

Abstract
GSK1265744 is a new HIV integrase strand transfer inhibitor (INSTI) engineered to deliver efficient antiviral activity with a once-daily, low-milligram dose that does not require a pharmacokinetic booster. The in vitro antiviral profile and mechanism of action of GSK1265744 were established through integrase enzyme assays, resistance passage experiments, and cellular assays with site-directed molecular (SDM) HIV clones resistant to other classes of anti-HIV-1 agents and earlier INSTIs. GSK1265744 inhibited HIV replication with low or subnanomolar efficacy and with a selectivity index of at least 22,000 under the same culture conditions. The protein-adjusted half-maximal inhibitory concentration (PA-EC50) extrapolated to 100% human serum was 102 nM. When the virus was passaged in the presence of GSK1265744, highly resistant mutants with more than a 10-fold change (FC) in EC50 relative to that of the wild-type were not observed for up to 112 days of culture. GSK1265744 demonstrated activity against SDM clones containing the raltegravir (RAL)-resistant Y143R, Q148K, N155H, and G140S/Q148H signature variants (FC less than 6.1), while these mutants had a high FC in the EC50 for RAL (11 to >130). Either additive or synergistic effects were observed when GSK1265744 was tested in combination with representative anti-HIV agents, and no antagonistic effects were seen. These findings demonstrate that, similar to dolutegravir, GSK1265744 is differentiated as a new INSTI, having a markedly distinct resistance profile compared with earlier INSTIs, RAL, and elvitegravir (EVG). The collective data set supports further clinical development of GSK1265744.
INTRODUCTION
The morbidity and mortality of HIV infection and AIDS have dramatically decreased since the introduction of highly active antiretroviral therapy (HAART), now commonly referred to as combination antiretroviral therapy (cART). Thus, HIV infection has become a manageable chronic infectious disease. This significant clinical advancement has resulted from progress in the discovery and development of anti-HIV drugs that exploit novel mechanisms of action, improve efficacy and durability with less inherent toxicity, and are amenable to more convenient dosing regimens for patients. However, there still exist unmet medical needs, including those for patients who have experienced treatment failure, for prevention of transmission via a preexposure prophylaxis (PrEP) approach, and for patients with difficulties in maintaining adherence. The reasons for virologic failure can be complex but are often due to intrinsic characteristics of a drug, which may include the emergence of drug-resistant mutants and can in turn be exacerbated by low drug levels due to pharmacokinetic or adherence issues.
The first integrase strand transfer inhibitor (INSTI), raltegravir (RAL) (MK-0518) (1, 2), was approved by the U.S. FDA in 2007. The second INSTI, elvitegravir (EVG) (GS-9130, formerly JTK-303), was approved in 2012 as a component of “Stribild,” which is a fixed-dose combination of EVG, cobicistat, tenofovir disoproxil fumarate, and emtricitabine (3, 4, 5). INSTIs are now recognized as a safe and highly effective class of anti-HIV drugs (6). However, clinical resistance to RAL and EVG has been observed, and a high degree of cross-resistance between these two agents has been demonstrated (7, 8, 9, 10, 11, 12). Furthermore, dosing of RAL is done twice daily, while once-daily administration of EVG requires a pharmacokinetic (PK) booster, such as ritonavir or cobicistat. Because these two agents are inhibiters of drug metabolism, their use raises concerns about the levels of concomitantly used drugs. Ritonavir also raises long-term safety concerns (13). Therefore, new INSTIs should have attributes that address these unmet needs.
The Shionogi and GlaxoSmithKline research collaboration initiated in 2002 has made considerable progress in engineering INSTIs with a distinct resistance profile and low-dose once-daily unboosted regimens. Dolutegravir (DTG) (S/GSK1349572, brand name TIVICAY) was approved by the U.S. FDA in 2013, and GSK1265744 (formerly S/GSK1265744), with the generic name cabotegravir (USAN approved), is in phase 2 clinical trials. They contain a two-metal binding pharmacophore consisting of a carbamoyl pyridone moiety (see Fig. 1) and were optimized to deliver the attributes that would differentiate them as new INSTIs (14, 15). Clinical data for GSK1265744 administered to healthy subjects and HIV patients present a PK profile supporting once-daily oral administration from a low-milligram dose, with low PK variability and excellent short-term safety/tolerability, as well as highly effective anti-HIV activity from 10 days of monotherapy (16).
FIG 1.
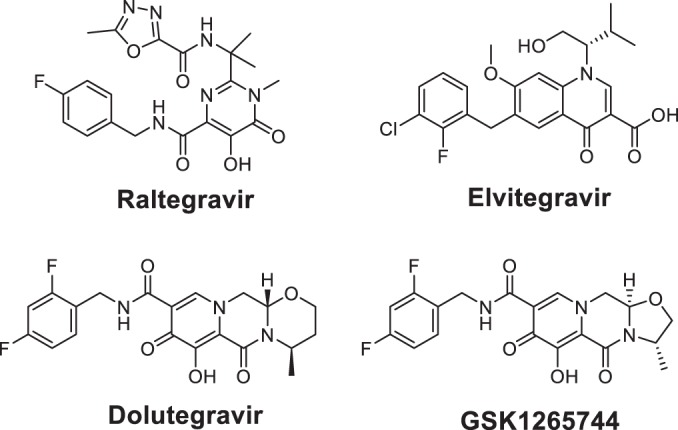
Chemical structures of the HIV-1 INSTIs used in this study. Raltegravir and Elvitegravir are earlier INSTIs. Dolutegravir and GSK1265744 are newer INSTIs.
The focus of the development of GSK1265744 was a long-acting (LA) injectable formulation due to its favorable attributes: low solubility (0.015 mg/ml in pH 6.8 phosphate buffer at 20°C) and low plasma clearance (0.32 ml/min/kg of body weight in monkeys) for HIV treatment and prevention. The data from a phase 1 single-dose GSK1265744 LA study in healthy volunteers supported once-monthly to once-quarterly dosing (17, 18). Recently, a repeat-dose coadministration study of GSK1265744 LA and TMC278 LA in healthy subject results was described (19). LA development is attractive for both treatment and use as a preventative measure in a preexposure prophylaxis (PrEP) setting. Although Truvada (emtricitabine and tenofovir disoproxil fumarate) was approved by FDA for PrEP, a PrEP clinical study data of Truvada suggested that daily compliance was a complicating factor for healthy high-risk individuals, ultimately leading to reduced efficacy of the approach (20).
We report herein the in vitro antiretroviral properties of GSK1265744, including its mechanism of action and in vitro resistance characteristics essential for clinical development.
MATERIALS AND METHODS
Compounds.
GSK1265744, RAL, and EVG were synthesized at GlaxoSmithKline, Research Triangle Park, NC. Efavirenz (EFV) and lamivudine were purchased from Sequoia Research Products, Ltd, Pangbourne, United Kingdom. See Fig. 1 for the structures of GSK1265744, DTG, RAL, and EVG.
Cells and viruses.
MT-4 cells, a human T-cell leukemia virus type 1 (HTLV-1)-transformed human T-cell line, were maintained as described previously (21). 293T cells were maintained in Dulbecco's modified Eagle medium (DMEM)-F12 medium containing 10% fetal bovine serum (FBS). Peripheral blood mononuclear cells (PBMCs) were derived from whole-blood samples obtained from HIV-negative donors. PBMCs were separated from whole blood by density gradient centrifugation with Ficoll-Paque Plus (GE HealthCare, Waukesha, WI) according to the manufacturer's instructions and were stimulated by addition of either 20 U/ml of interleukin-2 (IL-2) or 10% natural T-cell growth factor (ZeptoMetrix, Buffalo, NY) plus 5 to 10 μg/ml of phytohemagglutinin (PHA). Molt-4 cells persistently infected with HIV-1 strain IIIB and MT-2 cells (22) were obtained from S. Harada (Kumamoto University, Kumamoto, Japan). HeLa-CD4 cells containing an HIV-1 long terminal repeat (LTR)-driven β-galactosidase reporter gene was described previously (23). HIV-1 strain IIIB was derived from cell-free supernatants of cultures of the chronically infected cell line H93B (H9/HTLV-IIIB). HIV-1 strain Ba-L was purchased from Advanced Biotechnologies Inc. (Columbia, MD) and was expanded in PHA-activated PBMCs, while HIV-1 NL432 (24) was obtained from A. Adachi (Tokushima University, Tokushima, Japan). Plasmid pGJ3-Luci, containing a replication-defective HIV lentivirus vector expressing luciferase (25), was licensed from Christian Jassoy (University of Leipzig) and used to create stocks of vesicular stomatitis virus glycoprotein (VSV-G)-pseudotyped self-inactivating pseudo-HIV (PHIV) lentiviral vector by cotransfecting along with plasmid pVSV-G (Clontech) into CIP4 cells (a derivative of the 293T human renal epithelial cell line that expresses macrophage scavenger receptor SRA-I to improve adherence to plastic) and harvesting the cell-free supernatant.
