

Abstract
Biofilms are complex communities of microorganisms that attach to surfaces and are embedded in a self-produced extracellular matrix. Since these cells acquire increased tolerance against antimicrobial agents and host immune systems, biofilm-associated infectious diseases tend to become chronic. We show here that the molecular chaperone DnaK is important for biofilm formation and that chemical inhibition of DnaK cellular functions is effective in preventing biofilm development. Genetic, microbial, and microscopic analyses revealed that deletion of the dnaK gene markedly reduced the production of the extracellular functional amyloid curli, which contributes to the robustness of Escherichia coli biofilms. We tested the ability of DnaK inhibitors myricetin (Myr), telmisartan, pancuronium bromide, and zafirlukast to prevent biofilm formation of E. coli. Only Myr, a flavonol widely distributed in plants, inhibited biofilm formation in a concentration-dependent manner (50% inhibitory concentration [IC50] = 46.2 μM); however, it did not affect growth. Transmission electron microscopy demonstrated that Myr inhibited the production of curli. Phenotypic analyses of thermosensitivity, cell division, intracellular level of RNA polymerase sigma factor RpoH, and vulnerability to vancomycin revealed that Myr altered the phenotype of E. coli wild-type cells to make them resemble those of the isogenic dnaK deletion mutant, indicating that Myr inhibits cellular functions of DnaK. These findings provide insights into the significance of DnaK in curli-dependent biofilm formation and indicate that DnaK is an ideal target for antibiofilm drugs.
INTRODUCTION
Biofilms are communities of microorganisms enclosed in a self-produced polymeric matrix of extracellular polymer substances (EPS). These matrices contribute to bacterial accumulation in multiple layers and protect the embedded cells from antimicrobial agents and host immune systems (1). Therefore, once biofilms are formed on tissues or implanted medical devices (e.g., catheters and orthopedic devices), it becomes difficult to eradicate them by chemotherapeutic treatment. Biofilm-associated infections (e.g., catheter-related bloodstream infections, prosthetic-joint infections, and artificial-valve infections) tend to be intractable and chronic (2). To eradicate biofilm-associated infections, effective antimicrobial agents and novel strategies based on conceptual advances in understanding the mechanisms underlying biofilm development are needed.
Bacterial biofilm development proceeds in three steps: initial attachment to a surface, maturation, and dispersal. Biofilm-forming bacteria produce EPS such as extracellular polysaccharides, proteins, DNA, and others (3). These components play crucial roles in cell-to-surface adhesion for initial attachment and cell-to-cell cohesion during maturation. The composition of EPS varies depending on environmental conditions (e.g., temperature and salt concentration) and genetic background (4). After biofilm maturation, dispersal of biofilm-embedded cells occurs via self-produced EPS-destructing factors (e.g., d-amino acids, proteases, and phenol-soluble modulins) (5–7) and other yet-uncharacterized mechanisms. Consequently, dispersed cells can move to different niches in the body or in the environment.
Curli is the extracellular functional amyloid produced by many Enterobacteriaceae, such as Escherichia coli and Salmonella enterica (4). In concert with other EPS, such as type I pili (8), colanic acids (9), cellulose (10), and poly-N-acetylglucosamine (11), curli contributes to the initial attachment to a surface and to cell-to-cell cohesion (8). Particularly, curli-dependent biofilm is involved in urinary tract infectious diseases (12–14). Considering that curli, like amyloid fibrils in general, is extremely stable against proteolytic enzymes and detergents such as sodium dodecyl sulfate (SDS), a biofilm depending on curli biosynthesis is stable and its removal can be difficult. Therefore, the development of countermeasures against curli-dependent biofilms is an important challenge.
The components of curli biosynthesis are encoded by two curli-specific gene (csg) operons, csgBAC and csgDEFG (15). The structural components of curli, CsgA and CsgB, are synthesized in the cytoplasm, probably in an unfolded, soluble state, translocated to the periplasm through the inner membrane via the Sec translocon, and subsequently exported to the extracellular milieu by the CsgG channel embedded in the outer membrane (15). CsgE and CsgF support the transport of CsgA and CsgB. The exported CsgB anchors to the cell envelope and converts the unfolded state of CsgA to a β-sheet-rich amyloid polymer (16). Expression of the csg operons requires at least two major regulatory proteins, CsgD and RNA polymerase sigma factor RpoS. CsgD is the master transcriptional regulator for curli biosynthesis and is required for the expression of the csgBAC operon (17). Expression of the csgDEFG operon is positively regulated by the stationary-phase-specific sigma factor RpoS (18). Therefore, CsgD, RpoS, and other positive regulators that work upstream of the curli biosynthesis could be potential drug targets to combat curli-dependent biofilms.
Molecular chaperone DnaK, also known as heat shock protein 70 (Hsp70) in bacteria, plays important roles in protein folding and refolding of denatured and aggregated proteins in cooperation with cochaperones DnaJ and GrpE (19). DnaK consists of two domains, the N-terminal nucleotide-binding domain (NBD) and the C-terminal substrate-binding domain (SBD), that are connected by a highly conserved linker (19). DnaJ binds to the NBD of DnaK and stimulates the rate of ATP hydrolysis by DnaK (20, 21). GrpE also binds to the NBD at a site different from DnaJ binding (22) and accelerates the release of ADP from the NBD and of substrate peptides or proteins captured in the SBD (23). Through these actions, DnaK contributes to diverse cellular functions, including stress responses (24, 25), cell division (26), motility (27), and pathogenesis (28). However, there is controversy over the role of DnaK in biofilm formation. Singh et al. reported that loss of functional DnaK caused a reduction in the ability of the major pathogenic biofilm producer Staphylococcus aureus to form biofilms or adhere to eukaryotic cells (29). These results were consistent with those observed in Streptococcus mutans (30). On the other hand, deletion of the dnaK gene only slightly affected biofilm formation and curli production in E. coli (31). According to the results of previous studies (32, 33), DnaK probably controls the quality and/or quantity of RpoS and CsgD, both of which are essential for curli-dependent biofilm formation. Therefore, a contribution of DnaK to curli and biofilm production seems reasonable. However, more precise reexamination is necessary to clarify the role(s) of DnaK in curli biosynthesis and biofilm formation of E. coli, aimed at the development of antibiofilm drugs.
In this report, we demonstrate that DnaK plays an important role in biofilm formation by regulating curli biosynthesis. We also show that chemical inhibition of DnaK cellular functions reduces curli-dependent biofilm formation. Our findings indicate that DnaK is a potential target for drug treatment of biofilm-related chronic infections.
