fragments of from 360 to 540 bp were size selected on a 5 XT DNA 750 chip via the use of a Lab Chip XT system (Caliper Life Sciences). Finally, a second PCR step using Klentaq LA DNA polymerase (Sigma-Aldrich) and 80% A/T deoxynucleoside triphosphates (dNTPs) was performed with the outer sequencing adaptors for 6 cycles (with an extension at 60°C for 6 min) to enrich for library fragments that were primed for sequencing. Preceding cluster generation, library concentrations were confirmed by quantitative PCR (qPCR) using Nextera adaptor sequences.
Pooled genomic DNA libraries at 2 nM each were sequenced using an Illumina HiSeq 2500 system. Cluster generation was performed via the use of cBot HiSeq Cluster kit v2 from Illumina at a final concentration of 6 to 8 pM and density of >400 k/mm2. Raw reads, consisting of 131-nt paired-end sequences, were filtered for low-quality reads using the PriceSeqFilter module, available as a component of the PRICE metagenomic assembler software package and publicly available at http://derisilab.ucsf.edu/software/price/index.html (21). Reads with an average per-base quality score of <1% probability of an incorrect base were retained and then mapped to the 3D7 reference genome (PlasmoDB release 9.3) using the short-read-alignment software bowtie, keeping reads that were uniquely mapping with no more than a single mismatch and excluding five nucleotides trimmed from both ends of each read. Following mapping, alignments were evaluated for the presence of copy number variants (CNVs) and mutations. For CNVs and coverage calculations, the R-package “CNV-Seq” was used in conjunction with SAMtools to detect amplifications or deletions with a P value of <0.001, using a 3-kb sliding window relative to the parental strain used to initiate each drug selection (22, 23). For mutation detection, a custom written python package (available upon request) was used to compare each bowtie alignment in the drug-selected strain to the parental strain and the 3D7 reference genome. For each detected variant, the number of reads featuring that variant is reported, as well as the percentage of the total reads for that nucleotide position and variant. Only variants covered by a depth of 30 or more reads were considered reliable.
Sanger sequencing.
The plasmodium IspD gene was PCR amplified from genomic DNA using primer pair 0.5F and 8R. PCR mixtures contained 0.5 μM primers, 0.2 mM dNTPs (80% AT), 0.04 U/μl Phusion polymerase, and 1.5 ng/μl gDNA. Thermocycling conditions were 94°C for 3 min; 30 cycles of 94°C for 30 s, 55°C for 30 s, and 60°C for 5 min; and final elongation of 60°C for 20 min. The resulting IspD PCR product was Sanger sequenced. Primer sequences were as follows: for 0.5F, 5′-GGCATAAATATATGCACACAC-3′; for 1R, 5′-TAC ACA TTT ATA CTG TGG AT-3′; for 2F, 5′-GAG AGA AGT ATC AGA AAA TG-3′; for 3F, 5′-TAT TAT GTG GAG GTA TAG GA-3′; for 3R, 5′-TTC CAC TTT CTA CAA TTT TT-3′; for 4F, 5′-ATT TTT CAT GTA TCA TCC AC-3′; for 4R, 5′-ATT AAC ATC TTC AAA TTC GT-3′; for 5F, 5′-AAT GTG ATC AAG ATG AAA AA-3′; for 6F, 5′-ACG AAT TTG AAG ATG TTA AT-3′; for 6R, 5′-TTT AAT AAA AGG GCT AGT TG-3′; for 7F, 5′-TTC AAA AGC TAC AGA TAC TA-3′; and for 8R, 5′-CATATCGAATTTAAGAATGCG-3′.
Cloning of a truncated P. falciparum IspD fusion protein for expression in Escherichia coli.
The plasmodium IspD gene was PCR amplified from wild-type W2 genomic DNA as described above for Sanger sequencing using the following primer pair: 5′-CAAGCATATGCTCGAGGGGATGCATTTTGTTCATACG-3′ and 5′-TTAGCAGCCGGATCCTCATTTTGAAGAATAATAAAATTTGTG-3′.
The resulting PCR product was purified using a Zymo DNA Clean & Concentrator-5 column (Zymo Research) and cloned using an In-Fusion PCR cloning kit (Clontech Laboratories, Inc.) into the XhoI site of a pET-19b T7 expression vector. The first 178 amino acids of IspD were then truncated using a QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies) and the following primer pair: 5′-GACGACAAGCATATGCTCGAGGGGTTTAATAAATATAATACAAAACAAT-3′ and 5′-ATTGTTTTGTATTATATTTATTAAACCCCTCGAGCATATGCTTGTCGTC-3′.
The truncated IspD was then PCR amplified from the pET-19b vector using the following primer pair: 5′-TACTTCCAATCCAATTTTAATAAATATAATACAAAACAATATG-3′ and 5′-TTATCCACTTCCAATTCATTTTGAAGAATAATAAAATTTGTG-3′.
The purified PCR product was In-Fusion cloned into the SspI site of vector 1C from the QB3 Macrolab (http://qb3.berkeley.edu/qb3/macrolab/), encoding an N-terminal His6-maltose binding protein (MBP)-N10-tobacco etch virus (TEV) protease site fusion tag (Pf-IspD-1C). This T7 expression system vector containing the IspD fusion was then transformed into BL21-CodonPlus (DE3)-RIL competent cells (Agilent Technologies, Inc.).
Expression of the PfIspD fusion protein in E. coli.
Overnight cultures were used to inoculate a 1-liter Luria-Bertani (LB)-kanamycin (KAN)-chloramphenicol (CHL) culture for protein expression at 37°C. Induction was initiated with 1 mM IPTG (isopropyl-β-d-1-thiogalactopyranoside) at an optical density at 600 nm (OD600) of 0.5, and protein expression was allowed to proceed for 3.5 h.