In vitro strand transfer assay.
The inhibitory concentrations of GSK1265744 and other INSTIs were measured in a strand transfer assay using recombinant HIV IN as previously described (26). A complex of integrase and biotinylated donor DNA-streptavidin-coated scintillation proximity assay (SPA) beads was formed by incubating 2 μM purified recombinant integrase with 0.66 μM biotinylated donor DNA–4 mg/ml streptavidin-coated SPA beads in 25 mM sodium morpholinepropanesulfonic acid (MOPS) (pH 7.2), 23 mM NaCl, and 10 mM MgCl2 for 5 min at 37°C. These beads were spun down and preincubated with diluted INSTIs for 60 min at 37°C. Next, 3H-labeled target DNA substrate was added to give a final concentration of 7 nM substrate, and the strand transfer reaction mixture was incubated at 37°C for 25 to 45 min, which allowed for a linear increase in strand transfer of donor DNA to radiolabeled target DNA. The signal was read using a Wallac MicroBeta scintillation plate reader.
Antiviral assay in MT-4 cells.
MT-4 cells growing exponentially at a density of (5 or 6) × 105 cells/ml were infected with HIV-1 strain IIIB at a viral multiplicity of infection of 0.001 or a 50% tissue culture infectious dose of 4 to 10. The cells were then aliquoted to 96-well plates in the presence of various concentrations of compounds. After incubation for 4 or 5 days, the antiviral activity was measured in a cell viability assay that either measured bioluminescence with a CellTiter-Glo luminescent reagent (Promega Corporation, Madison, WI) or absorbance at 560 and 690 nm using the yellow tetrazolium MTT reagent [(3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide)].
PHIV assay.
The antiviral activities of compounds were measured in a single-round assay using a self-inactivating PHIV lentiviral vector. CIP4 cells (2 × 104 cells/well) were infected with PHIV sufficient to produce approximately 50,000 relative light units in the assay. The infected cells were added to 96-well, black, clear-bottom plates with various concentrations of GSK1265744 and incubated for 2 days. Luciferase activity was measured in a luminometer using the Steady-Glo reagent (Promega Corporation).
Antiviral assay in PBMCs.
In one 96-well culture plate, PHA- and IL-2-stimulated PBMCs (4 × 105 cells/well) were preincubated with a test compound for 1 h while HIV-1 strain Ba-L was mixed with the same compound in a second plate. An aliquot of the Ba-L–compound mixture was then transferred to the PBMC-compound mixture and incubated for 7 days. In a separate experiment, PHA–IL-2–stimulated PBMCs (1 × 106 cells/well) were preincubated with HIV-1 strain NL432 for 2 h at room temperature. After incubation, the cells were resuspended and transferred to a 96-well culture plate containing a test compound and thoroughly mixed, and the mixture was incubated for 4 days. In both experiments, after incubation for 4 or 7 days, supernatants were assayed for reverse transcriptase (RT) activity by incorporating [methyl-3H]deoxythymidine-5′-triphosphate to measure viral replication as previously described (27).
Effect of human serum and serum proteins.
The effect of the presence of human serum albumin (HSA) (20 or 40 mg/ml), α1-acid glycoprotein (AAG) (2 mg/ml), and human serum (HS) (using up to 30% or 50% and extrapolated to 100%) on the antiviral activity of GSK1265744 was evaluated in PHIV and MT-4 assay systems. To estimate the effects of protein binding, antiviral activity was tested with the addition of various concentrations of human serum to an HIV replication assay in MT-4 cells as previously described (27). The protein-adjusted half-maximal effective concentration (PA-EC50) was estimated by multiplying the EC50 in PBMCs by the fold-shift value.
Cytotoxicity assays.
In vitro growth inhibition (cytotoxicity) studies were conducted with GSK1265744 in proliferating human leukemia and lymphoma cell lines (IM-9, U-937, MT-4, and Molt-4) as well as stimulated and unstimulated human PBMCs. As a surrogate of cell growth, ATP levels were quantified using the CellTiter-Glo luciferase reagent.
Mechanistic cellular studies.
To determine if GSK1265744 was inhibiting HIV replication in cellular assays through an integrase inhibition mechanism, the effect on the synthesis of HIV NL432 DNA species in MT-4 cells was measured in a single-round infection assay using quantitative PCR methods in the presence of INSTI or nonnucleoside reverse transcriptase inhibitor (NNRTI) as described previously with minor modifications (27). Briefly, 293T cells were transfected with the NL432 plasmid to generate infectious virus, and the supernatant was filtered through 0.45-μm-pore-size filters and treated with DNase I. MT-4 cells were infected with HIV-1 NL432 virus for 1 h with diluted compound and collected after 6 or 18 h of incubation. All cells were incubated with a 0.5 μM concentration of the protease inhibitor (PI) ritonavir to limit HIV replication to a single cycle. Total-DNA PCR to detect late RT products was performed with the samples after 6 h of incubation. Nested Alu-PCR to detect integrated provirus and two-LTR PCR to detect two-LTR circles were performed with the samples taken at 18 h of incubation. Reactions were analyzed using the ABI Prism 7900HT-3 sequence detection system (Applied Biosystems, Carlsbad, CA).
Isolation of drug-resistant viruses.
Isolation of drug-resistant viruses was performed according to the previously described protocol (28). Briefly, the virus for initiating passage work was prepared by coculturing MT-2 cells with Molt-4 cells persistently infected with HIV-1 strain IIIB for 3 days. Fresh MT-2 cells were dispensed into each well of a 24-well tissue culture plate. Three wells of each culture containing several concentrations of a compound were used initially, and the virus, prepared as described above, was added to each well. Every 3 or 4 days, the cells were passaged with or without addition of fresh MT-2 cells. When a cytopathic effect was observed, the supernatants were used to infect fresh MT-2 cells, and the concentration of the compound was held constant or increased 5-fold. Every 2 weeks, when replication of viruses was ascertained by observing the cytopathic effect, the infected cells were collected and used for genotypic and phenotypic analyses. To analyze mutations, DNA was extracted from the infected cells using the DNeasy blood and tissue kit (Qiagen, Hilden, Germany), and the integrase region of HIV proviral DNA was amplified by PCR with specific primers (M-poli7, AACAAGTAGATAAATTAGTCAGT; M-poli8, TAGTGGGATGTGTACTTCTGAAC). Sequencing of the products was provided by the Operon Biotechnologies sequencing service. The sequence of the integrase region derived from isolated viruses was compared with that of wild-type IIIB, and amino acid substitutions were identified.
Construction of IN region-recombinant HIV-1 molecular clones.
We constructed IN region-recombinant HIV-1 molecular clones as described previously (14). Briefly, the XbaI-EcoRI fragment from pNL-IN301 (pNL432 [24] inserted the XbaI site into the 5′ end of the integrase region) was cloned into the XbaI-EcoRI site of cloning vector pUC18. To introduce integrase-resistant mutations, in vitro mutagenesis of the integrase gene was performed with the QuikChange site-directed mutagenesis kit (Stratagene, a division of Agilent Technologies, Santa Clara, CA) using a pUC18 clone containing the integrase region as a template. The mutated XbaI-EcoRI fragment was amplified and ligated into pNL-IN301 to construct a recombinant HIV-1 molecular clone. Plasmids were subsequently transfected into 293T cells to generate infectious virus. Supernatants were harvested after 2 days of culture and stored as cell-free culture supernatants at −80°C.
Cross-resistance profiling of GSK1265744.
GSK1265744 was evaluated against molecular clones with mutations in the IN-, RT-, and protease (PR)-coding regions. INSTI-, nucleoside reverse transcriptase inhibitor (NRTI)-, and NNRTI-resistant mutants were analyzed by the reporter assay based on HeLa-CD4 cells, while PI-resistant mutants were analyzed by infectivity in MT-4 cells, monitoring RT activity as described previously (27). The HIV-1 wild-type infectious molecular clone pNL432 was used for site-directed mutagenesis to generate HIV clones containing mutations. Fifty INSTI-resistant mutants were constructed. The molecular clones with K101E, K103N, E138K, Y181C, M184I, M184V, Y188L, K101E/M184I, E138K/M184I D67/K70R/T215Y, and R4 (V75I/F77L/F116Y/Q151M) substitutions within the RT coding region were used as NRTI or NNRTI-resistant viruses, and PI-resistant mutants carrying the M46I/I47V/I50V and L24I/M46I/L63P/A71V/G73S/V82T mutations with the protease coding region were used. 293T cells were subsequently transfected with the plasmids to generate infectious virus using Lipofectamine 2000 (Invitrogen Corporation, Carlsbad, CA). Supernatants were harvested 2 to 3 days after transfection, stored as cell-free culture supernatants at −80°C, and used for each assay.
Combination antiviral activity assay in MT-4 cells.