MATERIALS AND METHODS
Bacterial strains and plasmids.
E. coli strains, S. aureus strains, and plasmids used in this study are listed in Table 1.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Source or reference |
|---|---|---|
| E. coli strains | ||
| BW25113 | Δ(araD-araB)567 ΔlacZ4787(::rrnB-3) λ− rph-1 Δ(rhaD-rhaB)568 hsdR514; K-12 wild-type strain | NBRC, 38 |
| JW0013 | BW25113 ΔdnaK::Kmr | NBRC, 38 |
| JW1025 | BW25113 ΔcsgA::Kmr | NBRC, 38 |
| JW1024 | BW25113 ΔcsgB::Kmr | NBRC, 38 |
| JW4277 | BW25113 ΔfimA::Kmr | NBRC, 38 |
| JW4283 | BW25113 ΔfimH::Kmr | NBRC, 38 |
| S. aureus strains | ||
| SH1000 | S. aureus strain 8325-4 with functional rsbU | 59 |
| MR2 | MRSA strain isolated in the Jikei Hospital | Sato et al., unpublished |
| MR4 | MRSA strain isolated in the Jikei Hospital | Sato et al., unpublished |
| MR11 | MRSA strain isolated in the Jikei Hospital | Sato et al., unpublished |
| MR23 | MRSA strain isolated in the Jikei Hospital | 60 |
| MS3 | MSSA strain isolated in the Jikei Hospital | Sato et al., unpublished |
| MS4 | MSSA strain isolated in the Jikei Hospital | Sato et al., unpublished |
| RN4220 | Laboratory MSSA strain, restriction-negative derivative of 8325-4 | 61 |
| Plasmid | ||
| pFtsZ-GFPuv | ftsZ cloned upstream of gfpuv in pGFPuv; Apr | 34 |
Apr, ampicillin resistance; Kmr, kanamycin resistance.
DnaK inhibitors.
Myricetin (Myr), pancuronium bromide (PaBr), telmisartan (Tel), and zafirlukast (Zaf) were purchased from Tokyo Kasei (Tokyo, Japan), Santa Cruz Biotechnology (Santa Cruz, CA, USA), AK Scientific (Union City, CA, USA), and Cayman Chemical (Ann Arbor, MI, USA), respectively. Stock solutions were prepared at 100 mM in 100% dimethyl sulfoxide (DMSO). These compounds were diluted with 100% DMSO to the 100-times-higher concentrations than the indicated final concentrations. These poststock solutions were diluted 100-fold in culture media to yield the indicated concentrations. Therefore, tested media contained 1% DMSO.
Biofilm formation.
E. coli strains were grown in Lennox broth (LB; 1% [wt/vol] peptone, 0.5% [wt/vol] yeast extract, and 0.5% [wt/vol] NaCl; Merck, Darmstadt, Germany) at 30°C overnight, and the overnight cultures were 1,000-fold diluted in YESCA medium (1% [wt/vol] Casamino Acids [Becton Dickinson, Franklin Lakes, NJ, USA], 0.1% [wt/vol] yeast extract [Becton Dickinson, Franklin Lakes, NJ, USA]), supplemented with or without DnaK inhibitors (as above) at the indicated concentrations. The subcultures were incubated in 96-well polystyrene plates at 30°C. After 7 days, culture broth and planktonic cells were removed carefully, and the biofilms bound to the surface of the wells were rinsed with phosphate-buffered saline (PBS) and stained with 0.05 or 0.2% crystal violet for 5 min. Stained biofilms were rinsed twice with PBS and extracted with 70% ethanol. Absorbance of the extracted solutions was measured at 595 nm by using a spectrophotometer (Wallac 1420 ARVO MX; PerkinElmer, Boston, MA, USA). If required, photographs of biofilms were taken before ethanol extraction. We estimated the 50% inhibitory concentration (IC50) using the following formula: IC50 = 10log(A/B) × (50 – C)/(D – C) + Log(B), where A is the corresponding concentration of test compound directly above 50% inhibition, B is the corresponding concentration of test compound directly below 50% inhibition, C is the percentage of inhibition directly below 50% inhibition, and D is the percentage of inhibition directly above 50% inhibition.
S. aureus biofilms were produced according to recently reported procedures with minor modifications (29). Briefly, S. aureus strains were grown in brain heart infusion (BHI) medium (Becton Dickinson, Franklin Lakes, NJ, USA) at 37°C overnight and 1,000-fold diluted in tryptic soy broth (Becton Dickinson, Franklin Lakes, NJ, USA) containing 3% NaCl and 0.5% (wt/vol) glucose (TSBNG medium), supplemented with the indicated concentrations of Myr. The cultures were incubated in 96-well polystyrene plates at 37°C for 24 h. To monitor cell growth, optical density at 595 nm was recorded using Infinite F200 Pro (Tecan, Männedorf, Switzerland). After removal of media and planktonic cells, biofilms were washed twice with PBS and stained with 0.05% crystal violet for 1 min. The plates were washed once with PBS, and the absorbance of each well was measured at 595 nm using Infinite F200 Pro (Tecan).
Congo red-binding assay.
E. coli strains were grown in LB medium overnight. Five microliters of the overnight cultures were spotted onto YESCA plates containing 2% agar, 10 μg/ml Congo red, and 10 μg/ml Coomassie brilliant blue G-250. The plates were incubated at 30°C for 2 days. Photographs of colonies were taken with a flatbed scanner (GT-X970; Epson, Tokyo, Japan).
Thermosensitivity assay.
E. coli wild-type and ΔdnaK cells were grown at 30°C in LB medium overnight. Overnight cultures were serially diluted 10-fold, and 5 μl of these dilutions were spotted onto LB agar plates with or without 500 μM Myr. These plates were incubated at 30°C or 44°C for 36 h.
Vancomycin susceptibility assay.
E. coli wild-type and ΔdnaK cells were grown in LB medium in the presence or absence of 500 μM Myr at 30°C overnight. Overnight cultures were 1,000-fold diluted in LB medium supplemented with or without 500 μM Myr and/or the indicated concentrations of vancomycin. Cultures were further incubated at 30°C for 24 h. Aliquots (5 μl) of 10-fold serial dilutions were spotted onto LB plates and incubated at 30°C. After 24 h, photographs of colonies were taken by a scanner.
Fluorescence microscopy.
