

Abstract
Tedizolid, a novel oxazolidinone antibacterial, was administered to Long Evans rats by oral gavage once daily for up to 9 months at doses near the maximum tolerated dose (MTD) to evaluate for potential neurotoxicity. Mean plasma exposures of tedizolid at the low-, medium-, and high-dose levels (7.5, 15, and 30 mg/kg of body weight/day for males; 2.5, 5, and 10 mg/kg/day for females) were similar between males and females and were 1.8-, 3.9-, and 8.0-fold greater than exposures in patients at the therapeutic dose (200 mg once daily). Evaluated endpoints included survival, clinical observations, body weight, and food consumption. At 1, 3, 6, and 9 months, ophthalmic examinations, functional observational batteries, and locomotor activity measures were conducted, brain weights/sizes were recorded, and perfusion-fixed tissues were collected from 12 rats/sex/group/time point. A detailed morphological assessment was conducted on brain, eyes, optic nerve/tract, spinal cord, peripheral nerves (includes sciatic, sural, tibial, peroneal, trigeminal), and skeletal muscle. At the end of 9 months, less body weight gain was seen in high-dose males (−6.7%) and females (−5.8%) compared with that seen in controls. There were no tedizolid-related adverse neurobehavioral effects or tedizolid-related histopathologic changes in the central/peripheral nervous systems, including the optic nerve. Results of this study indicate that tedizolid was not neurotoxic when administered long term to pigmented rats at doses near the MTD, which were up to 8-fold higher than the human therapeutic exposure.
INTRODUCTION
Tedizolid is a novel antibacterial agent belonging to a class of drugs called oxazolidinones with potent activity against a wide range of Gram-positive pathogens, including resistant strains such as methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci (1–4). The antibiotic activity of oxazolidinones is mediated through the inhibition of bacterial protein synthesis via binding to the 50S ribosomal subunit (1, 5–7). Linezolid, an oxazolidinone antibacterial agent approved by the U.S. Food and Drug Administration (FDA) in 2000, has been shown to induce peripheral and optic neuropathy in humans, adverse effects that can limit the clinical use of linezolid during long-term therapy (8–10). Similarly, peripheral neuropathy developed after 3 months in rats given linezolid, and optic and peripheral neuropathies developed after 6 months at plasma levels similar to those of humans at the therapeutic dose (10).
Tedizolid phosphate, recently approved by the FDA for management of adult acute bacterial skin and skin structure infection (11), is the prodrug of tedizolid, which is rapidly and extensively converted to the active moiety tedizolid by endogenous phosphatases. Tedizolid was tested in a full battery of nonclinical safety studies to identify potential toxicities. No neuropathological effects were observed during routine animal toxicologic testing in studies up to 3 months in duration (12). To provide a more in-depth assessment of the potential for tedizolid to cause neurotoxicity, a special neuropathology study that used sensitive morphological procedures was conducted with tedizolid in rats, a species used for general toxicologic testing and previously shown to be predictive of oxazolidinone-induced neuropathy (10). In addition, the rat model is established, widely used in the investigation of optic nerve diseases (13), and the choice for a number of approaches to assess neurotoxicity (14–16).
Tedizolid was administered in this neuropathology study at doses near the maximum tolerated dose (MTD), well above the human therapeutic plasma dose. The duration of this study (9 months) exceeded that of the linezolid 6-month rat study (10) and used high doses relative to the human clinical dose to increase the potential for inducing neurotoxicity. Furthermore, detailed histologic techniques in perfusion-fixed peripheral and central nervous system tissue collected at multiple times throughout a 9-month study period, coupled with a battery of neurobehavioral and clinical tests, were used to enhance the detection of neurotoxicity in this rat model.
(Data from rat long-term neurotoxicity studies of tedizolid were presented in part at the 53rd International Conference on Antimicrobial Agents and Chemotherapy [17] and also in part at the 53rd Annual Meeting of the Society of Toxicology, Phoenix, AZ, 23 to 27 March 2014. A top-line discussion on the potential long-term neurotoxicity of tedizolid in rats was included in part in reference 18.)
MATERIALS AND METHODS
Test materials.
The test article tedizolid phosphate (99.6% pure) was stable throughout the dosing period. Before dose administration, tedizolid phosphate was dissolved in vehicle (25 mM disodium hydrogen phosphate, pH 7.5) at concentrations ranging from 0.25 to 3 mg/ml. Dose solution concentrations were verified using a validated high-performance liquid chromatography method with UV absorbance detection.
Study design.