The in vitro combination activity relationships of GSK1265744 were determined as previously described (29). Multiple concentrations of GSK1265744 were tested in a checkerboard dilution fashion in the presence and absence of dilutions of approved representative anti-HIV drugs, adefovir or ribavirin. The interaction of compounds was analyzed by dosewise additivity-based calculations. Wells containing the top concentration of compounds by themselves were compared to wells with the top concentration of both compounds to show that combination effects were due to the drugs used and not to toxicity. Assays with the MT-4 system format were run as described previously (27). Fractional inhibitory concentration (FIC) values in the range of −0.1 to −0.2 indicated weak synergy, values that approached −0.5 indicated strong synergy, and positive values of 0.1 to 0.2 indicated weak antagonism. The effects of the anti-hepatitis B virus (HBV) and anti-hepatitis C virus (HCV) agents adefovir and ribavirin on the GSK1265744 EC50 were examined using linear regression as described previously (30). Since the HIV-1 IIIB MT-4 system is CXCR4 based, the CCR5 inhibitor Maraviroc was evaluated in a checkerboard dilution format using HIV-1 Ba-L-infected MAGI-CCR5 cells with Gal-Screen reagent (Tropix, Bedford, MA) for the chemiluminescent endpoint. Data were analyzed as described by Prichard (31) using the MacSynergy II program. Synergy volumes in the range of −50 to 50 defined additivity, <−50 defined antagonism, and >50 defined synergy.
RESULTS
Inhibition of recombinant HIV integrase and HIV replication by GSK1265744.
As with other INSTIs, GSK1265744 has a two-metal binding scaffold and is in the same structural class as DTG (Fig. 1). However, the differences in the structure of GSK1265744 give it properties distinct from those of DTG, including high protein binding. The biochemical strand transfer inhibitory activity and anti-HIV activity of GSK1265744 in the absence of human serum were similar to those of DTG (Table 1).
TABLE 1.
Inhibition of recombinant HIV integrase and HIV replication by GSK1265744
| INSTIa | Integrase inhibition, strand transfer IC50 (nM) | Antiviral activity |
|||
|---|---|---|---|---|---|
| PBMC EC50 (nM) | Potency shift with 100% HS (FC)b | PA-EC50 (nM) | PA-EC90 (nM) | ||
| GSK1265744 | 3.0 | 0.25c | 408 | 102 | 408 |
| Dolutegravir | 2.7 | 0.51 | 75 | 38 | 152 |
| Raltegravir | 3.3 | 2.0 | 4.7 | 5.6 | 23 |
| Elvitegravir | 6 | 2.0 | 22 | 20 | 78 |
Data for dolutegravir, raltegravir, and elvitegravir are from reference 14.
Protein-binding fold shift (FC, fold change) was estimated from antiviral activity with various concentrations of human serum and extrapolated to a 100% human serum condition.
Mean from 11 experiments with HIV-1 strain Ba-L and 5 experiments with HIV-1 NL432.
GSK1265744 inhibited the HIV-1 integrase catalyzed strand transfer reaction with an IC50 of 3.0 nM in vitro. The antiviral EC50 against HIV-1 Ba-L was 0.22 nM, and that against NL432 was 0.34 nM in PBMCs, 0.57 nM using CellTiter-Glo and 1.3 nM using MTT in MT-4 cells, and 0.5 nM in the PHIV assay, which uses a pseudotyped self-inactivating virus. Calculations of the EC50 to 90% effective concentration (EC90) fold shift showed a range from 3-fold to a maximum of 4-fold. Therefore, the EC90 was conservatively estimated by multiplying the EC50 by 4. The low-nanomolar GSK1265744 efficacy was similar to the efficacies of DTG, RAL, and EVG (Table 1). In the MT-4 antiviral assay, the estimated potency shift due to plasma protein binding was 408-fold when extrapolated to 100% human serum and 84-fold in the presence of 20 mg/ml HSA. In the PHIV assay, the potency shift was 63-fold with 40 mg/ml HSA and 1.6-fold in the presence of 2 mg/ml AAG. The extrapolated potency shift of 408-fold for GSK1265744 in the presence of 100% human serum was also applied to the EC50 and EC90 in PBMC, resulting in PA-EC50 and PA-EC90 values of 102 nM and 408 nM, respectively. The potency shift of GSK1265744 was higher than those of the other three approved INSTIs, with consequently higher PA-EC50 and PA-EC90 values (Table 1).
The 50% cytotoxic concentrations (CC50) for GSK1265744 in proliferating IM-9, U-937, MT-4, and Molt-4 cell lines were 6.4, 5.0, 9.2, and 13 μM, respectively. In unstimulated and stimulated peripheral blood lymphocytes (PBLs) (both from the same four donors), the CC50 were 120 μM and 42 μM, respectively. Based on the EC50 of GSK1265744 versus HIV-1 in PBLs (0.22 nM), the cell-based therapeutic index was at least 22,000.
Cellular mechanistic studies.
As expected for an INSTI and as shown in Fig. 2, GSK1265744 inhibited integration of viral DNA (Fig. 2a) with a concomitant increase in 2-LTR circles (Fig. 2b) and no effect on viral DNA production (Fig. 2c). Furthermore, the concentration dependency of the effects was within the margin of error for the potency observed in the inhibition of viral replication in PBMCs and MT-4 cells. These effects were similar to those observed with RAL and DTG in our previous study (14, 27) and were in contrast to the effects of the NNRTI EFV.
FIG 2.
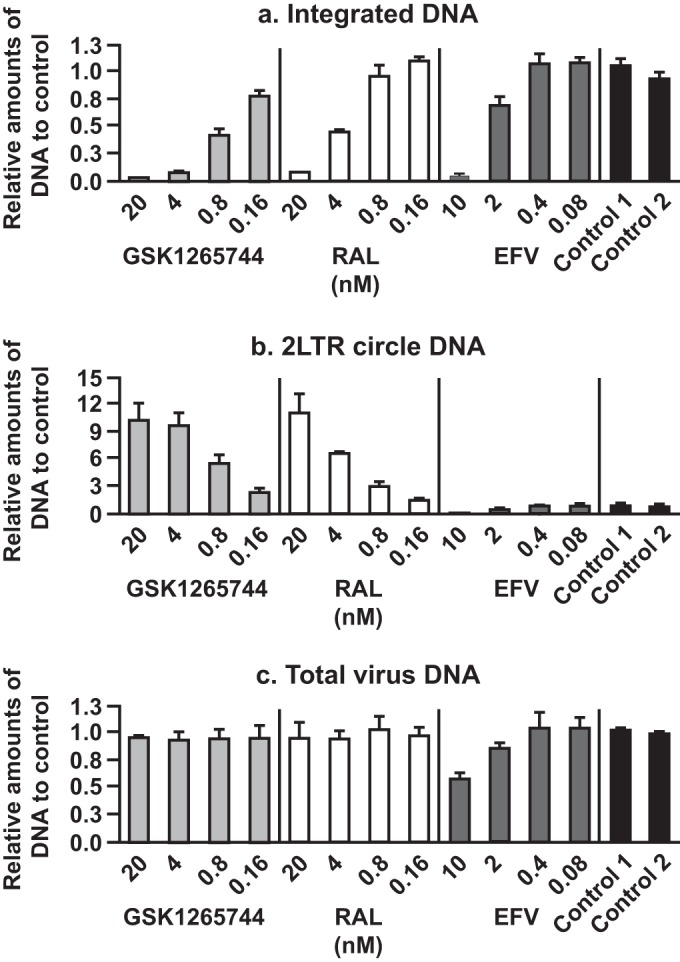
Effects of inhibitors on various forms of viral DNA were evaluated separately with HIV-1 NL432-infected MT-4 cells to determine the amounts of integrated viral DNA (a), 2-LTR circular viral DNA (b), or total viral DNA (c). The y axis of each graph represents amounts of DNA relative to those of the control. Each bar represents the mean value of results from three independent experiments. Error bars represent standard deviations.
Cross-resistance profiling of GSK1265744.
When tested against HIV strains resistant to marketed NNRTIs or NRTIs, GSK1265744 showed efficacy against five different NNRTI-resistant or NRTI-resistant viruses, with activity equivalent to that against wild-type virus (fold change [FC] values ranged from 0.9 to 1.4). Because rilpivirine (RPV) is being investigated in combination with GSK1265744 in LA clinical studies (19, 32), the FC values of six different RPV signature mutants ranged from 1.0 to 1.3. Likewise, GSK1265744 showed efficacy against two different PI-resistant viruses (the M46I/I47V/I50V and L24I/M46I/L63P/A71V/G73S/V82T viruses), with activity equivalent to that against wild-type virus (FC of 0.22 and 0.36, respectively).
Cellular combination studies.
GSK1265744 was tested in combination studies with backbone NRTIs (lamivudine, tenofovir, and emtricitabine) and the NNRTI RPV. There was no antagonism observed with any combination, and there was no enhanced cytotoxicity observed among the concentrations tested for antiviral activity. In combination with NRTIs, GSK1265744 was weakly synergistic with lamivudine, tenofovir, and emtricitabine. In combination with RPV, GSK1265744 was weakly synergistic.