To observe Z-ring formation, the E. coli wild-type strain was transformed with pFtsZ-GFPuv (where GFP is green fluorescent protein) (34), and the resulting transformants were grown in LB medium supplemented with or without the indicated concentrations of Myr at 30°C overnight. The overnight cultures were harvested by centrifugation at 10,000 × g for 5 min at room temperature, and the pelleted cells were resuspended in PBS containing 2 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) (Dojin, Kumamoto, Japan). Stained cells were mounted onto a microscope slide, and cell morphology, Z-ring formation, and nucleoids were analyzed using a fluorescence and phase-contrast microscope (Nikon Corporation, Tokyo, Japan).
For analysis of membrane integrity and vancomycin permeability, the E. coli K-12 wild type and the isogenic ΔdnaK strain were grown in LB medium at 30°C overnight. The overnight culture was 1,000-fold diluted in LB medium and incubated at 30°C. When the optical density at 660 nm of the culture reached approximately 0.5, cells were harvested by centrifugation at 10,000 × g for 5 min at room temperature and resuspended into PBS, supplemented with 1.5 μM FM4-64 dye (Life Technologies, Tokyo, Japan) and 2 μg/ml DAPI or with 40 μM BODIPY FL-conjugated vancomycin (BODIPY-VAN) (Life Technologies, Tokyo, Japan). After incubation at 30°C for 20 min, cells were analyzed with a fluorescence and phase-contrast microscope. For analysis of BODIPY-VAN binding, cells were washed with PBS to remove unbound BODIPY-VAN and resuspended in PBS, and the localization of BODIPY-VAN was analyzed by fluorescence microscopy using a green fluorescent protein filter (Eclipse E600; Nikon Corporation, Tokyo, Japan).
Transmission electron microscopy (TEM) analysis.
To detect curli produced in the extracellular milieu, E. coli strains were grown on YESCA plates at 30°C. After 2 days, colony biofilm cells were resuspended in PBS, applied to a carbon-coated copper grid, and stained with 2% uranyl acetate. Samples were analyzed using a transmission electron microscope (H-7500; Hitachi, Tokyo, Japan) at a voltage of 100 kV.
Western blotting.
For detection of RpoH, E. coli strains were grown in LB medium at 30°C overnight. Overnight cultures were 1,000-fold diluted in 20 ml YESCA medium. After 2 days of cultivation at 30°C, cells were collected by centrifugation (8,000 × g, 15 min, 4°C) and resuspended in PBS. Gel electrophoresis was carried out using 5%-to-20% gradient SDS-polyacrylamide gels. Western blotting was performed as previously described (35).
For detection of CsgA, E. coli strains were grown in LB medium at 30°C overnight, and 500-μl samples were spread and grown on YESCA plates containing 2% agar at 30°C. After 3 days, cells were collected using a scraper, resuspended in PBS, and centrifuged (8,000 × g, 15 min, 4°C). Aliquots of 50 μl of 90% formic acid (FA) were added to 5 mg (wet weight) of cells, vortexed, and incubated on ice for 30 min. After vacuum drying, 100 μl of 8 M urea was added to the tube and mixed well by vortexing. An equal volume of SDS sample buffer (34) was added, and 5 μl of the mixture was applied to a 5%-to-20% gradient SDS-polyacrylamide gel. Western blotting was performed as previously described (36).
RESULTS
DnaK is important for curli production and curli-dependent biofilm formation in E. coli.
A previous study demonstrated that deletion of the dnaK gene only modestly affected biofilm formation of E. coli, indicating that DnaK might be dispensable for curli production (31). In a large number of E. coli mutants, biofilm had been formed after 24 h of incubation in LB medium at 25°C. When grown under such conditions, type I pili, but not curli, seemed to be required for biofilm formation (31, 37). However, curli production was analyzed using only the Congo red plate assay, instead of more direct methods, such as Western blotting or transmission electron microscopy (TEM). To assess the role of molecular chaperone DnaK in curli-dependent biofilm formation more accurately, we used an E. coli K-12 single-gene knockout mutant library (Keio collection) (38) and culture conditions in YESCA medium at 30°C as previously described (37). As shown in Fig. 1A and B, E. coli ΔcsgA and ΔcsgB mutants did not form a biofilm, while ΔfimA and ΔfimH mutants, defective in production of type I pili, formed a robust biofilm equivalent to that formed by the K-12 wild-type strain (BW25113). Therefore, when grown in YESCA medium at 30°C, curli, but not type I pili, was required for biofilm formation. This observation was consistent with a previous report (37). Under these conditions, the ΔdnaK mutant did not form a biofilm, resembling the ΔcsgA and ΔcsgB mutants (Fig. 1A and B), and the biofilm phenotype was restored by the plasmid-born DnaK (see Fig. S1 in the supplemental material). These results indicate that DnaK could play a key role in curli-dependent biofilm formation.
FIG 1.

Molecular chaperone DnaK is essential for curli-dependent biofilm formation in E. coli. (A) Biofilm formation of E. coli K-12 wild type (WT) and its isogenic mutants was analyzed using 96-well plates. Biofilms were formed in YESCA medium at 30°C for 7 days and stained with 0.2% crystal violet (CV). (B) The CV-stained biofilms were extracted with 70% ethanol and quantified by measuring absorbance at 595 nm. The means and standard deviations from triplicate determinations are represented. (C) Congo red binding of E. coli cells was analyzed with YESCA agar plates containing Congo red and CBB. (D) Production of curli by E. coli strains grown on YESCA plates at 30°C for 2 days was analyzed by Western blotting to detect CsgA monomers after formic acid treatment. The wild-type strain was used as a positive control.
We subsequently analyzed curli production by the Congo red-binding assay, using Congo red-containing YESCA agar plates. Wild-type, ΔfimA, and ΔfimH cells formed red colonies, while ΔcsgA and ΔcsgB mutants did not, indicating that the red colony phenotype depends on curli production, as reported (17). In contrast to the data published previously (31), the ΔdnaK mutant did not form a red colony, equivalent to the ΔcsgA and ΔcsgB mutants (Fig. 1C), indicating that the ΔdnaK mutant produces smaller amounts of or no curli under the conditions used. To substantiate this assumption, curli production was evaluated by Western blotting using an anti-CsgA peptide antibody. After formic acid (FA) treatment to depolymerize curli amyloid fibrils associated with the cell surface, CsgA monomers were detected in wild-type, ΔfimA, and ΔfimH cells. In contrast, no signal corresponding to CsgA monomers was detected in ΔdnaK, ΔcsgA, or ΔcsgB cells (Fig. 1D). TEM demonstrated that wild-type, ΔfimA, and ΔfimH cells produced curli fibrils in the extracellular milieu, while ΔdnaK, ΔcsgA, and ΔcsgB cells did not (Fig. 2). These results indicate that DnaK is crucial for curli production.