Bacterial cells were harvested by centrifugation at 7,000 × g, and the cell pellet was suspended in 50 mM phosphate buffer–300 mM NaCl–20 mM imidazole (pH 8.0) containing lysozyme (1 mg/ml) and a Complete EDTA-free protease inhibitor tablet (Roche) (one tablet per 25 ml buffer). Cells were incubated for 30 min at room temperature (RT) and then disrupted by sonication (20% amplitude, 6 bursts of 10 s with cooling in an ice bath for 10 s between bursts). After centrifugation at 20,000 × g for 30 min at 4°C, the supernatant was recovered and loaded onto a HisTrap HP 1-ml column (GE Healthcare Life Sciences) equilibrated in 50 mM phosphate buffer–300 mM NaCl–20 mM imidazole using an AKTApure chromatography system. The column was washed with the same buffer and eluted using an imidazole gradient (20 to 500 mM). Fractions were monitored for protein content by measuring absorbance at 280 nm and analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and silver staining. Fractions containing the crude desired IspD fusion were pooled and concentrated to 250 μl using an Amicon Ultra-4 centrifugal filter with a 30-molecular-weight cutoff (MWCO) (EMD Millipore). The crude protein was loaded onto a HiLoad 16/60 Superdex 200 gel filtration column (GE Healthcare) preequilibrated with 100 mM Tris-HCl (pH 8.0) buffer. The column was eluted at a flow rate of 1 ml/min, and fractions were monitored for protein content by measuring absorbance at 280 nm and analyzed by SDS-PAGE and silver staining. The fraction containing the desired IspD fusion was isolated, the concentration was determined using a bicinchoninic acid (BCA) assay kit (Pierce Biotechnology, Inc.), and the reaction mixture was stored at 4°C until further use. The recombinant Pf IspD protein sequence was confirmed by peptide sequencing with liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of trypsin-digested gel bands from the preparation.
Activity measurements of P. falciparum IspD fusion protein.
The enzymatic activity of IspD was measured using an EnzChek pyrophosphate assay kit (Life Technologies) adapted for a 96-well plate microplate format, allowing continuous monitoring of enzyme activity at pH 8.0 by measuring absorbance (360 nm) on a SpectraMax plate reader. Assay components were combined with enzyme (70.8 nM) and CTP (1 mM) in a 200-μl reaction volume and allowed to preequilibrate to 37°C for 10 min. The reaction was initiated by addition of 2C-MEP (methyl-d-erythritol-4-phosphate) (0 to 400 μM). Initial rates and kinetic parameters were calculated using GraphPad Prism (GraphPad Software). Enzyme activity at saturating MEP concentrations was also assayed in the presence of various concentrations of MMV-08138 stereoisomers which were added to the reaction mixture before preequilibration.
RESULTS
IPP rescue of MMV-08138 inhibition does not result in apicoplast loss.
The Medicines for Malaria Venture (MMV) Open-Access Malaria Box is a diverse library of 200 drug-like antimalarial compounds and 200 probe-like compounds compiled from 20,000 hits generated from previously reported large-scale screens (13–15). Previously, a screen for Malaria Box compounds showing an IPP rescue phenotype identified MMV-08138, indicating that it specifically targets an apicoplast pathway (Fig. 1A) (20; https://www.ebi.ac.uk/chembldb/doc/inspect/CHEMBL2448809). We confirmed the IPP rescue phenotype of MMV-08138, which had an EC50 of 772 nM (664 to 741 nM) against P. falciparum W2 strain in the absence of IPP but in the presence of IPP was at least 50-fold less potent, with an EC50 of ≥50 μM (Fig. 1B). The robust IPP rescue phenotype was also demonstrated by monitoring the growth of drug-treated parasites over several intraerythrocytic cycles. During the first cycle of treatment, MMV-08138 blocked the maturation of trophozoite parasites to schizonts and then subsequent reinvasion at 24 to 48 h (Fig. 1C). In contrast, in the presence of IPP, drug-treated parasites underwent continued growth and replication (Fig. 1C). Of note, parasites treated with MMV-08138 and rescued with IPP do not become dependent on IPP for growth after removal of the drug (Fig. 1C) or show a decrease in the apicoplast/nuclear genome ratio (see Fig. S1 in the supplemental material). Localization of apicoplast-targeted green fluorescent protein (GFP) expressed in transgenic D10 ACPL-GFP parasites was also unaffected (see Fig. S2) (25). Thus, unlike the results seen with prokaryotic translation inhibitors such as doxycycline and chloramphenicol, neither organellar genome expression nor organelle replication is affected by MMV-08138. The “immediate” death phenotype of MMV-08138 and maintenance of the apicoplast during drug treatment and IPP rescue are most consistent with an inhibition of apicoplast metabolic pathways rather than housekeeping, protein import, or organelle replication functions (18, 26).
FIG 1.
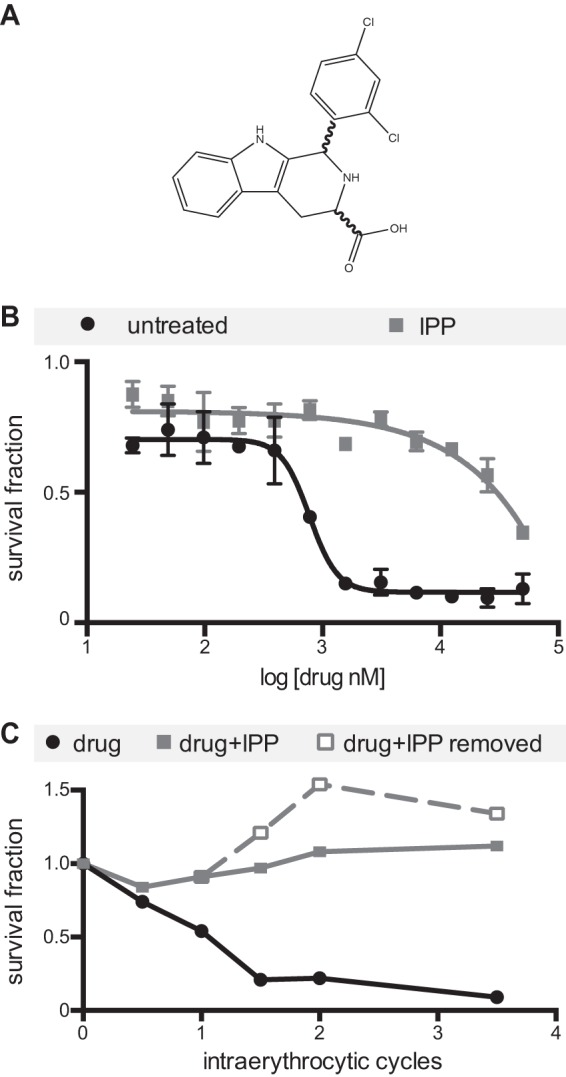
IPP rescue of growth inhibition by MMV-08138. (A) Chemical structure. (B) EC50 curves in the absence and presence of IPP. (C) Time course of growth through the intraerythrocytic cycle. Parasites were treated with drug only, drug plus IPP, or drug plus IPP followed by removal of drug and IPP after the first reinvasion. Parasitemia is normalized to that of an untreated control.