Susceptibilities of INSTI-resistant molecular clones to GSK1265744.
A panel of 49 INSTI-resistant site-directed mutants (SDMs) was constructed and tested for susceptibilities to GSK1265744 and other INSTIs (Table 2). Many of the DTG, RAL, EVG, and EFV susceptibilities have been reported (14). These mutants were isolated during in vitro passage studies with INSTIs and/or clinical trials of RAL, while a few were derived from the literature. EFV was used as a control, with a maximum FC against the INSTI-resistant mutants of 2.9-fold. Therefore, the viruses with an FC of ≥3 were considered to be resistant in this study. For descriptive purposes, we define an FC of more than 10 as “high resistance.”
TABLE 2.
Fold change for molecular clones with INSTI resistance substitutions
| Description of resistant virus | Mean fold change in resistance vs. wild-type valuea |
||||
|---|---|---|---|---|---|
| GSK1265744 | Dolutegravir | Raltegravir | Elvitegravir | Efavirenz | |
| Wild type | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| T66A | 0.31 ± 0.03 | 0.26 ± 0.010 | 0.61 ± 0.090 | 4.1 ± 0.83 | 1.3 ± 0.026 |
| T66I | 0.33 ± 0.08 | 0.26 ± 0.090 | 0.51 ± 0.10 | 8.0 ± 1.7 | 1.5 ± 0.23 |
| T66K | 2.7 ± 0.44 | 2.3 ± 0.35 | 9.6 ± 1.3 | 84 ± 29 | 2.1 ± 0.28 |
| E92I | 1.8 ± 0.21 | 1.5 ± 0.19 | 2.1 ± 0.62 | 8.0 ± 1.9 | 1.0 ± 0.30 |
| E92Q | 1.5 ± 0.11 | 1.6 ± 0.12 | 3.5 ± 1.4 | 19 ± 8.1 | 1.2 ± 0.22 |
| E92V | 1.5 ± 0.15 | 1.3 ± 0.20 | 1.4 ± 0.18 | 8.3 ± 1.5 | 0.93 ± 0.29 |
| G118S | 1.1 ± 0.35 | 1.1 ± 0.21 | 1.2 ± 0.30 | 4.9b | 0.73b |
| F121Y | 1.0 ± 0.28 | 0.81 ± 0.12 | 6.1 ± 1.3 | 36 ± 23 | 2.1 ± 0.14 |
| T124A | 0.97 ± 0.15 | 0.95 ± 0.19 | 0.82 ± 0.080 | 1.2 ± 0.29 | 0.95 ± 0.12 |
| G140S | 0.81 ± 0.11 | 0.86 ± 0.30 | 1.1 ± 0.22 | 2.7 ± 0.63 | 1.1 ± 0.12 |
| Y143C | 1.1 ± 0.29 | 0.95 ± 0.26 | 3.2 ± 0.57 | 1.5 ± 0.46 | 1.2 ± 0.23 |
| Y143H | 1.1 ± 0.21 | 0.89 ± 0.11 | 1.8 ± 0.38 | 1.5 ± 0.047 | 1.3 ± 0.32 |
| Y143R | 1.4 ± 0.22 | 1.4 ± 0.29 | 16 ± 3.9 | 1.8 ± 0.16 | 0.98 ± 0.041 |
| P145S | 0.43 ± 0.03 | 0.49 ± 0.080 | 0.87 ± 0.20 | >350 | 2.9 ± 0.28 |
| Q146L | 2.1 ± 1.2 | NDc | 0.93 ± 0.08 | 13 ± 2.5 | 1.5 ± 0.2 |
| Q146R | 1.7 ± 0.25 | 1.6 ± 0.17 | 1.2 ± 0.26 | 2.8 ± 0.72 | 0.94 ± 0.40 |
| Q148H | 0.86 ± 0.12 | 0.97 ± 0.67 | 13 ± 5.0 | 7.3 ± 2.3 | 1.4 ± 0.83 |
| Q148K | 5.6 ± 0.61 | 1.1 ± 0.19 | 83 ± 6.6 | >1700 | 2.1 ± 0.26 |
| Q148R | 5.1 ± 1.8 | 1.2 ± 0.21 | 47 ± 9.3 | 240 ± 91 | 1.9 ± 0.21 |
| I151L | 2.5 ± 0.48 | 3.6 ± 3.6 | 8.4 ± 4.7 | 29b | 2.9 ± 0.67 |
| S153Y | 2.0 ± 0.63 | 2.5 ± 1.1 | 1.3 ± 0.19 | 2.3 ± 0.49 | 1.9 ± 0.18 |
| M154I | 0.87 ± 0.1 | 0.93 ± 0.27 | 0.82 ± 0.18 | 1.1 ± 0.18 | 1.4 ± 0.14 |
| N155H | 1.7 ± 0.18 | 0.99 ± 0.094 | 8.4 ± 1.8 | 25 ± 7.8 | 1.1 ± 0.044 |
| N155S | 1.5 ± 0.29 | 1.4 ± 0.36 | 6.2 ± 1.9 | 68 ± 26 | 1.7 ± 0.09 |
| N155T | 1.5 ± 0.26 | 1.9 ± 0.32 | 5.2 ± 2.0 | 39b | 1.5 ± 0.43 |
| T66I/L74 M | 0.38 ± 0.11 | 0.35 ± 0.080 | 2.0 ± 0.81 | 14b | 1.2 ± 0.015 |
| T66I/E92Q | 1.0 ± 0.38 | 1.2 ± 0.19 | 18 ± 3.6 | 190 ± 100 | 2.0 ± 0.20 |
| T66K/L74 M | 6.3 ± 1.8 | 3.5 ± 0.94 | 40 ± 13 | 120 ± 33 | 2.0 ± 0.25 |
| L74 M/N155H | 2.5 ± 0.041 | 0.91 ± 0.17 | 28 ± 12 | 42 ± 8.6 | 1.1 ± 0.24 |
| E92Q/N155H | 5.3 ± 2.8 | 2.5 ± 1.2 | >130 | 320 ± 39 | 1.9 ± 0.71 |
| T97A/N155H | 2.9 ± 0.27 | 1.1 ± 0.46 | 26 ± 7.9 | 37 ± 6.9 | 1.1 ± 0.14 |
| F121Y/T125K | 1.4 ± 0.55 | 0.98 ± 0.35 | 11 ± 0.49 | 34b | 1.5b |
| T124A/S153Y | 2.4 ± 0.1 | NDc | 1.5 ± 0.44 | 4 ± 0.4 | 1.9 ± 0.46 |
| E138A/Q148R | 13 ± 0.86 | 2.6 ± 0.47 | 110b | 260 ± 12 | 1.2 ± 0.090 |
| E138K/Q148H | 0.92 ± 0.08 | 0.89 ± 0.24 | 17 ± 5.9 | 6.7 ± 1.5 | 1.0 ± 0.47 |
| E138K/Q148K | 81 ± 24 | 19 ± 8.0 | 330 ± 75 | 371b | 1.2b |
| E138K/Q148R | 12 ± 3.4 | 4.0 ± 1.1 | 110 ± 37 | 460 ± 230 | 1.0 ± 0.31 |
| G140C/Q148R | 22 ± 4.9 | 4.9 ± 1.8 | 200 ± 42 | 485b | 2.8b |
| G140S/Q148H | 6.1 ± 0.75 | 2.6 ± 1.4 | >130 | >890 | 1.7 ± 0.99 |
| G140S/Q148K | 5.6 ± 2.5 | 1.5 ± 0.10 | 3.7 ± 1.3 | 94 ± 53 | 1.3 ± 0.39 |
| G140S/Q148R | 12 ± 3.5 | 8.4 ± 4.0 | 200 ± 5.3 | 267b | 1.5b |
| Y143H/N155H | 6.2 ± 1.9 | 1.7 ± 0.27 | 38b | 16 ± 4.6 | 1.5 ± 0.28 |
| Q148R/N155H | 61 ± 28 | 10 ± 1.4 | >140 | 390b | 1.7 ± 0.15 |
| N155H/G163K | 1.6 ± 0.26 | 1.4 ± 0.40 | 23 ± 7.2 | 35 ± 4.4 | 1.1 ± 0.10 |
| N155H/G163R | 1.9 ± 0.28 | 1.1 ± 0.18 | 17 ± 5.9 | 35 ± 12 | 1.1 ± 0.16 |
| N155H/D232N | 2.5 ± 0.39 | 1.4 ± 0.25 | 20 ± 3.9 | 36 ± 7.1 | 0.92 ± 0.050 |
| V72I/F121Y/T125K | 2.0 ± 0.61 | 1.3 ± 0.54 | 13 ± 7.1 | 58b | 1.5b |
| E138A/S147G/Q148R | 3.7 ± 0.98 | 1.9 ± 0.89 | 27 ± 3.7 | 130 ± 8.8 | 0.83 ± 0.032 |
| V72I/F121Y/T125K/I151V | 1.6 ± 0.66 | 1.2 ± 0.32 | 7.0 ± 2.8 | 37b | 1.1b |
Each value represents the mean and SD for 3 to 5 independent experiments, each performed in duplicate. FCs of >10-fold are highlighted in bold.
