

Abstract
We previously reported the development of a prototype antibiotic sensitivity assay to detect drug-resistant Mycobacterium tuberculosis using infection by mycobacteriophage to create a novel nucleic acid transcript, a surrogate marker of mycobacterial viability, detected by reverse transcriptase PCR (M. C. Mulvey et al., mBio 3:e00312-11, 2012). This assay detects antibiotic resistance to all drugs, even drugs for which the resistance mechanism is unknown or complex: it is a phenotypic readout using nucleic acid detection. In this report, we describe development and characteristics of an optimized reporter system that directed expression of the RNA cyclase ribozyme, which generated circular RNA through an intramolecular splicing reaction and led to accumulation of a new nucleic acid sequence in phage-infected bacteria. These modifications simplified the assay, increased the limit of detection from 104 to <102 M. tuberculosis cells, and correctly identified the susceptibility profile of M. tuberculosis strains exposed for 16 h to either first-line or second-line antitubercular drugs. In addition to phenotypic drug resistance or susceptibility, the assay reported streptomycin MICs and clearly detected 10% drug-resistant cells in an otherwise drug-susceptible population.
INTRODUCTION
Multidrug-resistant tuberculosis (MDR-TB) is caused by Mycobacterium tuberculosis strains resistant to at least two of the drugs, isoniazid (INH) and rifampin (RIF), in the recommended four-drug TB treatment regimen (1). The World Health Organization (WHO) estimates the prevalence of MDR-TB to be approximately 50 million people worldwide (2), expanding by nearly 500,000 new cases each year (3). A more disquieting development is the increase in and global distribution of extremely drug-resistant TB (XDR-TB) (M. tuberculosis resistant to INH, RIF, and key second-line drugs) (4). Early recognition of M/XDR-TB through the use of an accurate and sensitive antibiotic susceptibility test (AST) promises to improve patient outcomes and assist in TB control efforts.
Due to the notoriously slow doubling time of M. tuberculosis, a standard AST using culture takes several weeks to months. Culture-based assays are the gold standard for AST, however, because they measure every type of drug susceptibility and do not require knowledge of the genetics underlying resistance. However, they are slow or too insensitive to interrogate effects of antibiotics on small numbers of M. tuberculosis present in most clinical samples. Rapid molecular diagnostic tests such as Xpert MTB/RIF (Cepheid) and GenoType MTBDR (Hain Lifescience) are the emerging standard for rapid INH- or RIF-resistant M. tuberculosis detection, markers of MDR-TB, because their detection thresholds from patient specimens are comparable to culture (5, 6). These tests identify M. tuberculosis DNA in a patient sample and detect common mutations that confer resistance to INH (GenoType MTBDR) and/or RIF (GenoType MTBDR and Xpert MTB/RIF) in 2 h (Xpert MTB/RIF) or 2 days (GenoType MTBDR) from the initiation of the test. However, although these assays identify MDR-TB, they do not detect susceptibility to the other first- or second-line drugs. They are an excellent epidemiologic tool and an indicator of what drugs not to use, but they do not function as an AST should, guiding clinicians to the drugs that will be effective for treating an individual patient.
For antitubercular drugs other than RIF and INH, the various genes and genetic mutations responsible for clinical drug resistance are either unknown or too numerous to test in a simple assay. Clearly, a rapid technology capable of detecting phenotypic drug resistance from low numbers of M. tuberculosis is needed. To achieve this goal, we designed a nucleic acid detection system that measures the phenotypic effects of antimicrobials. This strategy combines the best attribute of nucleic acid detection methods, namely, the ability to rapidly detect low numbers of cellular nucleic acids, with the best attribute of phenotypic assays, namely, the ability to detect the biological effects antimicrobials exert on susceptible microorganisms.
The reporter system has two functional cassettes: (i) the SP6 RNA polymerase (SP6Pol) gene under transcriptional control of a promoter active in mycobacteria and (ii) the consensus SP6Pol promoter that directs expression of an SP6Pol-dependent transcript (7). The reporter system is incorporated into a mycobacteriophage for delivery into M. tuberculosis and the SP6Pol-dependent transcript is detected by reverse transcription-PCR (RT-PCR) in infected cells. In our previous publication describing the prototype system, the assay detected ≥104 M. tuberculosis cells and correctly identified drug-resistant M. tuberculosis strains. The SP6Pol-dependent transcript is called the surrogate marker locus (SML) since it is a nucleic acid that functions as a surrogate marker of bacterial metabolic capacity. The reporter system is called the SML generation module (SGM), and the reporter phage encoding the prototype SGM is phSGM1. The prototype SML expressed in cells infected with phSGM1 is a linear RNA copy of its cognate DNA locus in the phage genome. Because this RNA transcript is not present in the input phage, synthesis of the SML indicates the presence of metabolically active M. tuberculosis. Detection of this linear SML RNA, however, is laborious because phage DNA must be degraded prior to RT-PCR. Furthermore, the prototype phSGM1 assay could not detect <104 CFU of M. tuberculosis.
We describe here phSGM2, a second-generation, improved version of phSGM1. The SGM in phSGM2 was optimized for function in mycobacteria, encoded the RNA cyclase ribozyme under SP6Pol transcriptional control, and generated a circular SML RNA (cSML) resulting in a new nucleic acid sequence distinct form its cognate DNA locus. This simplified the assay and improved both limits of M. tuberculosis detection and ability to detect drug-resistant M. tuberculosis strains, including clinical isolates, by themselves, as well as when seeded into drug-sensitive populations.