MMV-08138 inhibition is stereospecific.
A set of 29 compounds, which were structurally related to MMV-08138 but with unknown stereochemistry, was also tested for antimalarial growth inhibition and IPP rescue (see Fig. S3 and Table S1 in the supplemental material). These compounds were all less active than MMV-08138, due either to alterations in important functional groups or to improper stereochemistry. Notably, MMV-08138 contains 2 stereocenters, resulting in 4 possible diastereomers of this compound. Evaluation of its activity had thus far been carried out with an unspecified racemic mixture that could contain both active and inactive diastereomers (Fig. 1). However, knowledge of the stereospecificity of the inhibitor will be important for further structure-activity optimization and in vivo studies. Therefore, we obtained all 4 chirally pure diastereomers and evaluated each for its IPP-rescued growth inhibitory activity (Table 1; see also Fig. S4). The most active compound was the 1R,3S conformer, with an EC50 of 110 nM, which showed IPP rescue at up to 25 μM. The 1R,3R conformer was at least 30-fold less active but still showed IPP-rescuable activity at up to 25 μM. The 1S,3R conformer inhibited growth at an EC50 of 18.6 μM which was minimally rescued with IPP. Finally, the 1S,3S conformer was completely inactive. The stereospecificity of the growth inhibition indicated drug binding to a specific cellular target.
TABLE 1.
Inhibition by diastereomers of MMV-08138 of parasite growth and PfIspD enzyme activity
| Stereoisomer | Parasite growth (EC50 [μM]) under indicated conditions |
PfIspD activity (IC50 [μM])a | |
|---|---|---|---|
| No rescuea | +IPP | ||
| 1S,3S | >50b | >50b | >25c |
| 1R,3S | 0.11 (0.10–0.12) | >25c | 0.0071 (0.0061–0.0083) |
| 1R,3R | 3.8 (2.7–5.3) | >25c | 0.17 (0.14–0.20) |
| 1S,3R | 18.6 (12.4–28.0) | >25c | 2.5 (1.9–3.1) |
Values represent means (95% confidence intervals).
No inhibition was observed at 50 μM, the highest concentration tested.
Estimated value based on observation of partial inhibition.
MMV-08138-resistant populations can be selected under conditions of drug pressure.
In order to clarify the mechanism of action of MMV-08138, parasites resistant to MMV-08138 were generated in 2 independent selections (Fig. 2A). In the first selection, susceptible blood-stage P. falciparum parasites were directly exposed to a lethal dose of a racemic mixture of MMV-08138. Resistant parasites emerged after 20 days of continuous drug exposure. The resistant population from this selection, designated 08138R1, was determined to have an EC50 that was 12.7-fold greater than the EC50 of the initial susceptible population against the 1R,3S diastereomer of MMV-08138 (Fig. 2B). In the second selection, susceptible parasites were exposed to the 1R,3S diastereomer of MMV-08138 at a dose equal to the IC75. Resistant parasites, designated 08138R2, emerged after 43 days of continuous drug exposure and were found to have an EC50 that was 3.5-fold greater than that of the initial susceptible population (Fig. 2B). These 08138R2 parasites were then exposed to a lethal dose of MMV-08138 to generate a population, 08138R3, with an EC50 that was 19.2-fold greater than that of the initial population after a total of 75 days of continuous drug exposure (Fig. 2B).
FIG 2.
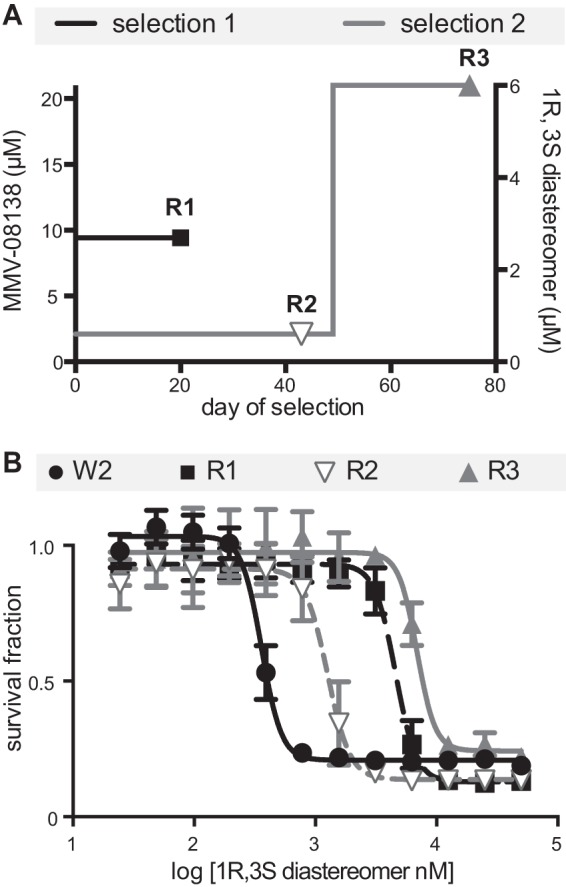
Selection of MMV-08138-resistant parasite populations. (A) Drug selection timeline. (B) EC50 curves of resistant populations.