The mean FCs from one or two experiments are shown. The SD value could not be analyzed.
ND, no data.
Among 25 single-amino-acid-substitution SDMs, most remained fully susceptible to GSK1265744. However, the Q148K (FC = 5.6) and Q148R (FC = 5.1) variants, which were RAL-resistant primary mutants, resulted in moderately reduced susceptibility to GSK1265744. Among 24 multimutated SDMs, 6 mutant viruses (the E138A/Q148R, E138K/Q148K, E138K/Q148R, G140C/Q148R, G140S/Q148R and Q148R/N155H variants) were highly resistant to GSK1265744, with FC values of >10. For comparison, of the 49 SDMs tested, there were 2 mutants highly resistant to DTG, 24 resistant to RAL, and 33 resistant to EVG, with FC values of >10. Thus, the resistance profile of GSK1265744 was different from those of RAL and EVG and similar to that of DTG.
Isolation of viruses resistant to GSK1265744 and RAL.
In vitro passage experiments were performed with GSK1265744 or RAL starting with wild-type HIV-1 IIIB. Dose escalation patterns of GSK1265744 and RAL are shown in Fig. 3a and b, respectively. Isolated genotype and phenotype assay results are shown in Tables 3 and 4. Each passage started from 0.256, 1.28, 6.4, and 32 nM GSK1265744 or RAL, and compound concentrations were increased in a stepwise manner when a cytopathic effect was observed (reflecting a loss of antiviral activity and a gain in viral replication). The final concentration of RAL increased up to 800 nM or 4,000 nM (Fig. 3b). In contrast, no replication was observed at concentrations of 32 nM or greater of GSK1265744 throughout the passage experiment, except for day 53 in the well culture which had started with 1.28 nM (Fig. 3a). Thus, the maximum concentration of GSK1265744 allowing replication was 6.4 nM in this assay.
FIG 3.
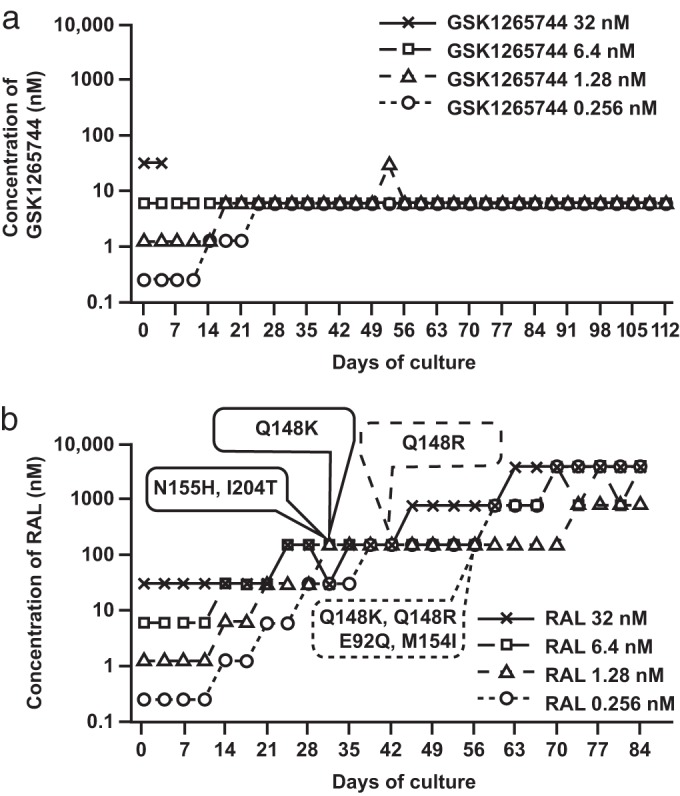
Long-term culture of infected MT-2 cells with escalating concentrations of INSTIs. MT-2 cells were infected with strain HIV-1 IIIB and passaged twice weekly in the presence of GSK1265744 (a) or RAL (b); the highest concentration at which the virus could replicate in cultured wells is indicated. The first points at which RAL signature mutants were observed in each RAL concentration are indicated by the oval callouts.
TABLE 3.
Genotype and phenotype assay results for GSK1265744a
| Initial conc (nM) | Concn (nM) | Substitution (FC in resistance) at day: |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 14 | 28 | 42 | 56 | 70 | 84 | 98 | 112 | ||
| 32 | 32 | Nonreplication | Nonreplication | Nonreplication | Nonreplication | Nonreplication | Nonreplication | Nonreplication | Nonreplication |
| 6.4 | 6.4 | None (1.8–2.0); T124A (1.9) | None (2.0); T124A (2.2) | None | None (4.2); S153Y (4.7); Q146L (3.3) | None (3.4); S153Y (4.7); Q146L (2.1) | None (4.2); S153Y (5.1); Q146L (2.9) | None (4.1); S153Y (4.7); Q146L (3.9) | None (4.9); S153Y (5.1); Q146L (4.6) |
| 1.28 | 6.4 | T124A (2.4–5.5) | None (3.3); T124A (2.7) | None (3.7–4.6); T124A (2.4) | None (5.1); T124A (4.6); S153Y (3.9) | T124A (4.9–5.1); S153Y (4.3) | T124A (4.6–5.2); S153Y (6.0) | T124A (3.3); S153Y (5.6) | |
| 1.28 | T124A (1.1–1.6) | T124A (1.4–1.7) | T124A (1.7–2.8) | T124A (1.1–2.0) | T124A (1.2–2.6) | T124A (2.4–2.5); T124A, I162M (2.8) | T124A (1.3–1.6) | T124A (1.4–1.5) | |
| 0.256 | 6.4 | T124A (2.5–2.8) | T124A (3.1–4.0) | T124A (3.7–4.6); T124A/S153Y (6.4) | T124A (2.8–3.7); T124A/S153Y (5.1) | T124A (4.3–5.7); T124A/S153Y (8.4) | T124A (3.9–4.5); T124A/S153Y (6.3) | T124A (3.3–5.9); T124A/S153Y (6.6) | |
| 1.28 | T124A (1.4–1.6) | T124A (1.9–2.3) | T124A (1.2–2.0) | T124A (1.8–2.5) | T124A (1.6–3.1) | T124A (1.3–2.4) | T124A (1.6–2.2) | T124A (1.1–1.9) | |
| 0.256 | T124A (1.2–1.4) | T124A (2.5–2.9) | T124A (<0.88) | ||||||
Fold changes (FCs) from the phenotype assay are shown in parentheses after genotype assay results. When FCs of two wells are different, the data are shown as a range.
TABLE 4.
Genotype and phenotype assay results for RALa
| Initial concn (nM) | Concn (nM) | Substitution (FC in resistance) at day: |
|||||
|---|---|---|---|---|---|---|---|
| 14 | 28 | 42 | 56 | 70 | 84 | ||
| 32 | 4,000 | G140C/Q148K/G163R (>160) | G140C/Q148K/G163R (>138) | ||||
| 800 | Q148K (111) | G140C/Q148K (>160) | G140C/Q148K/G163R (>138) | ||||
| 160 | Q148K (>23) | Q148K (11) | Q148K/G163R (38) | Q148K/G163R (149) | E138K/Q148K/G163R (>138) | ||
| 32 | T124A (1.5–2.2) | T124A (0.65–0.78); Q148K (20) | T124A (1.1); Q148K (8.0) | T124A (1.6); Q148K (29) | T124A (1.3); Q148K (56) | T124A (2.0); Q148K (38) | |
| 6.4 | 4,000 | G140S/Q148R (>160) | G140S/Q148R (>138); T124A/V151I/N155H (>138) | ||||
| 800 | G140S/Q148R (>160); T124A/V151I/N155H (65) | G140S/Q148R (>138); T124A/V151I/N155H (37) | |||||
| 160 | T124A (0.71–0.91) | N155H (19) | Q148R (11); N155H (9.7) | Q148R (11); V151I/N155H (5.4) | Q148R (9.5); V151I/N155H (13) | ||
| 32 | T124A (2.2–2.5) | N155H/I204T (20 ->23); T124A (1.1–1.3) | N155H/I204T (10); G59E (1.4) | Q148R (11); N155H/I204T (6.0) | Q148R (11–24); N155H/I204T (8.0) | N155H/I204T (6.4) | |
| 6.4 | T124A (1.1–1.5) | T124A (0.62–1.1) | T124A (0.93–1.1) | T124A (0.90–1.2) | T124A (1.0–1.6) | ||
| 1.28 | 800 | T124A (0.40); E138K/Q148R (28) | |||||
| 160 | Q148R (9.5); T124A (2.2–7.6) | Q148R (19); T124A (0.70) | Q148R (31) | Q148R (25) | |||
| 32 | T124A (0.66–0.71) | Q148R (8.4); T124A (2.2–7.6) | Q148R (18); T124A (2.0–3.5) | Q148R (8.4); T124A (1.6–1.7) | Q148R (15); T124A (1.5–2.4) | ||
| 6.4 | T124A (0.89–1.0) | T124A (0.64–0.79) | T124A (0.94–0.97) | T124A (0.90–1.1) | T124A (0.50–1.0) | ||
| 1.28 | T124A (0.68–0.76) | T124A (0.54–0.65) | T124A (0.84–0.91) | ||||
| 0.256 | 4,000 | E92Q/M154I (1.9) | E92Q/E138K/Q148K/M154I (34) | ||||
| 800 | N17S/Q148K (124); E92Q/M154I (63) | N17S/Q148K/G163R (75); E138K/Q148K (70) | |||||
| 160 | None (1.1) | Q148K (27); E92Q/M154I (3.1) | N17S/Q148K (78); E92Q/M154I (6.8) | N17S/Q148K/G163R (24); E138K/Q148K (40) | |||
| 32 | T124A (0.89–3.7) | T124A (0.99->61) | Q148R (8.8); T124A (4.6) | Q148R (6.8); T124A (1.0) | Q148R (11); T124A (1.2) | ||
| 6.4 | T124A (0.63–0.83) | T124A (0.43–0.84) | T124A (0.90–1.1) | T124A (0.50–0.70) | |||
| 1.28 | T124A (0.66–0.80) | T124A (0.64–0.70) | T124A (0.72–0.94) | ||||
| 0.256 | T124A (0.54–0.85) | T124A (0.54–0.64) | |||||
Fold changes from the phenotype assay are shown in parentheses after genotype assay results. When FCs of three wells are different, the data are shown as a range.