MATERIALS AND METHODS
Propagation of bacterial strains and phages.
M. smegmatis mc2155 and mc24502 (8) were obtained from William Jacobs, Albert Einstein College of Medicine. All mycobacteria were maintained in 7H9 broth supplemented with Middlebrook oleic acid-albumin-dextrose-catalase (OADC) and 0.01% Tween 80. Both the RIF-resistant (RIFr) and the ethambutol-resistant (EMBr) M. tuberculosis strains used in the present study were created and isolated from H37Rv by Sequella, Inc., and maintained as for H37Rv. The MICs of RIFr M. tuberculosis to RIF and EMBr M. tuberculosis to EMB were >32 μg/ml for each. Both resistant strains were susceptible to all other anti-TB drugs tested. The deidentified clinical isolates tested in here were previously obtained from the State of Maryland TB Lab and are maintained at Sequella. The drug susceptibility profile of each strain was determined by Bactec 460 or agar proportion method (data not shown). Recombinant mycobacteriophages phSGM2 and phPleft-Cyc were propagated using mc24502 as described previously (8).
Recombinant mycobacteriophage construction and production.
The SGM was synthesized de novo by GenScript, USA. To create phSGM2, we integrated the SGM into a mycobacteriophage TM4 shuttle phasmid between nucleotides 47314 and 50108 of the TM4 genome. SGM integration was performed by GeneBridges, GmbH, using Red/ET recombineering. To create phPleft-Cyc, we amplified by PCR the region in the SGM downstream of the SP6 promoter and upstream of the SP6MCS-T7-rrnC termination region. This product was then fused to the Pleft promoter and integrated into the mycobacteriophage TM4 shuttle phasmid between nucleotides 47314 and 50108 of the TM4 genome using Red/ET recombineering.
Shuttle phasmid DNA was then isolated and electroporated into mc24502 to create infectious phage, as described previously (9). Phage stocks were prepared by elution from confluent and lysed mc24502 agarose overlays using MP buffer (50 mM Tris, 150 mM NaCl, 10 mM MgCl2, 2 mM CaCl2 [pH 7.6]), followed by low-speed centrifugation to pellet insoluble debris and filtration to sterilize the preparation. Phage stocks were adjusted to 1010 PFU/ml in MP buffer, treated with 50 ng of RNase A per ml for 2 h at 37°C to degrade cSML produced during phage growth on mc24502 overlays, and then stored at 4°C.
Phage infection.
Mycobacteria were washed three times with 7H9 broth supplemented with OADC (7H9-A) to remove Tween 80. Mycobacterial suspensions (0.2 ml) were then added to the wells of a 48-well tissue culture dish and incubated at 37°C for 2 h (M. smegmatis mc2155) or a minimum of 16 h (M. tuberculosis). Phage (50 μl) in MP buffer was then added, and infected cells were incubated at 37°C.
RNA purification.
Total RNA was isolated by adding 0.75 ml of TRIzol-LS containing 1.3 μg/ml poly(I·C) to 250 μl of sample. The mixture was then transferred to a lysing matrix B tube (MP Biomedicals), and cells were disrupted by two 50-s rounds of bead beating on a Mini-Bead beater (BioSpec Products) set at 4,800 oscillations per min. Samples were cooled on ice for at least 1 min between bead-beating cycles. Chloroform-isoamyl alcohol (24:1; 0.2 ml) was then added, the aqueous fraction was combined with an equal volume of 70% ethanol, and the final suspension was added to an RNeasy silica spin column (Qiagen). RNA was then purified according to the manufacturer's directions and eluted in 50 μl of RNase-free water containing 4 U of RNaseOUT (Life Technologies).
One-step real-time RT-PCR to detect cSML.
A 20-μl reaction mix was prepared that contained 7.4 μl of purified total RNA, 10 μl of Express SuperScript qPCR SuperMix Universal, 2 μl of Express SuperScript Mix for One-Step qPCR, and 200 nM concentrations of oligonucleotides, specifically, P1 (5′-GCGTCGGTGACAAAGGCCACGA-3′), P2 (5′-CAATGGATTCTGAGGTTGCTGCTTTGG-3′), and a molecular beacon (5′-FAM-CGCACGATGTGTAAAGCCGGAGCTCACGTGCG-DABCYL-3′). Express One-Step reagents were purchased from Life Technologies, and oligonucleotides were synthesized by Eurofins/Operon MWG and maintained in 10 mM Tris-HCl (pH 8.0)–0.1 mM EDTA (pH 8.0). One-Step RT-PCR was performed on a PikoReal 24-well real-time PCR system (Thermo Fisher Scientific) under the following conditions: 50°C for 15 min, 94°C for 3 min, and then 45 cycles of 94°C for 30 s, 57°C for 30 s, and 72°C for 30s. FAM data acquisition was performed during the 57°C extension step.
RT-PCR to detect cSML and 16SrRNA using SYBR green.
A 20-μl reaction mix contained 5 μl of purified total RNA, Superscript III RT (Life Technologies), and 2 μM P1 or 16S-Reverse (5′-GCGACGCTCACAGTTAAGCCGTG-3′) oligonucleotides. Reaction mixtures were incubated at 42°C for 30 min and heat inactivated at 70°C for 15 min. Then, 2 μl of RT reaction mixture was added to a 20λ Power SYBR green PCR master mix (Life Technologies) containing 200 nM P2 or 16S-Forward (5′-GCTTTAGCGGTGTGGGATGAGCC-3′) oligonucleotides. PCR was performed under the following conditions: 94°C for 3 min, followed by 45 cycles of 94°C for 30 s, 57°C for 30 s, and 72°C for 30 s. SYBR green data acquisition was performed during the 57°C extension step.