Whole-genome sequencing of drug-resistant populations identifies mutations in IspD.
Each of the 3 drug-resistant populations 08138R1, 08138R2, and 08138R3 and the parent W2 strains used to begin each selection were subjected to whole-genome sequencing. After quality filtering and alignment to the reference sequence, the average coverage ranged from 136-fold to 366-fold for the 14 chromosomes and was >1,000-fold for the mitochondria and >300-fold for the plastid. Each data set was evaluated for the presence of copy number variants (CNVs), such as amplified regions. Other than the variable sequences proximal to the telomeres, no CNVs of significance (P < 0.001) were detected relative to the drug-sensitive parental strain (see Table S2 in the supplemental material). Unlike other examples of drug resistance in P. falciparum (27), this result suggests that a simple amplification of a drug target or resistance determinant was not responsible for the observed resistance in these three selections.
Using the mapped reads for each resistant population, genomic mutations were detected by comparison to the reference sequence and the parental strain. Our criteria for mutations relevant to the selection consisted of nonsynonymous mutations in which >90% of the reads at that position were “mutant” with respect to the parent and reference. Mutations relative to the reference strain, but which were identical in the parental strain and the selected populations, were not considered relevant. Using these criteria, we detected 3 nonsynonymous mutations in 08138R1, 3 in 08138R2, and 2 in 08138R3 (Table 2). In all three resistant populations, the only shared mutated gene was PF3D7_0106900, which encodes the putative 2-C methyl-d-erythritol 4-phosphate cytidylyltransferase (IspD) enzyme of the MEP isoprenoid precursor pathway (Fig. 3). One mutation found in 08138R2 was a change from glutamate to glutamine at position 688 in IspD. Another mutation, a change from leucine to isoleucine at position 244 of IspD, was found in both 08138R1 and 08138R3.
TABLE 2.
Summary of whole-genome sequencing resultsa
| Resistant population | Gene IDb | Descriptionc | nt position | Base call |
No. of reads |
% readsd |
AA changee | |||
|---|---|---|---|---|---|---|---|---|---|---|
| WT | Mut | WT | Mut | WT | Mut | |||||
| 08138R1 | PF3D7_0106900 | 2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase | 730 | T | A | 227 | 320 | 99.6 | 99.4 | L244I |
| PF3D7_1247500 | Putative protein kinase | 2632 | G | T | 110 | 73 | 97.3 | 100 | D878Y | |
| PF3D7_1456700 | Conserved Plasmodium protein | 1384 | G | A | 280 | 413 | 93.9 | 96.4 | E462K | |
| 08138R2 | PF3D7_0106900 | 2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase | 2062 | G | C | 85 | 69 | 100 | 91.3 | E688Q |
| PF3D7_0302900 | Putative exportin | 1620 | A | T | 154 | 91 | 61.7 | 97.8 | E540D | |
| PF3D7_1478600 | Plasmodium exported protein | 1241 | C | T | 66 | 59 | 100 | 96.6 | T414I | |
| 08138R3 | PF3D7_0106900 | 2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase | 730 | T | A | 151 | 188 | 100 | 100 | L244I |
| PF3D7_1417400 | Cyclic nucleotide binding protein pseudogene | 10270 | G | T | 112 | 94 | 55.3 | 100 | E3424* | |
AA, amino acid; Mut, mutant; nt, nucleotide; WT, wild type. WT calls match the 3D7 reference genome.
Gene ID, PlasmoDB gene identification number.
Basic gene description based on PlasmoDB functional assignments.
% reads, percentages of reads corresponding to the WT call or Mut call.
Asterisk indicates stop codon.
FIG 3.
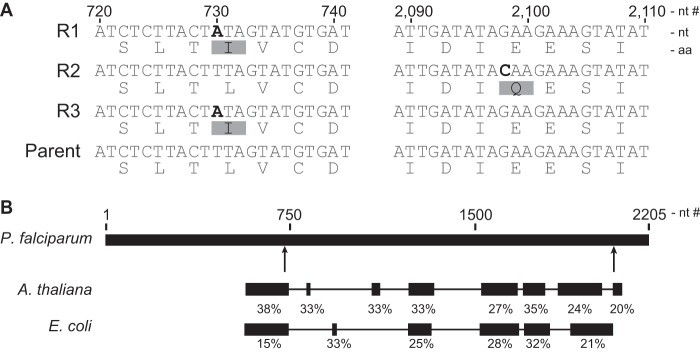
Identification of IspD mutations in MMV-08138-resistant parasites. (A) Mutations determined by whole-genome and Sanger sequencing of resistant populations 08138R1 (R1), 08138R2 (R2), and 08138R3 (R3). aa, amino acid; nt, nucleotide. (B) Block-level alignment of P. falciparum, A. thaliana, and E. coli IspD, with percent homology of each block. MMV-08138-resistant PfIspD mutation locations are denoted by arrows.
To confirm the presence of the identified mutations in IspD, the gene was PCR amplified from genomic DNA of the parent and resistant populations and the entire gene sequenced using the Sanger method (see Fig. S5 in the supplemental material). All the IspD mutations observed in the whole-genome sequencing data were validated by Sanger sequencing (Fig. 3A). In the case of population 08138R2, which was selected to only 91% mutant call purity, the PCR amplification product of IspD was cloned into a vector and transformed into E. coli, and 11 colonies were selected for sequencing. Ten colonies showed the E688Q mutation whereas one colony did not show the mutation, in agreement with the whole-genome-sequencing data. Several mutations relative to the reference strain were also shared by both the parental and selected populations (see Fig. S6). These data support the notion that the antiapicoplast action of MMV-08138 is due to inhibition of this critical isoprenoid precursor biosynthesis enzyme, given that mutations in this gene suppress its activity. A block-level amino acid alignment of IspD from P. falciparum with those from E. coli and Arabidopsis thaliana shows that PfIspD contains significant additional domains of unknown utility (Fig. 3B). However, both of the reported mutations do occur in positions proximal to conserved regions among the homologs of IspD.