Tedizolid phosphate dissolved in vehicle was administered via oral gavage to Long Evans pigmented rats once daily at control (vehicle), low-dose, medium-dose, and high-dose levels (i.e., 0, 7.5, 15, and 30 mg/kg of body weight/day [males] and 0, 2.5, 5, and 10 mg/kg/day [females], respectively). Male rats received higher doses than females because of a rat-specific sex difference in metabolic clearance of tedizolid. Doses were chosen with the intent to yield tedizolid plasma levels ranging from about one to eight times the systemic exposures achieved in patients at the therapeutic dose (200 mg once daily). The high doses of 30 mg/kg/day (males) and 10 mg/kg/day (females) were the MTD levels previously determined in a 3-month rat toxicity study, in which higher doses were not tolerated and exceeded the MTD (12). In addition, because the main purpose of the study was to assess the potential of tedizolid to induce neuropathies using sensitive morphological techniques, it was not considered necessary to include linezolid as a comparator, for which peripheral and optic neuropathies already have been well documented in rats at human therapeutic plasma exposures (10). Rats (12/sex/group) were evaluated for neurobehavioral changes before dosing and after 1, 3, 6, and 9 months of treatment. An additional set of rats (12/sex/group) dosed for 6 months also underwent a 3-month recovery (drug-free) period. Groups of up to 12 rats/sex were euthanized at 1, 3, 6, and 9 months for neuropathological assessment (low-dose rats were killed only at 3 and 6 months). Satellite animals (9 rats/sex in tedizolid groups; 5 rats/sex in vehicle control) were used for toxicokinetic blood draws with 3 rats/sex/time point bled on the first day of dosing and at the end of 1, 3, 6, and 9 months. The age of the rats at the start of dosing was approximately 9 weeks. Long Evans pigmented rats were used because this strain allows evaluation of potential neurotoxicity to drug-melanin binding structures (19), which is particularly important for assessing ocular structures (e.g., uveal tract). An overview of the experimental design is shown in Table 1.
TABLE 1.
Overview of experimental design
| Group | Tedizolid phosphate dose (mg/kg/day) |
No. of animals |
||||
|---|---|---|---|---|---|---|
| Neurotoxicity assessmenta |
Toxicokinetic evaluationb |
|||||
| Males | Females | Males | Females | Males | Females | |
| Control | 0 | 0 | 60 | 60 | 5 | 5 |
| Low dose | 7.5 | 2.5 | 36 | 36 | 9 | 9 |
| Medium dose | 15 | 5 | 60 | 60 | 9 | 9 |
| High dose | 30 | 10 | 60 | 60 | 9 | 9 |
Up to 12 rats/sex/group were killed at 1 month (control and medium- and high-dose groups), 3 months (all groups), 6 months (all groups), and 9 months (control and medium- and high-dose groups) of dose administration. In addition, up to 12 rats/sex/group (all groups) were killed after 6 months of dose administration and a 3-month recovery (no-dosing) period.
All surviving toxicokinetic animals were killed after 9 months of dose administration (3 rats/sex/time point were used for analysis).
Animals and animal husbandry.
Rats (Crl:LE; Charles River Laboratories, Inc., Raleigh, NC) were acclimated to the housing and animal room environment for 21 days and uniquely identified with the use of a subcutaneous microchip (BMDS System; BioMedic Data Systems, Inc., Seaford, DE) in the dorsoscapular area. Rats were weighed, assessed for health, and randomly assigned to groups to ensure that mean body weight in each treatment group was approximately the same. Animals were housed individually in clean, stainless steel, wire mesh cages suspended above cage board and were provided reverse osmosis-treated (on site) drinking water ad libitum, delivered via an automatic watering system, and a basal diet (LabDiet 5002; PMI Nutrition International, LLC, St. Louis, MO), except during periods of fasting, when food, but not water, was withheld. The room environment was controlled for temperature (19.7°C to 22.1°C), relative humidity (38.6% to 61.7%), and light cycle (0600 to 1800 h).
Animals were maintained in accordance with the Guide for the Care and Use of Laboratory Animals (20). The animal facilities at WIL Research are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Study parameters. (i) Mortality checks, clinical observation, body weights, food consumption.
Main study animals were checked twice daily for death, moribundity, and clinical signs of toxicity. Body weight and food consumption were recorded at least once weekly.
(ii) Ophthalmic examinations.
Ocular examinations were conducted on all animals before dosing and toward the end of 1, 3, 6, and 9 months. All ocular examinations were conducted by a board-certified veterinary ophthalmologist using an indirect ophthalmoscope and slit lamp biomicroscope preceded by pupillary dilation with a mydriatic agent.
(iii) Neurobehavioral evaluations.
The potential for neurotoxicity was evaluated using functional observational battery and locomotor activity measures before dosing and toward the end of 1, 3, 6, and 9 months (12 rats/sex/group). The functional observational battery measurements were based on previously developed protocols (21–26) and are shown in Table 2. Locomotor activity was assessed over 60 min within a black, enclosed, sound-attenuated room equipped with a white-noise generator and an automated computer device that measured photobeam breaks (horizontal and vertical).
TABLE 2.