Determining the dynamic range of cSML detection by one-step RT-PCR.
In vitro-transcribed cSML was generated by linearizing 100 ng of plasmid pSP6Pro-Cyc with HindIII, followed by addition to an in vitro transcription reaction mixture containing 1 U/μl purified SP6 RNA polymerase (New England BioLabs), 40 mM Tris-HCl (pH 7.9), 10 mM dithiothreitol, 2 mM spermidine, 4 mM concentrations of each nucleotide triphosphate, 8 mM MgCl2, and 0.8 U/μl RNaseOUT. The reaction mixture was incubated at 40°C for 1 h. DNA was then digested by adding RNase-free DNase I and DNase I reaction buffer (New England BioLabs), followed by incubation at 37°C for 2 h. EDTA was then added to a final concentration of 18 mM, and the sample was heat inactivated at 75°C for 10 min. Finally, duplicate 10-fold serial dilutions of in vitro-transcribed cSML were made in RNase-free water and added to a one-step RT-PCR. The average cycle detection threshold for each dilution was plotted to determine the dynamic range of the one-step RT-PCR cSML detection assay, which is described by the equation y = (109)−0.557x, where x is the cycle detection threshold, and y is the relative level of cSML in a sample.
Determination of the limit of detection.
H37Rv or mc2155 cells were washed and adjusted to an optical density at 600 nm (OD600) of 0.2. Serial 10-fold dilutions of cells were prepared in 7H9-A medium. A 0.2-ml volume of each dilution was added to duplicate wells of a 48-well dish and also plated on duplicate 7H10 agar dishes to determine the number of CFU. For the detection of mc2155, cells in 48-well culture dishes were incubated for 2 h at 37°C and infected with 50 μl of a 107-PFU/ml stock of phSGM2 for 4 h. For the detection of H37Rv, cells in 48-well culture dishes were incubated for 16 h at 37°C and infected with 50 μl of a 106-PFU/ml stock of phSGM2 for 4 h. RNA was then purified, cSML was detected by real-time RT-PCR, and the relative levels of cSML in samples were calculated.
Antibiotic susceptibility testing.
Washed mycobacterial cells were adjusted to an OD600 of 0.02 in 7H9-A medium containing antibiotics at the following concentrations: 1 μg/ml RIF, 0.1 μg/ml INH, 1 μg/ml streptomycin (STR), 5 μg/ml EMB, 500 μg/ml p-nitrobenzoic acid (PNB), 0.25 μg/ml moxifloxacin (MOX), 5 μg/ml ethionamide (ETH), 2 μg/ml para-amino salicylic acid (PAS), 4 μg/ml kanamycin (KAN), 1 μg/ml amikacin (AMK), 1.6 μg/ml SQ109, and 40 μg/ml cycloserine (CS). Cells (0.2 ml) were then added to wells of a 48-well dish, followed by incubation at 37°C for either 16 or 40 h. The phSGM2 diluted to 107 PFU/ml in MP buffer (50 μl) was then added, followed by incubation at 37°C for 4 h. The total RNA was then purified using TRIzol-LS and bead beating, the cSML and/or 16S rRNA was amplified and detected, and the relative levels of cSML in samples were calculated.
To determine RIF and STR MICs using Trek Sensititre MycoTB plates (Thermo Fisher Scientific), we added 100 μl of washed RIFr cells at an OD600 of 0.02 to the wells, incubated the plate at 37°C for 16 h, and then infected the wells with 25 μl of phSGM2 diluted to 107 PFU/ml in MP buffer for 8 h. We added 100 μl of 100% ethanol to the wells, and the mixture was transferred to a microcentrifuge tube containing 200 μl of 100% ethanol, followed by incubation at room temperature for 15 min to inactivate M. tuberculosis. Buffer RLT (425 μl; Qiagen) was then added, and the mixture was applied to an RNeasy silica spin column. The total RNA was then purified, the cSML was amplified and detected, and the relative levels of cSML in samples were calculated.
Susceptibility to an antibiotic was determined if drug treatment resulted in >10-fold reduction in cSML levels compared to the untreated control. Conversely, resistance was scored if drug treatment resulted in a ≤10-fold reduction in the cSML levels compared to the untreated control. These cutoff values were based on the extensive work performed by the Jacobs group to evaluate rapid AST using recombinant luciferase reporter phages (10).
RESULTS
Detection of mycobacteria by phSGM2.
Figure 1A and B describes the overall construction of the various components of phSGM2 and the actions and order of actions following infection of M. tuberculosis with the recombinant phage that results in the detection of viable bacterial cells. To determine the lowest number of cells detected by phSGM2, we made serial dilutions of M. smegmatis strain mc2155 or M. tuberculosis strain H37Rv (Fig. 1C). The dilutions were plated to determine the number of CFU and also infected with phSGM2. Uninfected 105 CFU and phage alone served as controls. At 4 h postinfection (p.i.), total RNA was purified and cSML measured by RT-PCR. The cSML was not detected in samples containing ∼105 CFU of either mc2155 or H37Rv (Fig. 1C) but was observed in phage alone controls. Despite the presence of cSML in the phage alone controls, cSML generation from as few as 18 CFU of mc2155 or 94 CFU of H37Rv infected with phSGM2 were detected with statistical significance (Fig. 1C).