MMV-08138 inhibits P. falciparum IspD activity in vitro.
To determine if MMV-08138 directly inhibits PfIspD, we heterologously expressed and purified His- and MBP-tagged PfIspD and measured its enzymatic activity using a pyrophosphate release assay. The purified enzyme was active, with a Km of 60.6 μM for MEP and a kcat of 0.16 s−1 (see Fig. S7 in the supplemental material). In the presence of various amounts of the 1R,3S diastereomer of MMV-08138, enzyme activity measured at saturating substrate concentrations was inhibited with an IC50 of 7.0 nM (Fig. 4 and Table 1). Inhibition by MMV-08138 of PfIspD activity was stereospecific, as the other diastereomers of MMV-08138 were all less potent (Fig. 4 and Table 1). The IC50s were similar when PfIspD activity was measured at substrate concentrations equal to Km, when the enzyme is not saturated (see Fig. S7).
FIG 4.
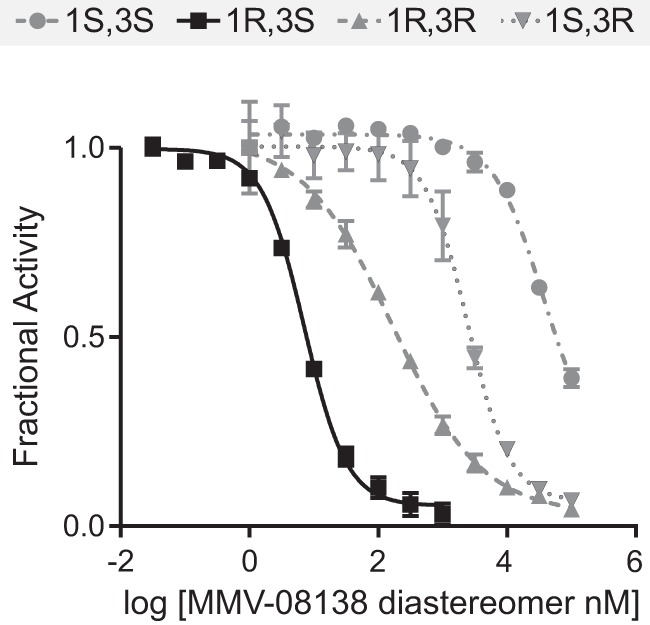
Inhibition of PfIspD enzyme activity by diastereomers of MMV-08138.
Interestingly, MMV-08138 inhibited PfIspD activity but did not inhibit E. coli, A. thaliana, or Plasmodium vivax IspD activity in vitro. We tested MMV-08138 in in vitro activity assays against MEP pathway enzyme homologs from various organisms. MMV-08138 showed no enzyme inhibition activity against E. coli DXS at up to a 100 μM concentration of the inhibitor, DXR/IspC at up 50 μM, or IspD, IspE, and IspF at 10 μM (see Fig. S8 in the supplemental material) (28–31). MMV-08138 also did not affect the enzyme activity of purified A. thaliana or P. vivax IspD at up to 1 mM inhibitor (see Fig. S9) (32, 33).
DISCUSSION
Both the genetic and biochemical data identify PfIspD, an enzyme in the key MEP isoprenoid precursor biosynthesis pathway in the apicoplast, as the molecular target of MMV-08138. The determinants of resistance to MMV-08138 were two mutations in IspD. The E688Q mutation was identified in a strain showing 3.5-fold (“low”) resistance. Meanwhile, two independent selections carried out with different protocols converged on the same L244I mutation, which was identified in both strains showing higher (13- to 19-fold) resistance. In all three resistant populations, the only mutated gene shared in common was the gene encoding IspD. Interestingly, the L244I mutation yielded the higher-resistance phenotype, and yet that mutation is a more subtle amino acid change than E688Q. Small amino acid changes can result in pronounced resistance phenotypes, as an isoleucine-to-leucine mutation in acetyl-coenzyme A (CoA) carboxylases has previously been demonstrated to confer herbicide resistance in plants (34, 35).
Based on the genetic results, further biochemical characterization demonstrated that MMV-08138 acts by directly binding and inhibiting IspD enzymatic activity. Consistent with this mechanism, the IC50 of the inhibitor against purified PfIspD was comparable to the EC50 against blood-stage P. falciparum parasites. The stereospecificity of the enzyme inhibition also paralleled that of cell growth inhibition. Surprisingly, MMV-08138 did not inhibit the enzyme activity of purified E. coli, A. thaliana, or P. vivax IspD (28, 32). This may be due to structural differences in IspD homologs in these species, as even the P. vivax IspD and P. falciparum IspD share only 31% identity. Further studies using the same detection assay and kinetic conditions to compare IspD homologs will be required to confirm the selectivity of MMV-08138 for IspD homologs from different species.
Further kinetic and structural characterization will be required to identify the mechanism of inhibition of PfIspD by MMV-08138. MMV-08138 may compete with either MEP or CTP for their respective substrate binding sites. Alternatively, MMV-08138 may bind at an allosteric site that affects enzyme activity. IspD may have important interactions with small molecules or proteins at allosteric sites to regulate its function. For example, interaction of IspD with other MEP enzymes may be important for the flux of intermediates through the pathway and final product formation. In addition, feedback regulation by MEP pathway intermediates and isoprenoid products has been shown for IspF, another pathway enzyme (36). Investigating how the L244I and E688Q mutations result in resistance to inhibition by MMV-08138 will also be revealing.