Functional observational battery used to screen tedizolid for potential neurotoxicity in rats
| Observation type | Indicators |
|---|---|
| Home cage | Posture, convulsions/tremors, feces consistency, biting, palpebral (eyelid) closure |
| Physiological | Catalepsy, body temperature, body weight |
| Neuromuscular | Hind limb extensor strength, hind limb foot splay, grip strength (hind limb and forelimb), RotaRod performance |
| Handling | Ease of removal from cage, lacrimation/chromodacryorrhea, piloerection, palpebral closure, eye prominence, red/crusty deposits, ease of handling animal in hand, salivation, fur appearance, respiratory rate/character, mucous membrane/eye/skin color, muscle tone |
| Open field | Mobility, rearing, convulsions/tremors, grooming, bizarre/stereotypic behavior, time to first step (in seconds), gait, arousal, urination/defecation, gait score, backing |
| Sensory | Approach response, startle response, pupil response, forelimb extension, air righting reflex, touch response, tail pinch response, eye blink response, hind limb response, olfactory response |
(iv) Neuropathological examinations.
At each scheduled necropsy (1, 3, 6, and 9 months and the recovery necropsy), all animals were weighed, anesthetized deeply with an intraperitoneal injection of sodium pentobarbital, and perfused in situ using a McDowell-Trump fixative (27). Fixed brain weights and brain dimensions (lengths [excluding olfactory bulbs] and widths) were recorded. Any observable gross changes, abnormal coloration, or lesions of the brain and spinal cord were recorded. Peripheral and central nervous system tissues (Table 3; Fig. 1) were collected from rats at the scheduled terminations and placed in 10% neutral buffered formalin fixative or in McDowell-Trump fixative, as appropriate. Of the rats perfused in situ, 10 randomly selected rats/sex in the control and high-dose groups from the 1-, 3-, 6-, and 9-month necropsies, including recovery necropsy, were selected for microscopic neuropathological evaluation. In general, the central nervous system tissue, eyes, and skeletal muscle were embedded in paraffin, and the peripheral nervous system tissue was embedded in plastic and thin sectioned. Alternating sections of the retrobulbar and intracranial optic nerves were embedded in paraffin. Ophthalmic examination findings were available at the time of enucleation, trimming of tissue, and the microscopic examination of ocular tissue. Tissue samples were prepared for qualitative histopathologic evaluation by being sectioned and stained with hematoxylin and eosin and, for duplicate sections of peripheral nervous system tissue, stained with toluidine blue. At completion of the histopathologic assessment, which included control and high-dose groups as per protocol, slides and data tables were independently evaluated (peer reviewed) by a board-certified anatomic pathologist with expertise in evaluating the nervous system (M. T. Butt).
TABLE 3.
Central and peripheral nervous system tissues evaluated at 1, 3, 6, and 9 months (including 3-month recovery after 6 months) of daily oral administration of tedizolid phosphate
| Nervous system tissue evaluated | Part(s) of tissue |
|---|---|
| Brain | Olfactory bulbs, cerebral cortex, hippocampus, basal ganglia, thalamus, hypothalamus, midbrain, cerebellum, pons and medulla oblongata, optic tract (including optic chiasm) |
| Optic nerves | Retrobulbar, intracranial |
| Eyes (with optic nerve) | |
| Spinal cord and nerves | At cervical swellings C3–C7, at lumbar swellings T13–L4 |
| Dorsal and ventral spinal roots | Ganglia and fibers at C3–C7 and T13–L4 |
| Spinal nerves | Cervical and lumbar regions |
| Trigeminal ganglia/nerves | Two sections (1 longitudinal) |
| Sciatic nerves | Mid-thigh region, sciatic notch |
| Sural nerves | |
| Tibial nerves | |
| Peroneal nerves | |
| Skeletal muscle, gastrocnemius |
FIG 1.

Illustration of central and peripheral nervous system tissues evaluated macroscopically and microscopically after perfusion fixation (excludes specific brain regions; see Table 3).
(v) Toxicokinetic evaluation.
Satellite animals were dosed similarly to main study animals, and blood samples were collected from 3 rats/sex at 1, 2, 4, 8, and 24 h after dose administration on the first day of dosing and at the end of 1, 3, 6, and 9 months. Control group blood samples were collected only 2 h after dosing at the approximate peak plasma exposure time point (Tmax). Blood was collected into chilled tubes containing potassium EDTA via the retroorbital sinus of rats anesthetized by isoflurane inhalation. Blood samples were maintained on wet ice during collection and processing. Plasma was isolated in a refrigerated centrifuge and stored at approximately −85°C to −65°C until analysis of tedizolid using a validated liquid chromatographic-tandem mass spectrometric (LC-MS/MS) procedure with a low limit of quantitation of 2.5 ng/ml. Toxicokinetic parameters and descriptive statistics (e.g., mean, standard deviation [SD]) were calculated using the WinNonLin version 5.2 software program (Pharsight Corporation, Mountain View, CA). These parameters were derived from the mean plasma concentration values for each sex, dosage level, and evaluation period. For this study, the area under the plasma concentration-time curve (AUC), dose proportionality, and plasma accumulation were determined.