FIG 1.
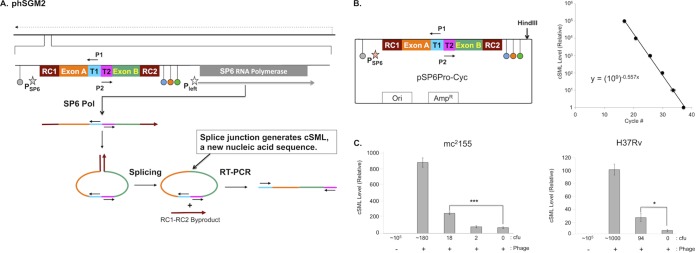
phSGM2 design, cSML detection assay dynamic range, and detection of M. smegmatis and M. tuberculosis. (A) The mycobacteriophage TM4 genome is depicted as a solid black line at the top of the figure. The direction of phage gene expression by host RNA polymerase is indicated by the dashed arrow above the phage genome. The site in the phage genome where the SGM is inserted is indicated and expanded. The SGM is comprised of two cassettes. The first is the SP6Pol ORF under transcriptional control of the mycobacteriophage L5 Pleft promoter (open star), which directs expression of SP6Pol during infection of mycobacteria. The SP6Pol ORF is codon optimized for efficient translation in M. tuberculosis, and translation initiation is directed by the Shine-Dalgarno sequence upstream of the TM4 major capsid subunit. The second cassette encodes the SP6 promoter (filled star) fused to a downstream sequence encoding several functional modules. The SP6 promoter cassette is flanked by transcription terminators. Upstream of the SP6 promoter is the E. coli rrnBT2 terminator (gray dot) (17), which precludes transcription of the SP6 promoter cassette by host RNA polymerase. We positioned three terminators downstream of the SP6 promoter cassette: phage SP6 major capsid subunit (SP6MCS; blue dot) (8), T7 (orange dot) (15), and the E. coli rrnC (green dot) (17) terminators. These terminators were designed to lower the frequency at which elongating SP6Pol transcribes the SP6Pol ORF. We also positioned several functional modules downstream of the SP6 promoter: T1 and T2 encoded sites to which primers P1 and P2 bind. These sites were oriented such that upon binding to T1 and T2, primers P1 and P2 are unable to generate a PCR amplification product. Adjacent to T1 and T2 were exons A and B. RC1 and RC2 were positioned at the 5′ of exon A and 3′ of exon B, respectively. RC1 and RC2 encode two halves of the RNA cyclase (RC) ribozyme (10). After transcription of this locus by SP6 Pol, a single-stranded RNA was synthesized that had one half of RC (RC1) fused to the 5′ end of exon A followed by T1, T2, and the other half of RC (RC2) fused to the 3′ end of exon B. Once RC2 was synthesized, RC1 and RC2 interacted and formed the active RC ribozyme, which mediated circularization of the single-stranded RNA between RC1 and RC2 by fusing exons A and B via a splicing reaction. Fusion of exons A and B created a new nucleic acid sequence distinct from the cognate DNA locus in the phage genome and constituted generation of the cSML. In addition, RC1 and RC2 fused to each other as a by-product. Finally, detection of the cSML was performed using P1 and P2 because the splicing of exons A and B generated an intervening sequence between the 3′ ends of P1 and P2, resulting in the creation of a template for RT-PCR amplification. The cSML was amplified by using a one-step, combined RT-PCR and detected by a molecular beacon. (B) The dynamic range was determined by cSML synthesis from HindIII-digested pSP6Pro-Cyc by in vitro transcription. Duplicate 10-fold serial dilutions were prepared, and cSML was amplified. The average cycle detection thresholds for each dilution were plotted, and the best-fit line through the data points is described by the equation y = (109)−0.557x, where x is the cycle detection threshold, and y is the relative level of cSML in a sample. This equation is used to determine the relative level of cSML present in a sample. (C) Determination of the lowest numbers of M. smegmatis mc2155 and M. tuberculosis H37Rv detected by the assay.
AST of M. tuberculosis using phSGM2 reporter phage.
To evaluate the ability of phSGM2 to determine M. tuberculosis antibiotic susceptibility, we left drug-susceptible H37Rv cells untreated or treated them with first-line anti-TB drugs RIF, INH, STR, or EMB. The cells were also treated with PNB, which exhibits antimycobacterial activity only against members of the TB complex and is used to verify that a sample does not contain nontuberculous mycobacteria. After either 16 or 40 h of antibiotic exposure, the cells were infected with phSGM2 for 4 h. The total RNA from each sample was then purified and cSML detected by RT-PCR. The level of cSML in untreated cells infected with phSGM2 was ∼1,000-fold higher than the phage alone controls. The level of cSML in infected cells exposed to antibiotics for either 16 or 40 h was reduced to background levels (Fig. 2A). Using >10-fold reduction in drug-treated samples compared to untreated controls as the cutoff for drug susceptibility, these data demonstrate that phSGM2 could report susceptibility to all first-line anti-TB antibiotics, excluding PZA (not shown) after less than 1 day of drug exposure. Furthermore, the dynamic range of the assay was ∼1,000-fold between drug-treated and untreated samples. The phSGM2 also reported susceptibility to all second-line anti-TB drugs tested (Fig. 2B). To determine whether the differences in cSML levels between untreated and drug-treated samples was due to effects on cSML generation or to global effects on cellular RNA, or differences in the number of cells added to each sample, we performed RT-PCR to detect cSML and 16S rRNA using purified total RNA from the samples in Fig. 2A (Table 1). In samples treated with antibiotics, cSML levels were reduced 12 to 15 cycles compared to the untreated control, whereas 16SrRNA was reduced by, at most, 4.4 cycles (RIF-treated sample). This demonstrated that the ∼1,000-fold differences in cSML levels observed between untreated and drug-treated cells infected with phSGM2 were not due to loading errors or pleiotropic effects of antibiotics on cellular RNA.