Under our isolation conditions as previously described, the M184V lamivudine-resistant virus emerged on day 14 (14). In the culture with GSK1265744 or RAL starting at any concentration except for GSK1265744 at 32 nM, T124A substitution was first observed on day 14 (Tables 3 and 4). However, IIIB contains ∼40% T124A at baseline, and the fold changes of GSK1265744 and RAL to T124A SDM were 0.97 and 0.82, respectively. Therefore, the T124A polymorphism has no impact on susceptibility to GSK1265744 or RAL. In the RAL-containing passage experiments, a Q148K RAL signature resistant virus emerged on day 28 when the culture was started at 32 nM (Fig. 3b and Table 4). An N155H/I204T variant emerged on day 28 when the culture was started at 6.4 nM. A Q148R variant emerged on day 42 when the culture was started at 1.28 nM. Q148K and Q148R variants emerged on day 56 when the culture was started at 0.256 nM. Continuous culture selected for additional mutations at all concentrations. Q148K and N155H/I204T variants emerged on day 28. Overall, the barrier to resistance of RAL was higher than that of lamivudine but lower than that of GSK1265744 in this assay.
In the case of GSK1265744, S153Y, Q146L (with culture started at 6.4 nM), and T124A/S153Y (with culture started at 0.256 nM) substitutions were observed on day 56, and S153Y (with culture started at 1.28 nM) was observed on day 70 (Table 3). Interestingly, no other secondary substitutions were added to each mutant virus up to day 112. The T124A/I162M substitution was observed only once, on day 84, in the well started with 1.28 nM, but this virus disappeared on day 88. We constructed a T124A/I162M SDM and have confirmed that this mutant has very low replication capacity and could not be evaluated for susceptibility to GSK1265744 (data not shown). The substitutions observed in the GSK1265744 passage study had no or minimal effects on susceptibility to GSK1265744 when they were introduced into a laboratory strain, NL432, by site-directed mutagenesis; FCs were 2.1 for the Q146L variant, 2.0 for the S153Y variant, and 2.4 for the T124A/S153Y variant (Table 2). Therefore, we confirmed that no virus highly resistant to GSK1265744 was isolated in vitro up to 112 days of passage.
DISCUSSION
INSTIs are generally recognized as a safe and effective class of anti-HIV drugs. DTG, RAL, and EVG have all been recommended as components for first-line therapy regimens in the most recent U.S. DHHS guidelines (13). Clinical data indicate that viruses resistant to the earlier INSTIs (RAL and EVG) have arisen (2, 8, 10). DTG and GSK1265744 are clearly differentiated as new INSTIs. They are highly effective against both wild-type and most INSTI-resistant mutants that have been identified to date. DTG has been clinically demonstrated with significantly less treatment-emergent INSTI or background therapy resistance than RAL in the SAILING study through 48 weeks (33). Thus, DTG could have a higher barrier to resistance clinically than RAL. GSK1265744 has in vitro characteristics similar to those of DTG which support its potential for having a high barrier to resistance. Herein, we define “high barrier to resistance” in vitro as a mutant virus which has more than a 3-fold change as SDM not emerging through more than 100 days in our in vitro passage study. While the chemical structure of GSK1265744 is related to that of DTG, GSK1265744 has unique attributes (high protein binding, low solubility, and low plasma clearance in animals). Consequently, the human oral PK data for GSK1265744 indicate a longer half-life (t1/2) of approximately 32 h) (16). Preliminary evaluation of GSK1265744 LA in animals suggested a possibility of parenteral once-monthly dosing in humans or possibly less frequently. Based on its resistance and pharmacokinetic profiles, GSK1265744 is in full clinical development as an LA formulation along with an oral tablet for use in the induction phase.
The potency shift for GSK1265744 with 100% human serum is 408-fold, which is consistent with a highly protein-bound molecule. Of note is the fact that achieving plasma concentrations above the PA-EC90 was still anticipated due to PK measurements (16) and high intrinsic antiviral efficacy. The clinical efficacy of GSK1265744 was demonstrated in a 10-day monotherapy trial at doses of 5 and 30 mg once daily (16); PK measurements showed that trough concentrations above the PA-EC90 were achievable with these very low oral doses.
The in vitro experiments described here confirmed that the mechanism of action of GSK1265744 is through inhibition of HIV IN at the strand transfer step of viral DNA insertion into host DNA. The inhibitor had no effect on total viral DNA synthesis in the infected cells but blocked the integration of viral DNA into host DNA with the same potency as its antiviral effect (Fig. 2). In addition, GSK1265744 increased the appearance of viral 2-LTR circles, a known by-product of integrase inhibition with the same potency as other INSTIs (14, 27, 34). As expected, GSK1265744 had efficacy against mutant viruses resistant to NRTIs, NNRTIs, and PIs, similar to its potency against the wild-type virus and consistent with its acting on a different antiretroviral target. GSK1265744 maintained activity against single and several multiple IN mutants but had decreased efficacy against a small subset of multimutated viruses resistant to other INSTIs (Table 2). In vitro combination studies confirmed that GSK1265744 has no antagonistic effect on all measured anti-HIV drugs across all classes, including likely combination agents in the NRTI and NNRTI classes.
It is striking that GSK1265744 had significant efficacy against most double or multiple integrase mutants in our panel of 24 mutants; only 6 mutant viruses (the E138A/Q148R, E138K/Q148K, E138K/Q148R, G140C/Q148R, G140S/Q148R, and Q148R/N155H viruses) had a FC of >10 (Table 2). This profile is similar to that of DTG, for which only 2 such mutants had a FC of >10. In contrast, RAL and EVG showed FC values of >10 with 20 and 22 such mutants, respectively. Therefore, we conclude that GSK1265744 has a new resistance profile similar to that of DTG.
Importantly, GSK1265744 showed potency against 25 INSTI-resistant single mutants in our panel of INSTI-resistant SDMs that was approximately equivalent to that of the wild type. The Q148 pathway confers a high level of resistance to RAL and EVG. The Q148K (FC = 5.6) and Q148R (FC = 5.1) mutants gave rise to a moderate loss of efficacy for GSK1265744. Given this moderate loss, we were curious as to whether Q148K/R would arise in a passage study. Neither mutant was observed. From these results, we surmise that the potential of resistance to GSK1265744 emerging may be very rare during clinical use for INSTI-naive patients.
Under our isolation conditions, the lamivudine-resistant M184V virus emerged as expected on day 14 (14). Our RAL data suggested that there was not sufficient pressure below 6.4 nM RAL to effectively select resistant viruses. However, above that concentration, RAL-resistant mutants were isolated, and the minimum culture period required for resistance to RAL to develop was 28 days under our experimental conditions.
In the case of GSK1265744, four mutant viruses (the Q146L, S153Y, T124A/S153Y, and T124A/I162Mvariants) were isolated (Table 3). Of note is the fact that T124A is polymorphic and has no effect on GSK1265744 activity and that it was present in the starting inoculum of the passage virus (35). However, as mentioned above, no Q148K/R or any other RAL/EVG signature mutant virus was isolated up to 112 days. Among the four isolated mutant viruses, we confirmed that three site-directed molecular clones maintained sensitivity to GSK1265744 (FCs were less than 2.9) (Table 2), although the T124A/I162M virus could not be evaluated because of its low replication capacity. These data were consistent with the fact that we could not maintain viruses in a GSK1265744 concentration at 32 nM (Fig. 3a). Interestingly, the Q146L virus was a new resistant variant for EVG (FC = 13), although Q146P has been reported (36).