Statistical methods.
Mean body weight, body weight gain, food consumption, measured functional observational battery values, and absolute brain weight, length, and width were analyzed by parametric one-way analysis of variance (ANOVA) (28) to determine intergroup differences. If the results of ANOVA were significant (P < 0.05), the Dunnett test (29) was applied to the data to compare the tedizolid-treated groups with the control group. The functional observational battery measurements that yielded scalar or descriptive data were analyzed by Fisher exact test (30). Locomotor activity was analyzed using repeated-measures ANOVA (RANOVA) as conducted by BioSTAT Consultants, Inc. (Portage, MI), using SAS software version 9.1 or higher (SAS Institute, Inc., Cary, NC). Necropsy and nongraded histopathologic findings of each tedizolid group were compared with those of the control group using the Fisher exact test (30).
RESULTS
Toxicokinetic analyses showed that tedizolid plasma exposure increased proportionally to dose, with 2- to 3-fold accumulation in plasma over the 9-month study period. At the end of the 9-month treatment period, the tedizolid steady-state mean plasma exposures for males and females were similar at the low, medium, and high doses throughout the study period. The overall mean plasma exposure for males and females at the high dose (206 μg · h/ml) was 8.0-fold higher than that observed in humans at the therapeutic dose (25.6 μg · h/ml) (11). Mean plasma exposures, AUC0–24 (μg · h/ml), throughout the 9-month study are shown in Table 4.
TABLE 4.
Plasma exposure of tedizolid after oral administration of tedizolid phosphate to rats over 9 months
| Dosing duration | AUC0–24a (μg · h/ml) by sex and dose (mg/kg/day) |
|||||
|---|---|---|---|---|---|---|
| Males |
Females |
|||||
| 7.5 | 15 | 30 | 2.5 | 5 | 10 | |
| 1 day | 16.9 | 32.4 | 69.9 | 24.9 | 55.7 | 95.6 |
| 1 mo | 20.9 | 45.8 | 85.2 | 26.1 | 52.0 | 97.8 |
| 3 mo | 27.2 | 81.8 | 145 | 34.3 | 82.6 | 147 |
| 6 mo | 34.1 | 82.7 | 204 | 45.8 | 97.1 | 174 |
| 9 mo | 41.9 | 101 | 222 | 51.7 | 97.2 | 189 |
AUC0–24, area under the concentration-time curve from time zero to 24 h.
No tedizolid-related death and no decrease in food intake were observed during the 9-month study period. Reduced body weight gain relative to that of controls was noted in the high-dose males (up to 6.7% lower) and females (up to 5.8% lower) throughout the study period. Body weight changes were considered related to tedizolid treatment and resulted in decreased mean body weight at the 1 (males only)-, 3-, 6-, and 9-month necropsy intervals. Body weight returned to control levels at the end of the 3-month recovery period, which followed 6 months of dosing. Test article-related observations of salivation (i.e., clear material around mouth) occurred in high-dose males and females up to 2 h after dosing throughout the study period, an indication that high-dose animals were under stress.
No tedizolid-related effects were observed at 1, 3, 6, and 9 months of dosing or after the 3-month recovery period. Specifically, no tedizolid-related effects were noted on functional observational battery measurements, locomotor activity, ophthalmic examination, brain weight and measurement, or macroscopic and microscopic neuropathological findings in any of the central or peripheral nervous system tissues examined in the high-dose group, including retinal and uveal tract tissue. No degeneration in optic nerves was observed after a full 9 months of dosing. Histopathological assessments were not conducted on either the low- or medium-dose groups based on the absence of findings at the high dose, per standard study protocol. Although brain length was slightly lower for female rats in the 5- and 10-mg/kg/day dose groups at 9 months (Table 5), this seemed to be a result of greater brain lengths in the control group and not directly because of tedizolid. When the recovery female control group killed at 9 months was used for comparison with the 9-month test article-treated females, no difference in brain length was observed. The severity (minimal to mild when present) of both spinal cord and peripheral nerve axonal degeneration was similar between control and high-dose animals (Fig. 2 and 3). In addition, these degenerative changes appear to be related to age, because the incidence of degeneration in the control and high-dose groups increased similarly over the 9-month study period (Fig. 2 and 3).
TABLE 5.