FIG 2.

phSGM2 accurately detected susceptibility and resistance to all classes of anti-TB drugs. (A and B) cSML generation in H37Rv cells infected by phSGM2 and exposed to first-line (A) and second-line (B) anti-TB drugs. (C) phSGM2 detected resistance to RIF and EMB in RIFr and EMBr H37Rv variants. (D and E) phSGM2 accurately reported the INH and RIF (D) and STR and KAN (E) susceptibility profiles of clinical isolates 386, 622, 7739, 8330, 8668, and 9016.
TABLE 1.
Cycle detection thresholds (CT) for SYBR green real-time RT-PCR detection of cSML and 16S rRNA from H37Rv cells treated with first-line anti-TB drugs and infected with phSGM2a
| Drug |
CTa |
|
|---|---|---|
| cSML | 16S rRNA | |
| No drug (no RT) | ND | 41.95 |
| No drug | 22.07 | 30.16 |
| RIF | 37.36 | 34.59 |
| INH | 34.95 | 32.20 |
| STR | 35.38 | 34.04 |
| EMB | 35.07 | 32.49 |
| PNB | 36.66 | 33.77 |
| Phage alone | 36.41 | ND |
ND, not detected. Data from one representative replicate are presented.
To determine whether phSGM2 reported drug resistance, we exposed two H37Rv variants to nothing (“no drug”) or to RIF, EMB, or PNB. One variant was susceptible to EMB and PNB but resistant to RIF (RIFr), and the other was susceptible to RIF and PNB, but resistant to EMB (EMBr). After 16 h of drug exposure, M. tuberculosis samples were infected with phSGM2 for 4 h, total RNA was purified, and cSML was detected by RT-PCR (Fig. 2D). cSML generation in untreated RIFr and EMBr cells infected with phSGM2 was robust, whereas exposure to PNB reduced cSML generation by between 100- and 1,000-fold, demonstrating that the organisms were M. tuberculosis. Treatment of the EMB-susceptible RIFr variant with EMB reduced cSML generation similarly to PNB. However, treatment with RIF did not significantly reduce cSML generation because the variant was resistant to RIF. Finally, treatment of the RIF-susceptible variant EMBr with RIF reduced cSML generation >100-fold. However, treatment with EMB did not significantly reduce cSML generation because the variant was resistant to EMB. These data demonstrate that cSML generation in phSGM2-infected cells could identify both drug susceptibility and drug resistance.
H37Rv is a standard, widely used M. tuberculosis laboratory strain that is highly metabolic compared to clinical isolates, especially M/XDR-TB isolates. To determine whether phSGM2 is able to report drug susceptibility and resistance in clinical isolates, several INH- and STR-resistant clinical isolates maintained at Sequella, Inc., were tested for susceptibility to INH and RIF (Fig. 2D) or to STR and KAN (Fig. 2E) after 40 h of drug exposure prior to phage infection. In all clinical isolates tested except strain 8330, cSML levels in the untreated control were elevated at least 100-fold more than the PNB background. Strain 8330 was the slowest growing of all isolates tested (M. Mulvey, unpublished observations), and the cSML levels in the untreated control were 16-fold higher than the in PNB background control (Fig. 2D), reflecting the low metabolic activity of this strain. Despite the low metabolic activity of strain 8330, phSGM2 correctly reported 8330 RIF susceptibility and INH resistance. In addition, phSGM2 correctly reported the susceptibility profile of the other clinical strains to INH, RIF, STR, and KAN.
Expression of cSML by SP6Pol is required to accurately report drug susceptibility.
Synthesis of cSML in phSGM2-infected cells could be mediated by host RNA polymerase rather than SP6Pol. Furthermore, the report of drug susceptibility by phSGM2 could simply reflect lower levels of phage gene expression by host RNA polymerase. In this situation, it would be far simpler to use wild-type phages as reporters and measure accumulation of an endogenous phage transcript. To determine whether cSML synthesis by SGM encoded SP6Pol was required for accurate reporting of drug susceptibility, we constructed phPleft-Cyc (Fig. 3A). Compared to phSGM2, phPleft-Cyc does not encode SP6Pol and the Pleft promoter directs host RNA polymerase to express cSML. As can be seen in Fig. 3B, H37Rv cells infected with phSGM2 synthesized dramatically higher levels of cSML compared to cells infected with phPleft-Cyc. This suggests that there was an SP6Pol-mediated positive feedback loop. Specifically, a subset of transcripts initiated at the SP6 promoter by SP6Pol continued through the triplet termination unit downstream of RC2 and transcribed the SP6Pol open reading frame (ORF), resulting in enhanced synthesis of SP6Pol compared to that expressed by host RNA polymerase, which in turn mediated increased cSML generation. The levels of cSML in RIF-treated cells infected with either phSGM2 or phPleft-Cyc were reduced to background levels. Furthermore, in phSGM2-infected cells treated with STR, the cSML levels were reduced to background, indicating STR susceptibility. However, cSML levels were unaffected in cells infected with phPleft-Cyc and treated with STR. These findings suggest that accurate reporting of susceptibility to bacteriostatic agents that target cellular protein synthesis must couple signal generation to translation.