FIG 2.

Deletion of the dnaK gene causes a defect in curli production. The indicated E. coli strains grown on YESCA agar plates for 2 days at 30°C were stained with uranyl acetate and were observed by TEM. The wild-type (WT) strain and ΔcsgA and ΔcsgB strains were used as a positive control and negative controls for curli production, respectively. ΔfimA and ΔfimH strains were used as curli-positive but type I pili-negative strains. Scale bars represent 500 nm.
Myricetin prevents curli production and curli-dependent biofilm formation.
Hence, we hypothesized that small molecules inhibiting DnaK cellular functions can be used to prevent the production of curli and consequent biofilm formation in E. coli. To address this assumption, we tested four DnaK inhibitors, including myricetin (Myr), pancuronium bromide (PaBr), telmisartan (Tel), and zafirlukast (Zaf), which have recently been reported to disturb DnaK functions in vitro (39). Briefly, Myr and Zaf bind to the NBD of DnaK and inhibit the formation of the DnaK-DnaJ complex but do not affect the DnaK-GrpE complex (39, 40). In contrast, PaBr and Tel inhibit only the formation of the DnaK-GrpE complex (39). In the present study, we used E. coli BW25113, the wild-type K-12 strain of the Keio collection (38), as an indicator strain to examine whether these small molecules inhibit curli-dependent biofilm formation in YESCA medium at 30°C after 7 days of incubation. Among these candidates, only Myr, a widely distributed flavonol, suppressed the biofilm formation in a concentration-dependent manner (Fig. 3A to C). The IC50 was estimated to be 46.2 μM. Notably, Myr did not suppress cell growth of E. coli at the concentrations tested (Fig. 3D). Myr also inhibited biofilm formation by E. coli wild-type strain MC4100 (see Fig. S2 in the supplemental material). In addition, we used TEM to analyze extracellular structures of E. coli cells grown on YESCA plates supplemented with or without Myr. As shown in Fig. 4, curli production was significantly reduced by the addition of Myr. These results indicate that Myr exerted its activity by inhibiting cellular functions of DnaK that contribute to curli-dependent biofilm formation.
FIG 3.

Myr suppresses curli-dependent biofilm formation in E. coli. (A) Chemical structure of Myr is shown. (B) Biofilm of E. coli wild-type strain was formed in YESCA medium supplemented with or without the indicated concentrations of Myr at 30°C for 7 days. A photograph of the 0.2% CV-stained biofilms is shown. (C) CV-stained biofilms shown in panel B were quantified as described for Fig. 1B and in Materials and Methods. (D) Growth of the E. coli wild-type strain was monitored in YESCA medium supplemented with the indicated concentrations of Myr. The means and standard deviations from triplicate determinations are represented.
FIG 4.

Myr suppresses curli production. The E. coli wild-type strain was grown on YESCA agar plates supplemented with or without Myr for 2 days at 30°C. The colony biofilm cells were stained with uranyl acetate and were observed by TEM. Scale bars represent 500 nm.
Myricetin can inhibit in vivo cellular functions of DnaK.
Myr was recently shown to inhibit the DnaJ-stimulated DnaK ATPase activity by blocking the DnaK-DnaJ complex formation in vitro (39, 40). However, its in vivo effects still remain elusive. In order to evaluate the hypothesis that Myr prevents DnaK functions in vivo, phenotypes of the E. coli K-12 wild-type strain (BW25113) under Myr treatment were compared with those of the isogenic ΔdnaK mutant.
First, we tested susceptibility of the strains to heat stress, since DnaK is essential for the growth of E. coli at temperatures above 43°C (41, 42) (Fig. 5). As expected, growth of the wild-type strain was significantly attenuated in the presence of Myr at 44°C. As a control, no detectable effect of Myr on the growth of wild-type and ΔdnaK strains on LB agar plates was observed at 30°C.
FIG 5.

Myr renders E. coli thermosensitive. Thermosensitivity of wild-type (WT) and ΔdnaK strains was assayed on LB agar plates supplemented with or without Myr. Tenfold serial dilutions of the overnight cultures were spotted on the plate. Plates were incubated at 30°C or 44°C for 36 h.
Second, we quantified the heat shock transcription factor RpoH (also known as σ32), a well-known substrate of DnaK, by Western blotting. Tatsuta et al. reported that DnaK promotes proteolysis of RpoH by the AAA (ATPases associated with diverse cellular activities) protease FtsH, and therefore the level of RpoH in ΔdnaK cells is much higher than that in wild-type cells (35). We demonstrated that the level of RpoH in the isogenic ΔdnaK strain was approximately 2-fold higher than that in the wild-type strain (Fig. 6). Similarly, the level of RpoH in wild-type cells grown in the presence of Myr was approximately 2-fold higher than that in the absence of Myr (Fig. 6).
FIG 6.

Cellular level of RpoH in wild-type cells increased in the presence of Myr. (A) Cellular levels of RpoH in wild-type (WT) cells in the presence and absence of Myr and those in ΔdnaK cells were detected by Western blotting using an anti-RpoH serum. (B) Band intensities in panel A were calculated with ImageJ. The level of RpoH in WT cells grown in the absence of Myr was defined as 100%. The means and standard deviations from at least triplicate determinations are represented.
Third, we assessed cell division of E. coli cells in the presence or absence of Myr, since the lack of DnaK functions reportedly results in defective cell division (26, 43). In many bacteria, FtsZ plays an essential role in cell division. Usually, FtsZ forms a ring-shaped structure, called Z-ring, at the midcell division site when cell division takes place, while it distributes to the cytoplasm or localizes on the cytoplasmic membrane, associating with membrane-anchored FtsA (44). In the present study, we introduced an FtsZ-GFPuv expression plasmid (34) into the E. coli wild-type strain and analyzed the effects of Myr on cell division and Z-ring formation. As shown in Fig. 7, many filamentous cells exhibiting abnormal Z-ring formation were observed when cultivated in the presence of Myr, equivalent to ΔdnaK cells and GrpE-overproducing cells, in which the DnaK chaperone system was disturbed (43).
FIG 7.

Cell division and localization of FtsZ are affected by Myr. To visualize localization of FtsZ, E. coli wild-type cells expressing FtsZ-GFPuv were cultured in the presence and absence of Myr. Nucleoid was stained with DAPI. Phase-contrast and fluorescence images (DAPI and GFP) were observed using a phase-contrast and fluorescence microscope. Merged images were constructed with Photoshop. Scale bars represent 5 μm.