Mean brain weights and dimensions of rats in control and medium- and high-dose tedizolid groups after 9 months of dose administrationa
| Group | Control or tedizolid phosphate dose (mg/kg/day) (n = no. of rats) | Brain wt (g) | Brain length (mm) | Brain width (mm) |
|---|---|---|---|---|
| Males | Control (n = 12) | 2.27 ± 0.10 | 22.04 ± 0.09 | 16.12 ± 0.09 |
| 15 (n = 11) | 2.31 ± 0.03 | 22.07 ± 0.07 | 16.06 ± 0.10 | |
| 30 (n = 10) | 2.32 ± 0.03 | 21.80 ± 0.19 | 16.36 ± 0.09 | |
| Females | Control (n = 11) | 2.11 ± 0.03 | 21.21 ± 0.10 | 15.35 ± 0.08 |
| 5 (n = 12) | 2.06 ± 0.03 | 20.70 ± 0.10b | 15.24 ± 0.08 | |
| 10 (n = 12) | 2.06 ± 0.03 | 20.69 ± 0.12b | 15.24 ± 0.09 |
Table shows means ± standard errors of the means. Female control group brain length was uncharacteristically high; the recovery female control group sacrificed at 9 months had a mean length of 20.79 ± 0.14 mm (n = 12 rats), which was similar to those of medium- and high-dose groups.
P < 0.05, compared to the control.
FIG 2.
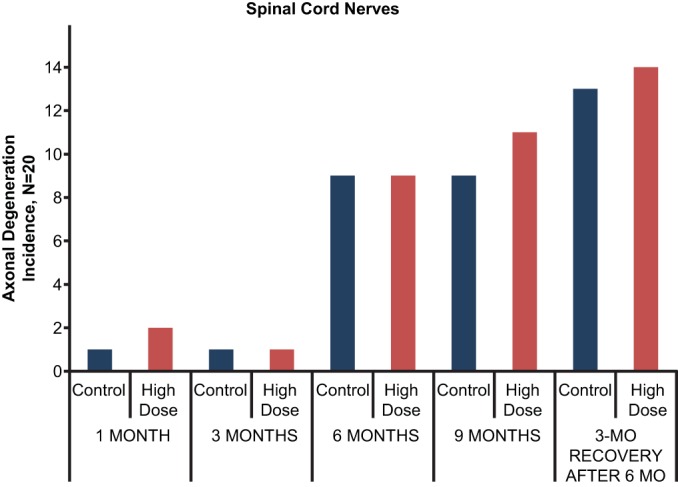
Summary of incidence of spinal cord axonal degeneration seen in control and high-dose groups (20 rats/group; 10 rats/sex/group) at 1, 3, 6, and 9 months (includes a 3-month recovery sacrifice after 6 months of dosing). When observed, the severity of degeneration was minimal to mild (score of 1 or 2) on a 4-point scale.
FIG 3.
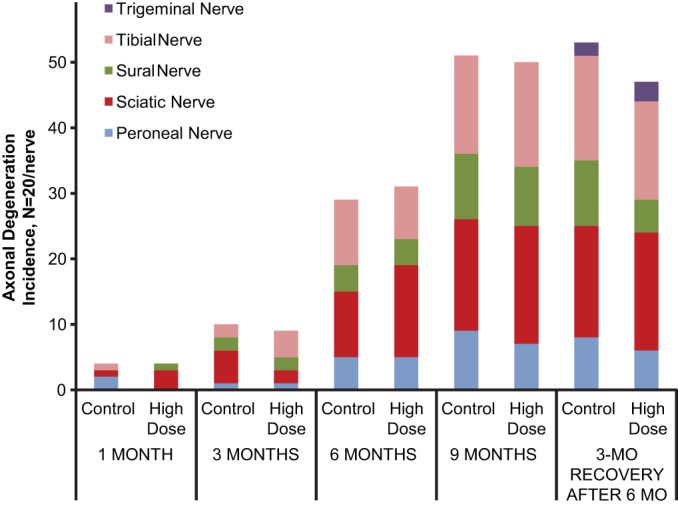
Summary of incidence of peripheral axonal degeneration seen in control and high-dose groups (20 rats/group; 10 rats/sex/group) at 1, 3, 6, and 9 months (includes a 3-month recovery sacrifice after 6 months of dosing). When observed, the severity of degeneration was minimal to mild (score of 1 or 2) on a 4-point scale.
DISCUSSION
In this study, a rigorous neuropathological evaluation was performed in rats given tedizolid for 1, 3, 6, and 9 months. The rat model is established and is widely used in the investigation of optic nerve diseases because of its ease of use and accessibility to key structures, particularly the optic nerve/tract (13). Rat models have been used in the investigation of optic neuritis and evaluation of traumatic optic neuropathy, ischemic optic neuropathy, and secondary neuronal degeneration that occurs beyond the initial lesion site after traumatic or ischemic injury (13). The rat is also the species of choice for most of the various approaches to assess neurotoxicity in experimental animals (16), and standard protocols have been developed for evaluation of neurotoxicity in adult (14) and developing (15) rats.