MMV-08138 has promise for drug development, with potent nM activity against infected red blood cells, drug-like properties, and synthetically accessible structural analogs to optimize in vivo activity (19). In particular, we showed that the 1R,3S diastereomer of MMV-08138 is most potent, which will be important for its structural optimization and for minimizing off-target effects in vivo. The apicoplast MEP pathway is a validated antimalarial target. While fosmidomycin has promise as a MEP pathway inhibitor against malaria, its development as a drug has been clinically hampered by its short half-life and high recrudescence rates (8). MMV-08138 has significant value as an alternative to fosmidomycin for development as an antimalarial MEP inhibitor. To date, few compounds targeting IspD in any organism have been developed, although the MEP pathway is a validated antibacterial and antimalarial target (32, 37) Notably, although humans do not have a functional MEP pathway, there is a human IspD homolog that is associated with Walker-Warburg syndrome, a congenital muscular dystrophy, and likely acts as a nucleotide-dependent glycosyltransferase (38, 39). Thus, it may be important to screen potential PfIspD inhibitors against binding to human IspD, although PfIspD and the human ISPD gene share minimal sequence similarity (20% identical at most over 102 residues that could be aligned by the Smith-Waterman algorithm). The heterologous expression and purification of active PfIspD will facilitate both high-throughput screening and structural biology efforts, which may further aid in the advancement of this target toward a preclinical lead compound.
An advantage of developing apicoplast-specific inhibitors is that the organelle is also essential in the liver and mosquito stages of infection, increasing the likelihood that the drug candidates will have activities against multiple stages of the parasite's complex life cycle (6, 40–42). Current MMV guidelines for compound development demand transmission blocking capability as part of target candidate profile 3, requiring a target vulnerable in the sexual stages and dormant liver stages (43). The efficacy of an IspD inhibitor against the sexual and liver stages will need to be determined. Furthermore, drug synergy with known apicoplast inhibitors, such as antibiotics or fosmidomycin, can also be explored (44). Notably, fosmidomycin-resistant parasites remained susceptible to MMV-08138 (20), and cross-resistance with fosmidomycin was not observed for the selected MMV-08138-resistant populations (see Fig. S10 in the supplemental material). Finally, as apicoplasts are found in other Apicomplexan parasites, developed drugs might also be used for treatment of diseases caused by Toxoplasma gondii and Babesia parasites.
In this work, the IPP chemical rescue phenotype and target elucidation identified an inhibitor that is active against a validated pathway with a known essential role in apicoplast biology. However, the same strategy has potential to reveal new pathways and targets we had not previously anticipated. Mechanism-of-action elucidation is facilitated by the availability of assays to interrogate specific apicoplast functions, in addition to unbiased methods such as drug resistance selection. Our previous work demonstrating that MEP isoprenoid precursor biosynthesis is the only essential function of the apicoplast in blood-stage P. falciparum had important implications for development of small-molecule inhibitors that are active against the apicoplast (18). Besides MEP isoprenoid precursor biosynthesis itself, there are few “classic” metabolic pathways to target. Instead, we need to pursue “nontraditional” pathways that are involved in maintaining organelle function or replicating the organelle during the Plasmodium life cycle. Unfortunately, our knowledge of these pathways from candidate proteins to mechanism is scarce. Due to the apicoplast's “exotic” evolution as a secondary plastid, there are few counterparts in other model organisms as starting points to tackle this unique but challenging biology. The forward chemical genetics approach described here is an opportunity to discover essential functions in the apicoplast that can be targeted by small molecules. These compounds will serve as starting points to (i) identify drug candidate leads with specificity for an apicoplast target, (ii) develop chemical tools to probe apicoplast pathways, and (iii) discover new pathway/protein targets in the apicoplast and novel modes of action.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Medicines for Malaria Ventures (MMV) for providing the Malaria Box compounds and making this valuable library freely available, as well as to Novartis for their screening efforts that first identified MMV-08138. We acknowledge Boris Illarionov and Markus Fischer (Universität Hamburg) for in vitro A. thaliana and P. vivax IspD assays, Katie Heflin and Caren Meyers (Johns Hopkins) for in vitro E. coli MEP enzyme assays, Giselle Knudsen and Michael Winter (UCSF Mass Spectrometry Facility) for mass spectrometry assistance, and Kip Guy (St. Jude Children's Hospital) for MMV-08138 structural analogs.
Funding support for this project was provided by the Stanford Consortium for Innovation, Design, Evaluation and Action (C-IDEA; E.Y.), NIH 1K08AI097239 (E.Y.), NIH 1DP5OD012119 (E.Y.), the Burroughs-Wellcome Fund (E.Y.), the Howard Hughes Medical Institute (J.L.D.), and a grant from the Grand Challenges Explorations, an initiative of the Bill & Melinda Gates Foundation (W.W.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03342-14.