In summary, these three lines of evidence strongly suggest that Myr inhibits cellular functions of DnaK in vivo.
Myricetin renders E. coli sensitive to vancomycin.
It is generally accepted that vancomycin kills diverse Gram-positive bacteria but does not affect the viability of Gram-negative bacteria such as E. coli because its large molecular size prevents it from permeating through the outer membrane. We found that wild-type cells were stained remarkably with FM4-64, a membrane-staining fluorescent probe (45), while ΔdnaK cells were not (Fig. 8A), indicating that the loss of the DnaK function affects membrane integrity. This finding led us to test the assumption that ΔdnaK cells would exhibit sensitivity to vancomycin. We used fluorescence probe-labeled vancomycin (BODIPY-VAN) and analyzed its binding to E. coli cells. As shown in Fig. 8B, wild-type cells did not display any detectable fluorescence, whereas ΔdnaK cells exhibited strong fluorescence, suggesting that BODIPY-VAN permeated through the outer membrane and bound to periplasm-localizing lipid II, a precursor of peptidoglycan and a known target of vancomycin (46). Next, we examined antimicrobial effects of vancomycin on E. coli wild-type and ΔdnaK cells in the presence or absence of Myr. Interestingly, growth of ΔdnaK cells was inhibited by vancomycin at a concentration of 50 μg/ml and above (Fig. 8C). Accordingly, wild-type cells exhibited sensitivity to vancomycin only in the presence of Myr at higher concentrations.
FIG 8.
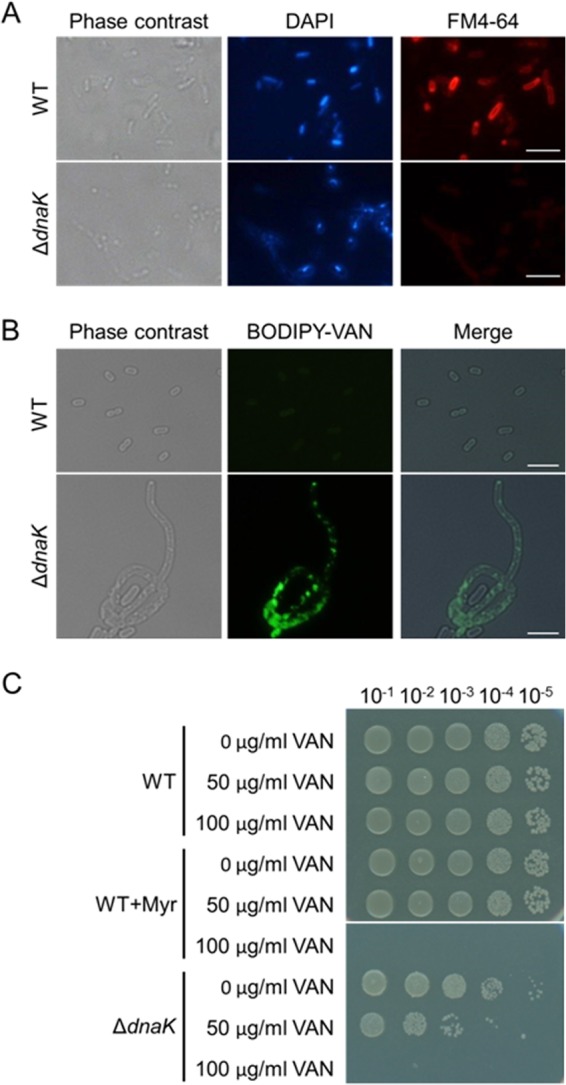
Myr renders E. coli sensitive to vancomycin. (A) Wild-type (WT) and ΔdnaK cells were stained with the membrane-staining dye FM4-64. Nucleoid was also stained with DAPI. (B) Exponential-phase cells of WT and ΔdnaK strains were stained with BODIPY-VAN. Phase-contrast and fluorescence images (DAPI, FM4-64, and BODIPY-VAN) were observed using a phase contrast and fluorescence microscope in panels A and B. Merged images in panel B were constructed using Photoshop. Scale bars represent 5 μm. (C) E. coli WT and ΔdnaK cells were grown in LB medium supplemented with or without vancomycin (VAN) at 30°C for 24 h. Myr (500 μM) was supplemented into medium as indicated. Serial 10-fold dilutions of the 24-h cultures were spotted on LB agar plates and incubated at 30°C for 24 h.
Myricetin suppresses biofilm formation of Staphylococcus aureus strains, including clinically isolated methicillin-resistant strains.
Staphylococcus aureus is a major bacterium found in catheter infection-related biofilms (47). We assessed whether Myr inhibits S. aureus biofilm formation using various methicillin-sensitive S. aureus (MSSA) and methicillin-resistant S. aureus (MRSA) strains, such as SH1000 and MR2. SH1000 is a laboratory MSSA strain, in which DnaK plays an important role for biofilm formation under the conditions reported previously (29). MR2 is a hospital-isolated MRSA strain also forming biofilms (F. Sato, S. Sugimoto, and Y. Mizunoe, unpublished data). These biofilms were examined via crystal violet staining. As shown in Fig. 9A and B, Myr inhibited biofilm formation of these S. aureus strains in a dose-dependent manner, while not inhibiting their growth at the indicated concentrations (Fig. 9C). In addition, Myr inhibited biofilm formation of various MRSA and MSSA strains (see Fig. S3 in the supplemental material). These results extend the potential applicability of Myr beyond a model biofilm of E. coli to authentic biofilms formed by the clinically important pathogenic bacteria S. aureus, including MRSA strains.
FIG 9.

Myr suppresses biofilm formation of S. aureus. (A) S. aureus biofilms were formed in TSBNG medium supplemented with the indicated concentrations of Myr at 37°C for 24 h. Biofilms were stained with 0.05% CV. (B) CV-stained biofilms in panel A were quantified by measuring absorbance at 595 nm. (C) Effects of Myr on S. aureus growth were also tested. The means and standard deviations from at least triplicate determinations are represented.
DISCUSSION
Curli is a major component of the extracellular matrix produced by many Enterobacteriaceae, such as Escherichia coli and Salmonella enterica (4). Here, we show for the first time that DnaK is indispensable for curli biosynthesis and biofilm formation (Fig. 1 and 2). The molecular mechanism of how DnaK contributes to curli biosynthesis is still unknown. Based on recent information, we speculate that DnaK may control quality and/or quantity of the biofilm master regulator CsgD and the sigma factor RpoS, both of which are essential for curli-dependent biofilm formation. In addition, we identified the DnaK inhibitor Myr as a potential antibiofilm drug. Phenotypic analyses demonstrate that Myr could prevent DnaK cellular functions in vivo (Fig. 5 to 7), leading to defective curli biosynthesis and biofilm formation (Fig. 3 and 4).