For linezolid, peripheral neuropathy was observed in rats after 3 months of treatment and both optic and peripheral neuropathies were observed after 6 months (10). Linezolid was administered to rats at 80 mg/kg/day for 3 and 6 months, a dose level with systemic exposures (AUCs) comparable to those in humans at the therapeutic dose. In that study, linezolid caused minimal to mild sciatic nerve degeneration, with changes observed as early as 3 months, and minimal to moderate optic nerve degeneration after 6 months (10). In the current study, tedizolid showed no evidence of neuropathological effects with long-term administration at doses near the MTD and at plasma exposures that were 8.0-fold greater than the human therapeutic plasma dose. No degeneration was seen in the optic nerve in either the control or the tedizolid group. Control and tedizolid groups both showed normal age-related increases in the incidence of spinal and peripheral axonal nerve degeneration, and, when observed, severity of the degeneration was always minimal to mild. Eisenbrandt et al. previously reported minimal axonal degeneration in spinal cord and peripheral nerves as a common background lesion in rats (31).
Although the linezolid findings cannot be compared directly with those of tedizolid, the differences in neuropathology profiles obtained for these two drugs using the same rat model suggest that tedizolid has a lower potential for inducing neuropathy than does linezolid. It has been hypothesized that the neuropathological effects of linezolid in rats and humans are caused by oxazolidinone-induced mitochondrial toxicity (8, 9). Many drugs, including various antibacterial agents—especially those that inhibit protein synthesis—are known to adversely influence mitochondrial function (32). Because of structural similarities between mitochondrial and prokaryotic ribosomes, oxazolidinones can also impair mammalian mitochondrial protein synthesis (MPS) (33–35), which is thought to be the underlying mechanism for peripheral and ocular neuropathies seen with prolonged use of the oxazolidinone linezolid (8, 9, 33, 36–42). Although tedizolid is a more potent inhibitor of MPS than linezolid on a molar basis, it has a lower potential to cause mitochondrion-related adverse events in vivo (such as myelosuppression, neuropathy, and lactic acidosis) than linezolid when each drug is assessed in the context of its respective therapeutic dosage (18).
The absence of neuropathy in rats administered tedizolid is consistent with results of previously conducted animal toxicology studies that demonstrate greater safety margins for tedizolid versus linezolid (Table 6). Studies conducted in rats and dogs for 1 month or 3 months showed that key target organs for adverse effects for both drugs were the gastrointestinal tract and the hematopoietic system (10, 12, 43–45). However, tedizolid had plasma exposure multiples 4 to 8 times the human-equivalent plasma exposure without adverse effects, whereas linezolid toxicities were evident at drug plasma levels comparable with human-equivalent doses, with no adverse effect levels below the human therapeutic plasma exposure (Table 6) (10, 12, 43).
TABLE 6.
Comparison of tedizolid and linezolid toxicology safety margins (animal-to-human plasma exposure ratios)
| Species/duration of oral toxicology study | Safety margina (fold therapeutic oral AUC0–24h) |
|
|---|---|---|
| Tedizolidb | Linezolidc | |
| Rat/1 mo | 5.3 | 0.18 |
| Rat/3 mo | 4.6 | 0.08 |
| Rat/6–9 mo (neuropathology) | 8.0 | Not reportedd |
| Dog/1 mo | 3.7 | 0.34 |
| Dog/3 mo | 5.3 | 0.37 |
Safety margins are based on comparison of mean plasma exposures (AUCs) at the animal no observed adverse-effect level (NOAEL) versus mean plasma exposures at the human therapeutic dose.
Human oral therapeutic exposure = 25.6 μg · h/ml for tedizolid (11); safety margins for 1- and 3-month tedizolid toxicology studies from tedizolid summaries (12).
Safety margins for linezolid from 2000 FDA Advisory Anti-Infective Drug Committee Meeting (43), except for neuropathology taken from the Zyvox label (10).
Neuropathology observed with linezolid at human therapeutic exposures after 3 and 6 months (no assessment at 9 months); dose causing an NOAEL was not reported (10).
In conclusion, Long Evans pigmented rats given tedizolid at plasma exposures near the MTD and up to 8-fold higher than the human therapeutic plasma exposure showed no behavioral abnormalities and no evidence of drug-related morphological changes in the brain, spinal cord, or peripheral or optic nerves, even after 9 months of treatment. The study results indicate that the potential for neuropathology is less with tedizolid than with linezolid when each drug is administered for extended periods at or above its therapeutic dose. These data add to the growing body of evidence that suggests that tedizolid may have a more favorable tolerability profile than linezolid when given over prolonged periods; however, further clinical studies are warranted.
ACKNOWLEDGMENTS
The long-term animal neurotoxicity study was funded by Trius Therapeutics (now part of Cubist Pharmaceuticals). The pharmacokinetic analyses were funded by Cubist Pharmaceuticals.
All authors had full access to the data. All authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development, and had final responsibility for the decision to submit for publication.
F. Oleson is an employee of Cubist Pharmaceuticals. M. J. Schlosser and M. T. Butt are consultants to Cubist Pharmaceuticals. H. Hosako, A. Radovsky, J. Vija, and D. Draganov conducted this research in the course of their employment, and their employer (WIL Research) received compensation from Cubist Pharmaceuticals.