FIG 3.
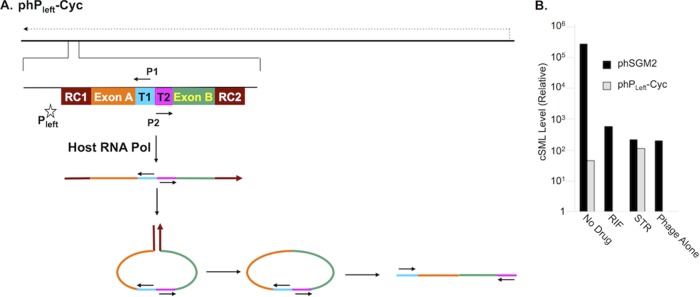
SP6Pol encoded in phSGM2 lead to increased cSML generation in infected cells and was required to report susceptibility to STR. (A) cSML expression was controlled by host RNA polymerase in cells infected with mycobacteriophage phPleft-Cyc. (B) cSML levels in H37Rv cells infected with phSGM2 accumulated to higher levels than in cells infected with phPleft-Cyc. Susceptibility to RIF was reported by both phages, but STR susceptibility was not reported by phPleft-Cyc.
phSGM2 reports STR MIC.
RIFr cells were added to wells of a Sensititre MycoTB dish containing STR, RIF, or excipient control to determine the MICs of STR and RIF. After overnight incubation, the cells were infected with phSGM2, and cSML generation was measured by RT-PCR. All STR concentrations of ≥0.5 μg/ml resulted in ∼1,000-fold reductions in cSML synthesis. However, once the STR concentration fell below 0.5 μg/ml, cSML generation was indistinguishable from that of the untreated controls. This identifies 0.5 μg/ml as the STR MIC against this M. tuberculosis strain, which agrees with Bactec 460 (data not shown). No MIC for RIF was identified because the strain was resistant to RIF.
phSGM2 detected RIFr M. tuberculosis in a drug-susceptible population.
To determine the level of drug-resistant bacteria that phSGM2 could detect, we mixed RIFr cells at various concentrations with M. tuberculosis strain H37Rv, which is susceptible to RIF. The mixtures were treated with RIF, STR, or PNB for 40 h and then infected with phSGM2, and cSML generation was detected by RT-PCR (see Fig. 5). No significant difference was observed in cSML levels in all samples containing H37Rv or RIFr M. tuberculosis exposed to STR or PNB. However, cSML generation in a population containing 10% RIFr cells was elevated >10-fold above the level of cSML generated in the STR- and PNB-treated samples. These data demonstrate that phSGM2 is able to detect the presence of 10% drug-resistant cells in an otherwise drug-susceptible population.
FIG 5.
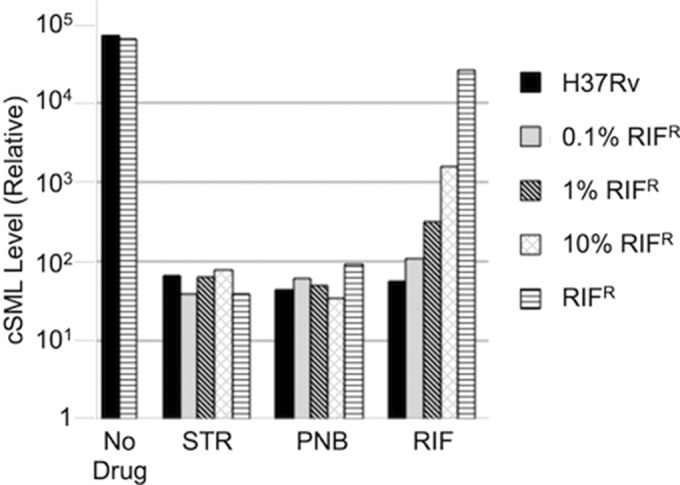
phSGM2 detected the presence of 10% RIFr cells in an otherwise drug-susceptible population. H37Rv cells, RIFr cells, or mixtures of both containing 0.1, 1, or 10% RIFr cells were either left untreated or exposed to RIF, STR, or PNB and then infected with phSGM2. A >10-fold increase in cSML generation above STR or PNB samples was detected in mixtures containing at least 10% RIFr cells and exposed to RIF.
DISCUSSION
We designed the SGM reporter as a bridge that connects the benefits of rapid molecular diagnostic technologies such as GeneXpert with those of phenotypic culture-based tests capable of identifying all forms of drug resistance and the presence of drug-resistant bacteria in an otherwise drug-susceptible population. To this end, we demonstrated that phSGM2 encoding the second-generation SGM detected <100 CFU (Fig. 1C) of M. tuberculosis, which compares favorably with the reported 131 CFU limit of detection for Xpert MTB/RIF (6). With the exception of PZA, phSGM2 accurately reported M. tuberculosis susceptibility to all antibiotics used to treat TB patients (Fig. 2A and B) in less than 1 day. Furthermore, the assay correctly reported each drug-resistant strain tested, including slow-growing clinical isolates (Fig. 2C to E), and clearly reported the STR MIC for an H37Rv-derived strain (Fig. 4). Finally, phSGM2 was able to clearly detect the presence of 10% drug-resistant cells in an otherwise drug-susceptible population (Fig. 5).
FIG 4.