Intensive efforts have been made to develop antibiofilm agents. Hydrolyzing enzymes degrading broad EPS, such as glycolytic enzymes (e.g., dispersin B) (48), proteases (e.g., the serine protease Esp) (49), and deoxyribonucleases (e.g., DNase I) (50), were proposed to eradicate biofilms produced by diverse bacteria. In addition, small molecule biofilm inhibitors that showed an inhibitory effect on specific EPS, such as type I pili (51, 52) and curli (37, 53), have also been reported. Currently, there is a strong demand for a significant advance in the search for new reagents that specifically target bacterial biofilms, similar to the antibiofilm peptide 1018 that functions by blocking guanosine pentaphosphate [(p)ppGpp], an important signal molecule upstream of biofilm development (54). The results described in the present study indicate that chemical inhibition of DnaK cellular functions is a potential strategy for treating biofilm-associated infectious diseases (Fig. 3 to 9). DnaK is known to play important roles under multiple stress conditions, such as heat, oxidative conditions, low pH, and salt stresses (55). DnaK may also play a crucial role in maintaining membrane integrity (Fig. 8). Our microarray analysis revealed that expression of several enzymes involved in phospholipid biogenesis was reduced in the ΔdnaK mutant (S. Sugimoto, K. Arita-Morioka, K. Yamanaka, T. Ogura, Y. Mizunoe, unpublished data). This observation might explain the diminished membrane integrity in the ΔdnaK mutant and in wild-type cells cultivated in the presence of Myr (Fig. 8). Usually, aminoglycoside antibiotics are ineffective against Gram-negative bacteria because they cannot penetrate the outer membrane owing to their large molecular size. For example, vancomycin does not act on the Gram-negative bacterium E. coli. However, we found that vancomycin inhibited growth of the ΔdnaK cells (Fig. 8C). This observation supports the hypothesis that DnaK plays an important role for membrane integrity. Interestingly, vancomycin, in combination with Myr, inhibited growth of the wild-type cells too (Fig. 8C). This finding provides a clinically significant impact on antimicrobial chemotherapy and may help to overcome the limitation of these antibiotics as their inefficiency against Gram-negative bacteria.
Acquired multidrug tolerance of biofilm-embedded cells makes therapeutic countermeasures difficult. In addition to protective roles of extracellular matrix, alteration of bacterial physiological states in a biofilm also contributes remarkably to this phenotype. Persister cells (persisters) are subpopulations of antibiotic-sensitive bacteria that exert slow or no growth and become tolerant to multiple antibiotics (56). Persistence also seems to be involved in chronic biofilm-related infections (1). Recently, Conlon et al. reported that deregulation of the ingeniously controlled AAA+ proteases ClpXP and ClpCP by activating ClpP protease with an acyldepsipeptide antibiotic (ADEP4) triggered the degradation of nonspecific proteins, including essential proteins, leading to bacterial cell death (57). Furthermore, treatment with ADEP4 in combination with other antibiotics eradicated S. aureus biofilms (57). Considering that DnaK contributes to persistence (58), inhibition of DnaK cellular functions by Myr may also be a potential strategy to treat persisters. As Myr repressed biofilm formation by E. coli and S. aureus, including diverse MRSA strains (Fig. 9; see also Fig. S3 in the supplemental material), Myr might be effective in eradicating various bacterial biofilms and persisters, since DnaK is highly conserved in diverse bacteria (55). These options need to be explored in the future. In addition, more active, wide-spectrum, and less toxic drugs should be developed using Myr as a lead compound. Employing a virtual “inverse” screening approach (e.g., in silico prediction based on the Myr-DnaK complex structure), we may identify potential Myr derivatives. This prediction will be validated by an in vitro ATPase activity assay, interaction with cochaperones and a model substrate peptide, inhibition of intracellular DnaK functions, as presented in this study, and a biofilm assay. These future efforts may be challenging but are expected to lead to the development of realistic countermeasures for biofilm-associated chronic infectious diseases.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the National BioResource Project (NIG, Japan) for Keio collection mutants. We thank M. Endo and E. Miyakawa for performing biofilm assay and all members of the Mizunoe lab and Ogura lab for stimulating discussions.
This work was supported in part by a Grant-in-Aid for Young Scientists (B) to S.S. from JSPS, by a grant to Y.M. from MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2012 to 2016, and by a grant to S.S. from Joint Usage/Research Center for Developmental Medicine, IMEG, Kumamoto University.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04465-14.