REFERENCES
- 1.WHO. 2013. World Malaria Report 2013. WHO, Geneva, Switzerland. [Google Scholar]
- 2.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NPJ, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Köhler S, Delwiche CF, Denny PW, Tilney LG, Webster P, Wilson RJ, Palmer JD, Roos DS. 1997. A plastid of probable green algal origin in Apicomplexan parasites. Science 275:1485–1489. doi: 10.1126/science.275.5305.1485. [DOI] [PubMed] [Google Scholar]
- 4.Janouskovec J, Horák A, Oborník M, Lukes J, Keeling PJ. 2010. A common red algal origin of the apicomplexan, dinoflagellate, and heterokont plastids. Proc Natl Acad Sci U S A 107:10949–10954. doi: 10.1073/pnas.1003335107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dahl EL, Shock JL, Shenai BR, Gut J, DeRisi JL, Rosenthal PJ. 2006. Tetracyclines specifically target the apicoplast of the malaria parasite Plasmodium falciparum. Antimicrob Agents Chemother 50:3124. doi: 10.1128/AAC.00394-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stanway R, Witt T, Zobiak B, Aepfelbacher M, Heussler V. 2009. GFP-targeting allows visualization of the apicoplast throughout the life cycle of live malaria parasites. Biol Cell 101:415–430. doi: 10.1042/BC20080202. [DOI] [PubMed] [Google Scholar]
- 7.Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Türbachova I, Eberl M, Zeidler J, Lichtenthaler HK, Soldati D, Beck E. 1999. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 285:1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- 8.Wiesner J, Borrmann S, Jomaa H. 2003. Fosmidomycin for the treatment of malaria. Parasitol Res 90(Suppl 2):S71–S76. [DOI] [PubMed] [Google Scholar]
- 9.Borrmann S, Issifou S, Esser G, Adegnika AA, Ramharter M, Matsiegui P-B, Oyakhirome S, Mawili-Mboumba DP, Missinou MA, Kun JFJ, Jomaa H, Kremsner PG. 2004. Fosmidomycin-clindamycin for the treatment of Plasmodium falciparum malaria. J Infect Dis 190:1534–1540. doi: 10.1086/424603. [DOI] [PubMed] [Google Scholar]
- 10.Borrmann S, Adegnika AA, Matsiegui P-B, Issifou S, Schindler A, Mawili-Mboumba DP, Baranek T, Wiesner J, Jomaa H, Kremsner PG. 2004. Fosmidomycin-clindamycin for Plasmodium falciparum infections in African children. J Infect Dis 189:901–908. doi: 10.1086/381785. [DOI] [PubMed] [Google Scholar]
- 11.Oyakhirome S, Issifou S, Pongratz P, Barondi F, Ramharter M, Kun JF, Missinou MA, Lell B, Kremsner PG. 2007. Randomized controlled trial of fosmidomycin-clindamycin versus sulfadoxine-pyrimethamine in the treatment of Plasmodium falciparum malaria. Antimicrob Agents Chemother 51:1869–1871. doi: 10.1128/AAC.01448-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanaspa M, Moraleda C, Machevo S, Gonzalez R, Serrano B, Macete E, Cistero P, Mayor A, Hutchinson D, Kremsner PG, Alonso P, Menendez C, Bassat Q. 2012. Inadequate efficacy of a new formulation of fosmidomycin-clindamycin combination in Mozambican children less than three years old with uncomplicated Plasmodium falciparum malaria. Antimicrob Agents Chemother 56:2923–2928. doi: 10.1128/AAC.00018-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jiménez-Díaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, Mazouni FE, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, DeRisi JL, Sullivan DJ, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Van Voorhis WC, Avery VM, Guy RK. 2010. Chemical genetics of Plasmodium falciparum. Nature 465:311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gamo F-J, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera J-L, Vanderwall DE, Green DVS, Kumar V, Hasan S, Brown JR, Peishoff CE, Cardon LR, Garcia-Bustos JF. 2010. Thousands of chemical starting points for antimalarial lead identification. Nature 465:305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 15.Rottmann M, McNamara C, Yeung BKS, Lee MCS, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, González-Páez GE, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck H-P, Brun R, Nosten F, Renia L, Dartois V, Keller TH, Fidock DA, Winzeler EA, Diagana TT. 2010. Spiroindolones, a potent compound class for the treatment of malaria. Science 329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Connor CJ, Laraia L, Spring DR. 2011. Chemical genetics. Chem Soc Rev 40:4332. doi: 10.1039/c1cs15053g. [DOI] [PubMed] [Google Scholar]
- 17.Kirk K. 2001. Membrane transport in the malaria-infected erythrocyte. Physiol Rev 81:495–537. [DOI] [PubMed] [Google Scholar]
- 18.Yeh E, DeRisi JL. 2011. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol 9:e1001138. doi: 10.1371/journal.pbio.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spangenberg T, Burrows JN, Kowalczyk P, McDonald S, Wells TNC, Willis P. 2013. The open access Malaria Box: a drug discovery catalyst for neglected diseases. PLoS One 8:e62906. doi: 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowman JD, Merino EF, Brooks CF, Striepen B, Carlier PR, Cassera MB. 18 November 2013. Anti-apicoplast and gametocytocidal screening to identify the mechanisms of action of compounds within the Malaria Box. Antimicrob Agents Chemother doi: 10.1128/AAC.01500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruby JG, Bellare P, Derisi JL. 2013. PRICE: software for the targeted assembly of components of (Meta) genomic sequence data. G3 (Bethesda) 3:865–880. doi: 10.1534/g3.113.005967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie C, Tammi MT. 2009. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics 10:80. doi: 10.1186/1471-2105-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reference deleted.