The GSK1265744 data herein are consistent with our definition of a high barrier to resistance in vitro. However, the high barrier to resistance ultimately must be proven in controlled clinical trials. In a clinical situation, there are many factors to consider, such as the drug plasma trough concentration (Ctau), drug binding affinity for the IN enzyme, and adherence to drug therapy. One of the key factors is the inhibitory quotient (IQ), which is defined as follows: IQ = Ctau/PA-EC90. In the case of DTG, the mean Ctau for a 50-mg once-daily dose measured in the SPRING-1 and SPRING-2 treatment-naive studies was 1.1 μg/ml (reference, Tivicay package insert); therefore, the IQ was 17. For GSK1265744, the 30-mg oral daily dose gave a geometric mean Ctau of 3.28 μg/ml, for an IQ of 19.8 (16). The IQ at 4 weeks after GSK1265744 LA 800-mg single-dose intramuscular injection was approximately 12 (16). Thus, because the IQ for GSK1265744 is similar to that of DTG, which has a proven clinically high barrier to resistance (33, 37–39), in conjunction with its encouraging in vitro resistance profile, we expect little if any clinical failure due to resistance for the GSK1265744 treatment regimens planned.
Recently, Quashie et al. reported that R263K and G118R were isolated in vitro as potential DTG resistant viruses (40). Also, although fold changes were less than 2 in a phenotype assay with isolated viruses, the R263K and R263R/K variants were identified from two DTG-treated virologic failure patients (33). Therefore, we evaluated the activity of GSK1265744 against these mutant viruses. Our preliminary data are that the fold change against the R263K variant was 1.3 ± 0.49 (n = 3) and the fold change against the G118R variant was 8.9 ± 0.86 (n = 3). We concluded that the R263K virus is not a resistant mutant for GSK1265744 and the G118R variant is a potential resistant mutant. However, the possibility of emergence of G118R during clinical use might be very low because the replication capacity of G118R virus is very poor (41).
In conclusion, GSK1265744 has a markedly more distinct profile than RAL and EVG in terms of cross-resistance and in vitro resistance passage studies. These characteristics in combination with its excellent antiviral efficacy and safety profile, as well as suitable PK from both oral and LA injection regimens, should provide a novel treatment option for patients. Taken together, the attributes of GSK1265744 may combine to allow for durable efficacy in difficult-to-treat patient populations, with improvement of compliance and quality of life, and provide an attractive new option for both treatment and PrEP indications.
ACKNOWLEDGMENTS
We recognize the entire Shionogi-GSK HIV integrase drug discovery team for the discovery and preclinical development of GSK1265744. In addition, we thank Marty St Clair, Alex Rinehart, William Spreen, Louise Martin-Carpenter, and Jim Demarest for critical reading.
REFERENCES
- 1.Markowitz M, Nguyen BY, Gotuzzo E, Mendo F, Ratanasuwan W, Kovacs C, Prada G, Morales-Ramirez JO, Crumpacker CS, Isaacs RD, Gilde LR, Wan H, Miller MD, Wenning LA, Teppler H. 2007. Rapid and durable antiretroviral effect of the HIV-1 integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled study. J Acquir Immune Defic Syndr 46:125–133. doi: 10.1097/QAI.0b013e318157131c. [DOI] [PubMed] [Google Scholar]
- 2.Markowitz M, Morales-Ramirez JO, Nguyen BY, Kovacs CM, Steigbigel RT, Cooper DA, Liporace R, Schwartz R, Isaacs R, Gilde LR, Wenning L, Zhao J, Teppler H. 2006. Antiretroviral activity, pharmacokinetics, and tolerability of MK-0518, a novel inhibitor of HIV-1 integrase, dosed as monotherapy for 10 days in treatment-naive HIV-1-infected individuals. J Acquir Immune Defic Syndr 43:509–515. doi: 10.1097/QAI.0b013e31802b4956. [DOI] [PubMed] [Google Scholar]
- 3.DeJesus E, Berger D, Markowitz M, Cohen C, Hawkins T, Ruane P, Elion R, Farthing C, Zhong L, Cheng AK, McColl D, Kearney BP. 2006. Antiviral activity, pharmacokinetics, and dose response of the HIV-1 integrase inhibitor GS-9137 (JTK-303) in treatment-naive and treatment-experienced patients. J Acquir Immune Defic Syndr 43:1–5. doi: 10.1097/01.qai.0000233308.82860.2f. [DOI] [PubMed] [Google Scholar]
- 4.Sax PE, DeJesus E, Mills A, Zolopa A, Cohen C, Wohl D, Gallant JE, Liu HC, Zhong L, Yale K, White K, Kearney BP, Szwarcberg J, Quirk E, Cheng AK. 2012. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet 379:2439–2448. doi: 10.1016/S0140-6736(12)60917-9 (Erratum, 380:730.) [DOI] [PubMed] [Google Scholar]
- 5.DeJesus E, Rockstroh JK, Henry K, Molina JM, Gathe J, Ramanathan S, Wei X, Yale K, Szwarcberg J, White K, Cheng AK, Kearney BP. 2012. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate versus ritonavir-boosted atazanavir plus co-formulated emtricitabine and tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet 379:2429–2438. doi: 10.1016/S0140-6736(12)60918-0. [DOI] [PubMed] [Google Scholar]
- 6.Arribas JR, Eron J. 2013. Advances in antiretroviral therapy. Curr Opin HIV AIDS 8:341–349. doi: 10.1097/COH.0b013e328361fabd. [DOI] [PubMed] [Google Scholar]
- 7.Marinello J, Marchand C, Mott BT, Bain A, Thomas CJ, Pommier Y. 2008. Comparison of raltegravir and elvitegravir on HIV-1 integrase catalytic reactions and on a series of drug-resistant integrase mutants. Biochemistry 47:9345–9354. doi: 10.1021/bi800791q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson VA, Calvez V, Gunthard HF, Paredes R, Pillay D, Shafer RW, Wensing AM, Richman DD. 2013. Update of the drug resistance mutations in HIV-1. Top Antivir Med 21:6–14. [PMC free article] [PubMed] [Google Scholar]
- 9.Geretti AM, Armenia D, Ceccherini-Silbestein F. 2012. Emerging patterns and implications of HIV-1 integrase inhibitor resistance. Curr Opin Infect Dis 25:677–686. doi: 10.1097/QCO.0b013e32835a1de7. [DOI] [PubMed] [Google Scholar]
- 10.Garrido C, Villacian J, Zahonero N, Pattery T, Garcia F, Gutierrez F, Caballero E, Van Houtte M, Soriano V, de Mendoza C. 2012. Broad phenotypic cross-resistance to elvitegravir in HIV-infected patients failing on raltegravir-containing regimens. Antimicrob Agents Chemother 56:2873–2878. doi: 10.1128/AAC.06170-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menéndez-Arias L. 2013. Molecular basis of human immunodeficiency virus type 1 drug resistance: overview and recent developments. Antiviral Res 98:93–120. doi: 10.1016/j.antiviral.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Huang W, Frantzell A, Fransen S, Petropoulos CJ. 2013. Multiple genetic pathways involving amino acid position 143 of HIV-1 integrase are preferentially associated with specific secondary amino acid substitutions and confer resistance to raltegravir and cross-resistance to elvitegravir. Antimicrob Agents Chemother 57:4105–4113. doi: 10.1128/AAC.00204-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panel on Antiretroviral Guidelines for Adults and Adolescents. 2013. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents, F-4, Table 5a. Department of Health and Human Services, Washington, DC: http://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf. [Google Scholar]
- 14.Kobayashi M, Yoshinaga T, Seki T, Wakasa-Morimoto C, Brown KW, Ferris R, Foster SA, Hazen RJ, Miki S, Suyama-Kagitani A, Kawauchi-Miki S, Taishi T, Kawasuji T, Jhons BA, Underwood MR, Garvey EP, Sato A, Fujiwara T. 2011. In vitro antiretoroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob Agents Chemother 55:813–821. doi: 10.1128/AAC.01209-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johns BA, Kawasuji T, Weatherhead JG, Taishi T, Temelkoff DP, Yoshida H, Akiyama T, Taoda Y, Murai H, Kiyama R, Fuji M, Tanimoto N, Jeffrey J, Foster SA, Yoshinaga T, Seki T, Kobayashi M, Sato A, Johnson MN, Garvey EP, Fujiwara T. 2013. Carbamoyl pyridone HIV-1 integrase inhibitors 3. A diastereomeric approach to chiral nonracemic tricyclic ring systems and the discovery of dolutegravir (S/GSK1349572) and (S/GSK1265744). J Med Chem 56:5901–5916. doi: 10.1021/jm400645w. [DOI] [PubMed] [Google Scholar]