REFERENCES
- 1.Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 2.De Pozo JL, Patel R. 2009. Clinical practice. Infection associated with prosthetic joints. N Engl J Med 361:787–794. doi: 10.1056/NEJMcp0905029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flemming HC, Wingender J. 2010. The biofilm matrix. Nat Rev Microbiol 8:623–633. doi: 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- 4.Mika F, Hengge R. 2014. Small RNAs in the control of RpoS, CsgD, and biofilm architecture of Escherichia coli. RNA Biol 11:494–507. doi: 10.4161/rna.28867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kolodkin-Gal I, Romero D, Cao S, Clardy J, Kolter R, Losick R. 2010. d-Amino acids trigger biofilm disassembly. Science 328:627–629. doi: 10.1126/science.1188628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boles BR, Horswill AR. 2008. agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog 4:e1000052. doi: 10.1371/journal.ppat.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Periasamy S, Joo HS, Duong AC, Bach TH, Tan VY, Chatterjee SS, Cheung GY, Otto M. 2012. How Staphylococcus aureus biofilms develop their characteristic structure. Proc Natl Acad Sci U S A 109:1281–1286. doi: 10.1073/pnas.1115006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pratt LA, Kolter R. 1999. Genetic analyses of bacterial biofilm formation. Curr Opin Microbiol 12:598–603. [DOI] [PubMed] [Google Scholar]
- 9.Danese PN, Pratt LA, Kolter R. 2000. Exopolysaccharide production is required for development of Escherichia coli K-12 biofilm architecture. J Bacteriol 182:3593–3596. doi: 10.1128/JB.182.12.3593-3596.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zogaj X, Nimtz M, Rohde M, Bokranz W, Römling U. 2001. The multicellular morphotypes of Salmonella Typhimurium and Escherichia coli produce cellulose as the second component of the extracellular matrix. Mol Microbiol 39:1452–1463. doi: 10.1046/j.1365-2958.2001.02337.x. [DOI] [PubMed] [Google Scholar]
- 11.Itoh Y, Wang X, Hinnebusch BJ, Preston JF III, Romeo T. 2005. Depolymerization of beta-1,6-N-acetyl-d-glucosamine disrupts the integrity of diverse bacterial biofilms. J Bacteriol 187:382–387. doi: 10.1128/JB.187.1.382-387.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kai-Larsen Y, Lüthje P, Chromek M, Peters V, Wang X, Holm A, Kádas L, Hedlund KO, Johansson J, Chapman MR, Jacobson SH, Römling U, Agerberth B, Brauner A. 2000. Uropathogenic Escherichia coli modulates immune responses and its curli fimbriae interact with the antimicrobial peptide LL-37. PLoS Pathog 6:e1001010. doi: 10.1371/journal.ppat.1001010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bian Z, Brauner A, Li Y, Normark S. 2000. Expression of and cytokine activation by Escherichia coli curli fibers in human sepsis. J Infect Dis 181:602–612. doi: 10.1086/315233. [DOI] [PubMed] [Google Scholar]
- 14.Kudinha T, Johnson JR, Andrew SD, Kong F, Anderson P, Gilbert GL. 2013. Genotypic and phenotypic characterization of Escherichia coli isolates from children with urinary tract infection and from healthy carriers. Pediatr Infect Dis J 32:543–548. doi: 10.1097/INF.0b013e31828ba3f1. [DOI] [PubMed] [Google Scholar]
- 15.Blanco LP, Evans ML, Smith DR, Badtke MP, Chapman MR. 2012. Diversity, biogenesis and function of microbial amyloids. Trends Microbiol 20:66–73. doi: 10.1016/j.tim.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammer ND, Schmidt JC, Chapman MR. 2007. The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization. Proc Natl Acad Sci U S A 104:12494–12499. doi: 10.1073/pnas.0703310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammar M, Arnqvist A, Bian Z, Olsén A, Normark S. 1995. Expression of two csg operons is required for production of fibronectin- and Congo red-binding curli polymers in Escherichia coli K-12. Mol Microbiol 18:661–670. doi: 10.1111/j.1365-2958.1995.mmi_18040661.x. [DOI] [PubMed] [Google Scholar]
- 18.Grantcharova N, Peters V, Monteiro C, Zakikhany K, Römling U. 2010. Bistable expression of CsgD in biofilm development of Salmonella enterica serovar Typhimurium. J Bacteriol 192:456–466. doi: 10.1128/JB.01826-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayer MP, Bukau B. 1998. Hsp70 chaperone systems: diversity of cellular functions and mechanism of action. Biol Chem 379:261–268. [PubMed] [Google Scholar]
- 20.Gässler CS, Buchberger A, Laufen T, Mayer MP, Schröder H, Valencia A, Bukau B. 1998. Mutations in the DnaK chaperone affecting interaction with the DnaJ cochaperone. Proc Natl Acad Sci U S A 95:15229–15234. doi: 10.1073/pnas.95.26.15229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B. 1999. Mechanism of regulation of Hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci U S A 96:5452–5457. doi: 10.1073/pnas.96.10.5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harrison CJ, Hayer-Hartl M, Di Liberto M, Hartl F, Kuriyan J. 1997. Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science 276:431–435. doi: 10.1126/science.276.5311.431. [DOI] [PubMed] [Google Scholar]
- 23.Langer T, Lu C, Echols H, Flanagan J, Hayer MK, Hartl FU. 1992. Successive action of DnaK, DnaJ and GroEL along the pathway of chaperone-mediated protein folding. Nature 356:683–689. doi: 10.1038/356683a0. [DOI] [PubMed] [Google Scholar]
- 24.Bukau B. 1993. Regulation of the Escherichia coli heat-shock response. Mol Microbiol 9:671–680. doi: 10.1111/j.1365-2958.1993.tb01727.x. [DOI] [PubMed] [Google Scholar]
- 25.Yura T, Nagai H, Mori H. 1993. Regulation of the heat-shock response in bacteria. Annu Rev Microbiol 47:321–350. doi: 10.1146/annurev.mi.47.100193.001541. [DOI] [PubMed] [Google Scholar]
- 26.Bukau B, Walker GC. 1989. Cellular defects caused by deletion of the Escherichia coli dnaK gene indicate roles for heat shock protein in normal metabolism. J Bacteriol 171:2337–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi W, Zhou Y, Wild J, Adler J, Gross CA. 1992. DnaK, DnaJ, and GrpE are required for flagellum synthesis in Escherichia coli. J Bacteriol 174:6256–6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chakrabarti S, Sengupta N, Chowdhury R. 1999. Role of DnaK in in vitro and in vivo expression of virulence factors of Vibrio cholerae. Infect Immun 67:1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh VK, Syring M, Singh A, Singhal K, Dalecki A, Johansson T. 2012. An insight into the significance of the DnaK heat shock system in Staphylococcus aureus. Int J Med Microbiol 302:242–252. doi: 10.1016/j.ijmm.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Lemos JA, Luzardo Y, Burne RA. 2007. Physiologic effects of forced down-regulation of dnaK and groEL expression in Streptococcus mutans. J Bacteriol 189:1582–1588. doi: 10.1128/JB.01655-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niba ET, Naka Y, Nagase M, Mori H, Kitakawa M. 2007. A genome-wide approach to identify the genes involved in biofilm formation in E. coli. DNA Res 14:237–246. doi: 10.1093/dnares/dsm024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rockabrand D, Livers K, Austin T, Kaiser R, Jensen D, Burgess R, Blum P. 1998. Roles of DnaK and RpoS in starvation-induced thermotolerance of Escherichia coli. J Bacteriol 180:846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niwa T, Ying BW, Saito K, Jin W, Takada S, Ueda T, Taguchi H. 2009. Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins. Proc Natl Acad Sci U S A 106:4201–4206. doi: 10.1073/pnas.0811922106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugimoto S, Yamanaka K, Nishikori S, Miyagi A, Ando T, Ogura T. 2010. AAA+ chaperone ClpX regulates dynamics of prokaryotic cytoskeletal protein FtsZ. J Biol Chem 285:6648–6657. doi: 10.1074/jbc.M109.080739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tatsuta T, Tomoyasu T, Bukau B, Kitagawa M, Mori H, Karata K, Ogura T. 1998. Heat shock regulation in the ftsH null mutant of Escherichia coli: dissection of stability and activity control mechanisms of sigma32 in vivo. Mol Microbiol 30:583–593. doi: 10.1046/j.1365-2958.1998.01091.x. [DOI] [PubMed] [Google Scholar]