We thank Ken Bartizal for his significant intellectual contributions to study conception and design.
REFERENCES
- 1.Shaw KJ, Poppe S, Schaadt R, Brown-Driver V, Finn J, Pillar CM, Shinabarger D, Zurenko G. 2008. In vitro activity of TR-700, the antibacterial moiety of the prodrug TR-701, against linezolid-resistant strains. Antimicrob Agents Chemother 52:4442–4447. doi: 10.1128/AAC.00859-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yum JH, Choi SH, Yong D, Chong Y, Im WB, Rhee DK, Lee K. 2010. Comparative in vitro activities of torezolid (DA-7157) against clinical isolates of aerobic and anaerobic bacteria in South Korea. Antimicrob Agents Chemother 54:5381–5386. doi: 10.1128/AAC.00728-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown SD, Traczewski MM. 2010. Comparative in vitro antimicrobial activities of torezolid (TR-700), the active moiety of a new oxazolidinone, torezolid phosphate (TR-701), determination of tentative disk diffusion interpretive criteria, and quality control ranges. Antimicrob Agents Chemother 54:2063–2069. doi: 10.1128/AAC.01569-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prokocimer P, Bien P, Surber J, Mehra P, De Anda C, Bulitta JB, Corey GR. 2011. Phase 2, randomized, double-blind, dose-ranging study evaluating the safety, tolerability, population pharmacokinetics, and efficacy of oral torezolid phosphate in patients with complicated skin and skin structure infections. Antimicrob Agents Chemother 55:583–592. doi: 10.1128/AAC.00076-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shinabarger D. 1999. Mechanism of action of the oxazolidinone antibacterial agents. Expert Opin Invest Drugs 8:1195–1202. doi: 10.1517/13543784.8.8.1195. [DOI] [PubMed] [Google Scholar]
- 6.Lin AH, Murray RW, Vidmar TJ, Marotti KR. 1997. The oxazolidinone eperezolid binds to the 50S ribosomal subunit and competes with binding of chloramphenicol and lincomycin. Antimicrob Agents Chemother 41:2127–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colca JR, McDonald WG, Waldon DJ, Thomasco LM, Gadwood RC, Lund ET, Cavey GS, Mathews WR, Adams LD, Cecil ET, Pearson JD, Bock JH, Mott JE, Shinabarger DL, Xiong L, Mankin AS. 2003. Cross-linking in the living cell locates the site of action of oxazolidinone antibiotics. J Biol Chem 278:21972–21979. doi: 10.1074/jbc.M302109200. [DOI] [PubMed] [Google Scholar]
- 8.Javaheri M, Khurana RN, O'Hearn TM, Lai MM, Sadun AA. 2007. Linezolid-induced optic neuropathy: a mitochondrial disorder? Br J Ophthalmol 91:111–115. doi: 10.1136/bjo.2006.102541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narita M, Tsuji BT, Yu VL. 2007. Linezolid-associated peripheral and optic neuropathy, lactic acidosis, and serotonin syndrome. Pharmacotherapy 27:1189–1197. doi: 10.1592/phco.27.8.1189. [DOI] [PubMed] [Google Scholar]
- 10.Pfizer Inc. 2014. Zyvox prescribing information. Pfizer, New York, NY. [Google Scholar]
- 11.Cubist Pharmaceuticals. 2014. Prescribing information SIVEXTRO. Cubist Pharmaceuticals, Lexington, MA. [Google Scholar]
- 12.FDA Advisory Committee. 2014. FDA briefing document anti-infective drug advisory committee meeting, March 31, 2014 morning session, NDAs 205-435 and 205-436: Sivextro (tedizolid phosphate) tablets and injection FDA, Washington, DC http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/ucm390788.htm. [Google Scholar]
- 13.Levkovitch-Verbin H. 2004. Animal models of optic nerve diseases. Eye 18:1066–1074. doi: 10.1038/sj.eye.6701576. [DOI] [PubMed] [Google Scholar]
- 14.OECD. 1997. Test no. 424: neurotoxicity study in rodents. OECD guidelines for the testing of chemicals, section 4. OECD Publishing, Paris, France. doi: 10.1787/9789264071025-en. [DOI] [Google Scholar]
- 15.OECD. 2007. Test no. 426: developmental neurotoxicity study. OECD guidelines for the testing of chemicals, section 4. OECD Publishing, Paris, France. doi: 10.1787/9789264067394-en. [DOI] [Google Scholar]
- 16.OECD. 2004. Series on testing and assessment no. 20. Guidance document for neurotoxicity testing, p 32–46. OECD Publishing, Paris, France. [Google Scholar]
- 17.Hosako H, Radovsky A, Draganov DI, Vija J, Bartizal K. 2013. Abstr 53rd Intersci Conf Antimicrob Agents Chemother, abstr A-017b American Society for Microbiology, Washington, DC. [Google Scholar]