- 16.Spreen W, Min S, Ford SL, Chen S, Lou Y, Bomar M, St Clair M, Piscitelli S, Fujiwara T. 2013. Pharmacokinetics, safety, and monotherapy antiviral activity of GSK1265744, an HIV integrase strand transfer inhibitor. HIV Clin Trials 14:192–203. doi: 10.1310/hct1405-192. [DOI] [PubMed] [Google Scholar]
- 17.Spreen WR, Margolis DA, Pottage JC Jr. 2013. Long-acting injectable antiretrovirals for HIV treatment and prevention. Curr Opin HIV AIDS 8:565–571. doi: 10.1097/COH.0000000000000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Underwood M, St Clair M, Johns BA, et al. 2010. S/GSK1265744: a next generation integrase inhibitor (INI) with activity against raltegravir-resistant clinical isolates, abstr MOAA0103 Twenty-Eighth Int AIDS Conf, Vienna, Austria. [Google Scholar]
- 19.Spreen W, Williams P, Margolis D, Ford S, Crauwels H, Lou Y, Gould E, Stevens M, Piscitelli S. 2013. First study of repeat dose co-administration of GSK1265744 and TMC278 long-acting parenteral nanosuspensions: pharmacokinetics, safety and tolerability in healthy adults, abstr WEAB0103. Seventh IAS Conf HIV Pathog Treat Prev, Kuala Lumpur, Malaysia http://www.ias2013.org/. [Google Scholar]
- 20.Plosker GL. 2013. Emtricitabine/tenofovir disoproxil fumarate: a review of its use in HIV-1 pre-exposure prophylaxis. Drugs 73:279–291. doi: 10.1007/s40265-013-0024-4. [DOI] [PubMed] [Google Scholar]
- 21.Daluge SM, Purifoy DJ, Savina PM, St Clair M, Parry NR, Dev IK, Novak P, Ayers KM, Reardon JE, Roberts GB, et al. 1994. 5-Chloro-2′,3′-dideoxy-3′-fluorouridine (935U83), a selective anti-human immunodeficiency virus agent with an improved metabolic and toxicological profile. Antimicrob Agents Chemother 38:1590–1603. doi: 10.1128/AAC.38.7.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harada S, Koyanagi Y, Yamamoto N. 1985. Infection of HTLV-III/LAV in HTLV-I-carrying cells MT-2 and MT-4 and application in a plaque assay. Science 229:563–566. doi: 10.1126/science.2992081. [DOI] [PubMed] [Google Scholar]
- 23.Isaka Y, Sato A, Miki S, Kawauchi S, Sakaida H, Hori T, Uchiyama T, Adachi A, Hayami M, Fujiwara T, Yoshie O. 1999. Small amino acid changes in the V3 loop of human immunodeficiency virus type 2 determines the coreceptor usage for CXCR4 and CCR5. Virology 264:237–243. doi: 10.1006/viro.1999.0006. [DOI] [PubMed] [Google Scholar]
- 24.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarmy G, Heinkelein M, Weissbrich B, Jassoy C, Rethwilm A. 2001. Phenotypic analysis of the sensitivity of HIV-1 to inhibitors of the reverse transcriptase, protease, and integrase using a self-inactivating virus vector system. J Med Virol 64:223–231. doi: 10.1002/jmv.1040. [DOI] [PubMed] [Google Scholar]
- 26.Boros EE, Johns BA, Garvey EP, Koble CS, Miller WH. 2006. Synthesis and HIV-integrase strand transfer inhibition activity of 7-hydroxy[1,3]thiazolo[5,4-b]pyridin-5(4H)-ones. Bioorg Med Chem Lett 16:5668–5672. doi: 10.1016/j.bmcl.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 27.Garvey EP, Johns BA, Gartland MJ, Foster SA, Miller WH, Ferris RG, Hazen RJ, Underwood MR, Boros EE, Thompson JB, Weatherhead JG, Koble CS, Allen SH, Schaller LT, Sherrill RG, Yoshinaga T, Kobayashi M, Wakasa-Morimoto C, Miki S, Nakahara K, Noshi T, Sato A, Fujiwara T. 2008. The naphthyridinone GSK364735 is a novel, potent human immunodeficiency virus type 1 integrase inhibitor and antiretroviral. Antimicrob Agents Chemother 52:901–908. doi: 10.1128/AAC.01218-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi M, Nakahara K, Seki T, Miki S, Kawauchi S, Suyama A, Wakasa-Morimoto C, Kodama M, Endoh T, Oosugi E, Matsushita Y, Murai H, Fujishita T, Yoshinaga T, Garvey E, Foster S, Underwood M, Johns B, Sato A, Fujiwara T. 2008. Selection of diverse and clinically relevant integrase inhibitor-resistant human immunodeficiency virus type 1 mutants. Antiviral Res 80:213–222. doi: 10.1016/j.antiviral.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 29.Selleseth DW, Talarico CL, Miller T, Lutz MW, Biron KK, Harvey RJ. 2003. Interactions of 1263W94 with other antiviral agents in inhibition of human cytomegalovirus replication. Antimicrob Agents Chemother 47:1468–1471. doi: 10.1128/AAC.47.4.1468-1471.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tukey JW, Ciminera JL, Heyse JF. 1985. Testing the statistical certainty of a response to increasing doses of a drug. Biometrics 41:295–301. doi: 10.2307/2530666. [DOI] [PubMed] [Google Scholar]
- 31.Prichard MN, Shipman C Jr. 1990. A three-dimensional model to analyze drug-drug interactions. Antiviral Res 14:181–205. doi: 10.1016/0166-3542(90)90001-N. [DOI] [PubMed] [Google Scholar]
- 32.Baert L, van 't Klooster G, Dries W, et al. 2009. Development of a long-acting injectable formulation with nanoparticles of rilpivirine (TMC278) for HIV treatment. Eur J Pharm Biopharm 72:502–508. doi: 10.1016/j.ejpb.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 33.Cahn P, Pozniak AL, Mingrone H, Shuldyakov A, Brites C, Andrade-Villanueva JF, Richmond G, Buendia CB, Fourie J, Ramgopal M, Hagins D, Felizarta F, Madruga J, Reuter T, Newman T, Small CB, Lombaard J, Grinsztejn B, Dorey D, Underwood M, Griffith S, Min S. 2013. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet 382:700–708. doi: 10.1016/S0140-6736(13)61221-0. [DOI] [PubMed] [Google Scholar]
- 34.Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. 2000. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 287:646–650. doi: 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- 35.Vavro C, Hasan S, Madsen H, Horton J, DeAnda F, Martin-Carpenter L, Sato A, Cuffe R, Chen S, Underwood M, Nichols G. 2013. Prevalent polymorphisms in wild-type HIV-1 integrase are unlikely to engender drug resistance to dolutegravir (S/GSK1349572). Antimicrob Agents Chemother 57:1379–1384. doi: 10.1128/AAC.01791-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimura K, Kodama E, Sakagami Y, Matsuzaki Y, Watanabe W, Yamataka K, Watanabe Y, Ohata Y, Doi S, Sato M, Kano M, Ikeda S, Matsuoka M. 2008. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J Virol 82:764–774. doi: 10.1128/JVI.01534-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stellbrink HJ, Reynes J, Lazzarin A, Voronin E, Pulido F, Felizarta F, Almond S, St Clair M, Flack N, Min S. 2013. Dolutegravir in antiretroviral-naive adults with HIV-1: 96-week results from a randomized dose-ranging study. AIDS 27:1771–1778. doi: 10.1097/QAD.0b013e3283612419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raffi F, Jaeger H, Quiros-Roldan E, Albrecht H, Belonosova E, Gatell JM, Baril JG, Domingo P, Brennan C, Almond S, Min S. 2013. Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial. Lancet Infect Dis 13:927–935. doi: 10.1016/S1473-3099(13)70257-3. [DOI] [PubMed] [Google Scholar]
- 39.Raffi F, Rachlis A, Stellbrink HJ, Hardy WD, Torti C, Orkin C, Bloch M, Podzamczer D, Pokrovsky V, Pulido F, Almond S, Margolis D, Brennan C, Min S. 2013. Once-daily dolutegravir versus raltegravir in antiretroviral-naive adults with HIV-1 infection: 48 week results from the randomised, double-blind, non-inferiority SPRING-2 study. Lancet 381:735–743. doi: 10.1016/S0140-6736(12)61853-4. [DOI] [PubMed] [Google Scholar]
- 40.Quashie PK, Mesplède T, Han YS, Oliveira M, Singhroy DN, Fujiwara T, Underwood MR, Wainberg MA. 2012. Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. J Virol 86:2696–2705. doi: 10.1128/JVI.06591-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bar-Magen Sloan RD, Donahue DA, Kuhl BD, Zabeida A, Xu H, Oliveira M, Hazuda DJ, Wainberg MA. 2010. Identification of novel mutations responsible for resistance to MK-2048, a second-generation HIV-1 integrase inhibitor. J Virol 84:9210–9216. doi: 10.1128/JVI.01164-10. [DOI] [PMC free article] [PubMed] [Google Scholar]