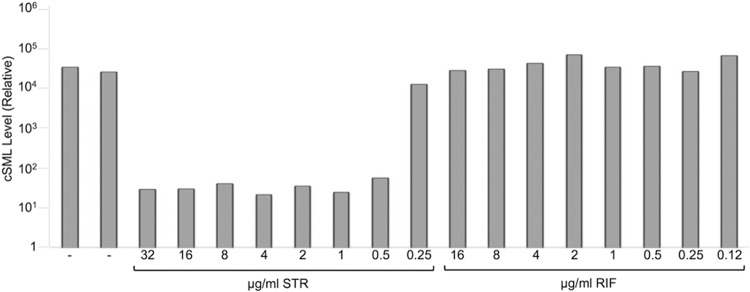
phSGM2 accurately reported the STR MIC of a RIFr H37Rv variant. RIFr cells were added to the positive control and to STR- and RIF-containing wells of a Trek Sensititre dish and then infected with phSGM2. cSML generation was reduced to background levels at STR concentrations at or above the STR MIC of the RIFr strain.
Other groups have reported detection of phenotypic AST using phage in combination with nucleic acid based detection: a phage amplification assay uses PCR to detect the increase in mycobacteriophage D29 genomes after low-multiplicity infection of cultured M. tuberculosis (11). Unlike the SGM assay, which produces a new nucleic acid signal during infection of M. tuberculosis, the phage amplification assay is unable to distinguish input phage genomes from those produced by phage replication. Therefore, multiple rounds of phage infection and replication are required to accumulate sufficient progeny phage genomes over the number of input phage.
Technologies that amplify and detect nucleic acids are the emerging standard for rapid pathogen identification directly from TB patient samples. Cepheid's Xpert MTB/RIF test system can diagnose TB infection within 2 h from initiation of the test, and with sensitivity similar to culture, which takes several days to weeks. Built into this system is simultaneous detection of RIF resistance, which is accomplished by four molecular beacons that span the RIF resistance-determining region (RRDR), an 81-bp region in the M. tuberculosis rpoB gene responsible for nearly 100% of clinically significant resistance to RIF. The presence of a single mutation in this region disrupts binding of one of the four molecular beacons to the RRDR amplicon, which indicates RIF resistance. This is a very powerful testing platform, but it is not scalable beyond the detection of RIF resistance because the number of mutations responsible for resistance to any of the other first-line and second-line drugs used to treat TB patients are far more numerous and spatially distributed throughout the M. tuberculosis genome. Furthermore, the strategy of detecting mutations by measuring the absence of molecular beacon binding requires that at least 65% of the M. tuberculosis genomes in a patient sample contain the mutation (12). Therefore, this approach does not meet the definition of drug resistance set by the gold standard agar proportion method, which is the presence of 1% drug-resistant cells in an otherwise drug-susceptible population (13, 14).
Detection of antigen 85B mRNA (short half-life) is another method that uses nucleic acid-based detection of drug phenotypic effects. In the present study, M. tuberculosis cultures treated with INH or RIF had rapid decreases in Ag85B mRNA and accurately reported drug susceptibility and resistance (15). Also, multiplexed quantitative detection of several M. tuberculosis mRNA targets using nCounter analysis (Nanostring Technologies) accurately reports phenotypic AST of cultured clinical strains (16). To our knowledge, the effectiveness of either of these approaches has not yet been validated using clinical samples, perhaps due to the complex mixture of viable, dying, and dead cells present in sputum samples that may complicate the relationship between drug effects and cellular mRNA turnover. Since the cSML is an entirely synthetic sequence not present in nature, interference of cellular RNA is not expected to be an issue.
Perhaps most importantly for TB patients and clinicians, we demonstrated that phSGM2 detected as few as 10% drug-resistant cells in an otherwise drug-susceptible population. This opens the door to developing the AST as a method to detect real-time development of drug resistance in patients undergoing TB therapy. A rapid test with this capability would indicate pending failure of the current treatment regimen much earlier than is currently possible, and allow the patient to switch to second-line treatment, while the drug-resistant M. tuberculosis biomass is substantially lower than it is in patients diagnosed with drug-resistant infection using current molecular or phenotypic tests. Such a test would identify patients likely to fail treatment, maximize the effectiveness of second-line drugs, and lower the incidence of MDR-TB. The current assay clearly detects the presence of 10% drug-resistant cells. There remains in the assay, however, background cSML resistant to RNase A and all RNases tested, which copurifies with infectious phSGM2 by CIM monolith chromatography and CsCl isopycnic gradient ultracentrifugation, and becomes a substrate for RNase A after phSGM2 is incubated at 70°C for 15 min (data not shown). We believe that a fraction of cSML produced during growth of phage stocks becomes incorporated inside phage capsids and is thus shielded from RNases. Future efforts will focus on developing an SGM that precludes the accumulation of the cSML during growth of phage stocks in M. smegmatis. Removal of background could facilitate detection of ≤0.01% drug-resistant bacteria.
In this report we demonstrated that SGM is a new genetic approach to detect microbial AST: it combines the sensitivity and accuracy of phenotypic AST (measures activity of any drug) with the power of nucleic acid tests to detect very few microorganisms in a matter of just hours. We are collaborating with TB clinical trial sites to assess the utility of this AST used directly on sputum samples and to determine its usefulness to identify drug resistance as it develops in patients.
ACKNOWLEDGMENT
This study was supported by NIAID grant 1R21AI096248-01A1 awarded to M.M.