- 25.Waller RF, Reed MB, Cowman AF, McFadden GI. 2000. Protein trafficking to the plastid of Plasmodium falciparum is via the secretory pathway. EMBO J 19:1794–1802. doi: 10.1093/emboj/19.8.1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramya TNC, Mishra S, Karmodiya K, Surolia N, Surolia A. 2007. Inhibitors of nonhousekeeping functions of the apicoplast defy delayed death in Plasmodium falciparum. Antimicrob Agents Chemother 51:307–316. doi: 10.1128/AAC.00808-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guler JL, Freeman DL, Ahyong V, Patrapuvich R, White J, Gujjar R, Phillips MA, DeRisi J, Rathod PK. 2013. Asexual populations of the human malaria parasite, Plasmodium falciparum, use a two-step genomic strategy to acquire accurate, beneficial DNA amplifications. PLoS Pathog 9:e1003375. doi: 10.1371/journal.ppat.1003375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Majumdar A, Shah MH, Bitok JK, Hassis-LeBeau ME, Freel Meyers CL. 2009. Probing phosphorylation by non-mammalian isoprenoid biosynthetic enzymes using 1H–31P–31P correlation NMR spectroscopy. Mol Biosyst 5:935. doi: 10.1039/b903513c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brammer LA, Smith JM, Wade H, Meyers CF. 2011. 1-Deoxy-D-xylulose 5-phosphate synthase catalyzes a novel random sequential mechanism. J Biol Chem 286:36522–36531. doi: 10.1074/jbc.M111.259747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brammer LA, Meyers CF. 2009. Revealing substrate promiscuity of 1-deoxy-D-xylulose 5-phosphate synthase. Org Lett 11:4748–4751. doi: 10.1021/ol901961q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith JM, Warrington NV, Vierling RJ, Kuhn ML, Anderson WF, Koppisch AT, Freel Meyers CL. 2014. Targeting DXP synthase in human pathogens: enzyme inhibition and antimicrobial activity of butylacetylphosphonate. J Antibiot (Tokyo) 67:77–83. doi: 10.1038/ja.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kunfermann A, Witschel M, Illarionov B, Martin R, Rottmann M, Höffken HW, Seet M, Eisenreich W, Knölker H-J, Fischer M, Bacher A, Groll M, Diederich F. 20 January 2014. Pseudilins: halogenated, allosteric inhibitors of the non-mevalonate pathway enzyme IspD. Angew Chem Int Ed Engl doi: 10.1002/anie.201309557. [DOI] [PubMed] [Google Scholar]
- 33.Witschel MC, Höffken HW, Seet M, Parra L, Mietzner T, Thater F, Niggeweg R, Röhl F, Illarionov B, Rohdich F, Kaiser J, Fischer M, Bacher A, Diederich F. 2011. Inhibitors of the herbicidal target IspD: allosteric site binding. Angew Chem Int Ed Engl 50:7931–7935. doi: 10.1002/anie.201102281. [DOI] [PubMed] [Google Scholar]
- 34.Christoffers MJ, Berg ML, Messersmith CG. 2002. An isoleucine to leucine mutation in acetyl-CoA carboxylase confers herbicide resistance in wild oat. Genome 45:1049–1056. doi: 10.1139/g02-080. [DOI] [PubMed] [Google Scholar]
- 35.Délye C, Wang T, Darmency H. 2002. An isoleucine-leucine substitution in chloroplastic acetyl-CoA carboxylase from green foxtail (Setaria viridis L. Beauv.) is responsible for resistance to the cyclohexanedione herbicide sethoxydim. Planta 214:421–427. [DOI] [PubMed] [Google Scholar]
- 36.Bitok JK, Meyers CF. 2012. 2C-Methyl-d-erythritol 4-phosphate enhances and sustains cyclodiphosphate synthase IspF activity. ACS Chem Biol 7:1702–1710. doi: 10.1021/cb300243w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao P, Yang Y, Xiao C, Liu Y, Gan M, Guan Y, Hao X, Meng J, Zhou S, Chen X, Cui J. 2012. Identification and validation of a novel lead compound targeting 4-diphosphocytidyl-2-C-methylerythritol synthetase (IspD) of mycobacteria. Eur J Pharmacol 694:45–52. doi: 10.1016/j.ejphar.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 38.Roscioli T, Kamsteeg EJ, Buysse K, Maystadt I, van Reeuwijk J, van den Elzen C, van Beusekom E, Riemersma M, Pfundt R, Vissers LE, Schraders M, Altunoglu U, Buckley MF, Brunner HG, Grisart B, Zhou H, Veltman JA, Gilissen C, Mancini GM, Delrée P, Willemsen MA, Ramadža DP, Chitayat D, Bennett C, Sheridan E, Peeters EA, Tan-Sindhunata GM, de Die-Smulders CE, Devriendt K, Kayserili H, El-Hashash OA, Stemple DL, Lefeber DJ, Lin YY, van Bokhoven H. 2012. Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan. Nat Genet 44:581–585. doi: 10.1038/ng.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Willer T, Lee H, Lommel M, Yoshida-Moriguchi T, de Bernabe DB, Venzke D, Cirak S, Schachter H, Vajsar J, Voit T, Muntoni F, Loder AS, Dobyns WB, Winder TL, Strahl S, Mathews KD, Nelson SF, Moore SA, Campbell KP. 2012. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat Genet 44:575–580. doi: 10.1038/ng.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagaraj VA, Sundaram B, Varadarajan NM, Subramani PA, Kalappa DM, Ghosh SK, Padmanaban G. 2013. Malaria parasite-synthesized heme is essential in the mosquito and liver stages and complements host heme in the blood stages of infection. PLoS Pathog 9:e1003522. doi: 10.1371/journal.ppat.1003522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vaughan AM, O'Neill MT, Tarun AS, Camargo N, Phuong TM, Aly AS, Cowman AF, Kappe SH. 2009. Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cell Microbiol 11:506–520. doi: 10.1111/j.1462-5822.2008.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu M, Kumar TR, Nkrumah LJ, Coppi A, Retzlaff S, Li CD, Kelly BJ, Moura PA, Lakshmanan V, Freundlich JS, Valderramos JC, Vilcheze C, Siedner M, Tsai JH, Falkard B, Sidhu AB, Purcell LA, Gratraud P, Kremer L, Waters AP, Schiehser G, Jacobus DP, Janse CJ, Ager A, Jacobs WR Jr, Sacchettini JC, Heussler V, Sinnis P, Fidock DA. 2008. The fatty acid biosynthesis enzyme FabI plays a key role in the development of liver stage malarial parasites. Cell Host Microbe 4:567–578. doi: 10.1016/j.chom.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burrows JN, van Huijsduijnen RH, Möhrle JJ, Oeuvray C, Wells TN. 2013. Designing the next generation of medicines for malaria control and eradication. Malar J 12:187. doi: 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiesner J, Henschker D, Hutchinson DB, Beck E, Jomaa H. 2002. In vitro and in vivo synergy of fosmidomycin, a novel antimalarial drug, with clindamycin. Antimicrob Agents Chemother 46:2889–2894. doi: 10.1128/AAC.46.9.2889-2894.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.