- 36.Chiba A, Sugimoto S, Sato F, Hori S, Mizunoe Y. 23 August 2014. A refined technique for extraction of extracellular matrices from bacterial biofilms and its applicability. Microb Biotechnol doi: 10.1111/1751-7915.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cegelski L, Pinkner JS, Hammer ND, Cusumano CK, Hung CS, Chorell E, Aberg V, Walker JN, Seed PC, Almqvist F, Chapman MR, Hultgren SJ. 2009. Small-molecule inhibitors target Escherichia coli amyloid biogenesis and biofilm formation. Nat Chem Biol 5:913–919. doi: 10.1038/nchembio.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cesa LC, Patury S, Komiyama T, Ahmad A, Zuiderweg ER, Gestwicki JE. 2013. Inhibitors of difficult protein-protein interactions identified by high-throughput screening of multiprotein complexes. ACS Chem Biol 8:1988–1997. doi: 10.1021/cb400356m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang L, Miyata Y, Ung PM, Bertelsen EB, McQuade TJ, Carlson HA, Zuiderweg ER, Gestwicki JE. 2011. Chemical screens against a reconstituted multiprotein complex: myricetin blocks DnaJ regulation of DnaK through an allosteric mechanism. Chem Biol 18:210–221. doi: 10.1016/j.chembiol.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Itikawa H, Ryu J. 1979. Isolation and characterization of a temperature-sensitive dnaK mutant of Escherichia coli B. J Bacteriol 138:339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paek KH, Walker GC. 1987. Escherichia coli dnaK null mutants are inviable at high temperature. J Bacteriol 169:283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sugimoto S, Saruwatari K, Higashi C, Sonomoto K. 2008. The proper ratio of GrpE to DnaK is important for protein quality control by the DnaK-DnaJ-GrpE chaperone system and for cell division. Microbiology 154:1876–1885. doi: 10.1099/mic.0.2008/017376-0. [DOI] [PubMed] [Google Scholar]
- 44.Ma X, Ehrhardt DW, Margolin W. 1996. Colocalization of cell division proteins FtsZ and FtsA to cytoskeletal structures in living Escherichia coli cells by using green fluorescent protein. Proc Natl Acad Sci U S A 93:12998–13003. doi: 10.1073/pnas.93.23.12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vida TA, Emr SD. 1995. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol 128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sheldrick GM, Jones PG, Kennard O, Williams DH, Smith GA. 1978. Structure of vancomycin and its complex with acetyl-d-alanyl-d-alanine. Nature 271:223–225. doi: 10.1038/271223a0. [DOI] [PubMed] [Google Scholar]
- 47.Marrie TJ, Costerton JW. 1984. Scanning and transmission electron microscopy of in situ bacterial colonization of intravenous and intraarterial catheters. J Clin Microbiol 19:687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaplan JB, Velliyagounder K, Ragunath C, Rohde H, Mack D, Knobloch JK, Ramasubbu N. 2004. Genes involved in the synthesis and degradation of matrix polysaccharide in Actinobacillus actinomycetemcomitans and Actinobacillus pleuropneumoniae biofilms. J Bacteriol 186:8213–8220. doi: 10.1128/JB.186.24.8213-8220.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dubin G, Chmiel D, Mak P, Rakwalska M, Rzychon M, Dubin A. 2001. Molecular cloning and biochemical characterisation of proteases from Staphylococcus epidermidis. Biol Chem 382:1575–1582. doi: 10.1515/BC.2001.192. [DOI] [PubMed] [Google Scholar]
- 50.Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. 2002. Extracellular DNA required for bacterial biofilm formation. Science 295:1487. doi: 10.1126/science.295.5559.1487. [DOI] [PubMed] [Google Scholar]
- 51.Cusumano CK, Pinkner JS, Han Z, Greene SE, Ford BA, Crowley JR, Henderson JP, Janetka JW, Hultgren SJ. 2011. Treatment and prevention of urinary tract infection with orally active FimH inhibitors. Sci Transl Med 3:109ra115. doi: 10.1126/scitranslmed.3003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lo AW, Van de Water K, Gane PJ, Chan AW, Steadman D, Waksman G, Remaut H. 2014. Suppression of type 1 pilus assembly in uropathogenic Escherichia coli by chemical inhibition of subunit polymerization. J Antimicrob Chemother 69:1017–1026. doi: 10.1093/jac/dkt467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andersson EK, Bengtsson C, Evans ML, Chorell E, Sellstedt M, Lindgren AE, Hufnagel DA, Bhattacharya M, Tessier PM, Wittung-Stafshede P, Almqvist F, Chapman MR. 2013. Modulation of curli assembly and pellicle biofilm formation by chemical and protein chaperones. Chem Biol 20:1245–1254. doi: 10.1016/j.chembiol.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de la Fuente-Núñez C, Reffuveille F, Haney EF, Straus SK, Hancock RE. 2014. Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog 10:e1004152. doi: 10.1371/journal.ppat.1004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sugimoto S, Mahin A, Sonomoto K. 2008. Molecular chaperones in lactic acid bacteria: physiological consequences and biochemical properties. J Biosci Bioeng 106:324–336. doi: 10.1263/jbb.106.324. [DOI] [PubMed] [Google Scholar]
- 56.Bigger JW. 1944. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet 244:497–500. doi: 10.1016/S0140-6736(00)74210-3. [DOI] [Google Scholar]
- 57.Conlon BP, Nakayasu ES, Fleck LE, LaFleur MD, Isabella VM, Coleman K, Leonard SN, Smith RD, Adkins JN, Lewis K. 2013. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 503:365–370. doi: 10.1038/nature12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hansen S, Lewis K, Vulić M. 2008. Role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrob Agents Chemother 52:2718–2726. doi: 10.1128/AAC.00144-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horsburgh MJ, Aish JL, White IJ, Shaw L, Lithgow JK, Foster SJ. 2002. σB modulates virulence determinant expression and stress resistance: characterization of a functional rsbU strain derived from Staphylococcus aureus 8325-4. J Bacteriol 184:5457–5467. doi: 10.1128/JB.184.19.5457-5467.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sugimoto S, Iwase T, Sato F, Tajima A, Shinji H, Mizunoe Y. 2011. Cloning, expression and purification of extracellular serine protease Esp, a biofilm-degrading enzyme, from Staphylococcus epidermidis. J Appl Microbiol 111:1406–1415. doi: 10.1111/j.1365-2672.2011.05167.x. [DOI] [PubMed] [Google Scholar]
- 61.Kreiswirth BN, Löfdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.