REFERENCES
- 1.Centers for Disease Control and Prevention. 2006. Emergence of Mycobacterium tuberculosis with extensive resistance to second-line drugs worldwide, 2000–2004. MMWR Morb Mortal Wkly Rep 55:301–305. [PubMed] [Google Scholar]
- 2.WHO/IUATLD. 2000. Antituberculosis drug resistance in the world: report no. 2, prevalence and trends. WHO/CDS/TB/2000.278 World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.Zignol M, Hosseini MS, Wright A, Weezenbeek CL, Nunn P, Watt CJ, Williams BG, Dye C. 2006. Global incidence of multidrug-resistant tuberculosis. J Infect Dis 194:479–485. doi: 10.1086/505877. [DOI] [PubMed] [Google Scholar]
- 4.Dorman SE, Chaisson RE. 2007. From magic bullets back to the magic mountain: the rise of extensively drug-resistant tuberculosis. Nat Med 13:295–298. doi: 10.1038/nm0307-295. [DOI] [PubMed] [Google Scholar]
- 5.Barnard M, Albert H, Coetzee G, O'Brien R, Bosman ME. 2008. Rapid molecular screening for multidrug-resistant tuberculosis in a high-volume public health laboratory in South Africa. Am J Respir Crit Care Med 177:787–792. doi: 10.1164/rccm.200709-1436OC. [DOI] [PubMed] [Google Scholar]
- 6.Helb D, Jones M, Story E, Boehme C, Wallace E, Ho K, Kop J, Owens MR, Rodgers R, Banada P, Safi H, Blakemore R, Lan NT, Jones-Lopez EC, Levi M, Burday M, Ayakaka I, Mugerwa RD, McMillan B, Winn-Deen E, Christel L, Dailey P, Perkins MD, Persing DH, Alland D. 2010. Rapid detection of Mycobacterium tuberculosis and rifampin resistance by use of on-demand, near-patient technology. J Clin Microbiol 48:229–237. doi: 10.1128/JCM.01463-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mulvey MC, Sacksteder KA, Einck L, Nacy CA. 2012. Generation of a novel nucleic acid-based reporter system to detect phenotypic susceptibility to antibiotics in Mycobacterium tuberculosis. mBio 3:e00312-11. doi: 10.1128/mBio.00312-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bardarov S Jr, Dou H, Eisenach K, Banaiee N, Ya S, Chan J, Jacobs WR Jr, Riska PF. 2003. Detection and drug-susceptibility testing of M. tuberculosis from sputum samples using luciferase reporter phage: comparison with the Mycobacteria Growth Indicator Tube (MGIT) system. Diagn Microbiol Infect Dis 45:53–61. doi: 10.1016/S0732-8893(02)00478-9. [DOI] [PubMed] [Google Scholar]
- 9.Hatfull G, Jacobs WR Jr. 2000. Molecular genetics of mycobacteria. American Society for Microbiology, Washington, DC. [Google Scholar]
- 10.Banaiee N, Bobadilla-Del-Valle M, Bardarov S Jr, Riska PF, Small PM, Ponce-De-Leon A, Jacobs WR Jr, Hatfull GF, Sifuentes-Osornio J. 2001. Luciferase reporter mycobacteriophages for detection, identification, and antibiotic susceptibility testing of Mycobacterium tuberculosis in Mexico. J Clin Microbiol 39:3883–3888. doi: 10.1128/JCM.39.11.3883-3888.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pholwat S, Ehdaie B, Foongladda S, Kelly K, Houpt E. 2012. Real-time PCR using mycobacteriophage DNA for rapid phenotypic drug susceptibility results for Mycobacterium tuberculosis. J Clin Microbiol 50:754–761. doi: 10.1128/JCM.01315-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blakemore R, Story E, Helb D, Kop J, Banada P, Owens MR, Chakravorty S, Jones M, Alland D. 2010. Evaluation of the analytical performance of the Xpert MTB/RIF assay. J Clin Microbiol 48:2495–2501. doi: 10.1128/JCM.00128-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canetti G, Froman S, Grosset J, Hauduroy P, Langerova M, Mahler HT, Meissner G, Mitchison DA, Sula L. 1963. Mycobacteria: laboratory methods for testing drug sensitivity and resistance. Bull World Health Organ 29:565–578. [PMC free article] [PubMed] [Google Scholar]
- 14.Rastogi N, Goh KS, David HL. 1989. Drug susceptibility testing in tuberculosis: a comparison of the proportion methods using Lowenstein-Jensen, Middlebrook 7H10 and 7H11 agar media and a radiometric method. Res Microbiol 140:405–417. doi: 10.1016/0923-2508(89)90016-8. [DOI] [PubMed] [Google Scholar]
- 15.Hellyer TJ, DesJardin LE, Hehman GL, Cave MD, Eisenach KD. 1999. Quantitative analysis of mRNA as a marker for viability of Mycobacterium tuberculosis. J Clin Microbiol 37:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barczak AK, Gomez JE, Kaufmann BB, Hinson ER, Cosimi L, Borowsky ML, Onderdonk AB, Stanley SA, Kaur D, Bryant KF, Knipe DM, Sloutsky A, Hung DT. 2012. RNA signatures allow rapid identification of pathogens and antibiotic susceptibilities. Proc Natl Acad Sci U S A 109:6217–6222. doi: 10.1073/pnas.1119540109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi W, Zhang X, Jiang X, Yuan H, Lee JS, Barry CE III, Wang H, Zhang W, Zhang Y. 2011. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 333:1630–1632. doi: 10.1126/science.1208813. [DOI] [PMC free article] [PubMed] [Google Scholar]