- 18.Flanagan S, McKee EE, Das D, Tulkens PM, Hosako H, Fiedler-Kelly J, Passarell J, Radovsky A, Prokocimer P. 2015. Nonclinical and pharmacokinetic assessments to evaluate the potential of tedizolid and linezolid to affect mitochondrial function. Antimicrob Agents Chemother doi: 10.1128/AAC.03684-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ings RMJ. 1984. The melanin binding of drugs and its implications. Drug Metab Rev 15:1183–1212. doi: 10.3109/03602538409033561. [DOI] [PubMed] [Google Scholar]
- 20.National Research Council. 1996. Guide for the care and use of laboratory animals. National Academies Press, Washington, DC. [Google Scholar]
- 21.Moser VC, McDaniel KL, Phillips PM. 1991. Rat strain and stock comparisons using a functional observational battery: baseline values and effects of amitraz. Toxicol Appl Pharmacol 108:267–283. doi: 10.1016/0041-008X(91)90117-W. [DOI] [PubMed] [Google Scholar]
- 22.Irwin S. 1968. Comprehensive observational assessment: Ia. A systematic, quantitative procedure for assessing the behavioral and physiologic state of the mouse. Psychopharmacologia 13:222–257. [DOI] [PubMed] [Google Scholar]
- 23.Gad SC. 1982. A neuromuscular screen for use in industrial toxicology. J Toxicol Environ Health 9:691–704. [DOI] [PubMed] [Google Scholar]
- 24.Moser VC, McCormick JP, Creason JP, MacPhail RC. 1988. Comparison of chlordimeform and carbaryl using a functional observational battery. Fundam Appl Toxicol 11:189–206. doi: 10.1016/0272-0590(88)90144-3. [DOI] [PubMed] [Google Scholar]
- 25.Haggerty GC. 1989. Development of tier I neurobehavioral testing capabilities for incorporation into pivotal rodent safety assessment studies. J Am Coll. Toxicol 8:53–69. doi: 10.3109/10915818909009093. [DOI] [Google Scholar]
- 26.O'Donoghue JL. 1989. Screening for neurotoxicity using a neurologically based examination and neuropathology. J Am Coll Toxicol 8:97–115. doi: 10.3109/10915818909009097. [DOI] [Google Scholar]
- 27.McDowell EM, Trump BF. 1976. Histologic fixatives suitable for diagnostic light and electron microscopy. Arch Pathol Lab Med 100:405–414. [PubMed] [Google Scholar]
- 28.Snedecor GW, Cochran WG. 1980. One-way classifications; analysis of variance, p 215–237. In Statistical methods, 7th ed. The Iowa State University Press, Ames, IA. [Google Scholar]
- 29.Dunnett CW. 1964. New tables for multiple comparisons with a control. Biometrics 20:482–491. doi: 10.2307/2528490. [DOI] [Google Scholar]
- 30.Steel RGD, Torrie JH. 1980. Principles and procedures of statistics: a biometrical approach, 2nd ed, p 504–506. McGraw-Hill Book Company, New York, NY. [Google Scholar]
- 31.Eisenbrandt DL, Mattsson JL, Albee RR, Spencer PJ, Johnson KA. 1990. Spontaneous lesions in subchronic neurotoxicity testing of rats. Toxicol Pathol 18:154–164. [DOI] [PubMed] [Google Scholar]
- 32.Barnhill AE, Brewer MT, Carlson SA. 2012. Adverse effects of antimicrobials via predictable or idiosyncratic inhibition of host mitochondrial components. Antimicrob Agents Chemother 56:4046–4051. doi: 10.1128/AAC.00678-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leach KL, Swaney SM, Colca JR, McDonald WG, Blinn JR, Thomasco LM, Gadwood RC, Shinabarger D, Xiong L, Mankin AS. 2007. The site of action of oxazolidinone antibiotics in living bacteria and in human mitochondria. Mol Cell 26:393–402. doi: 10.1016/j.molcel.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 34.McKee EE, Ferguson M, Bentley AT, Marks TA. 2006. Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrob Agents Chemother 50:2042–2049. doi: 10.1128/AAC.01411-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagiec EE, Wu L, Swaney SM, Chosay JG, Ross DE, Brieland JK, Leach KL. 2005. Oxazolidinones inhibit cellular proliferation via inhibition of mitochondrial protein synthesis. Antimicrob Agents Chemother 49:3896–3902. doi: 10.1128/AAC.49.9.3896-3902.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palenzuela L, Hahn NM, Nelson RP Jr, Arno JN, Schobert C, Bethel R, Ostrowski LA, Sharma MR, Datta PP, Agrawal RK, Schwartz JE, Hirano M. 2005. Does linezolid cause lactic acidosis by inhibiting mitochondrial protein synthesis? Clin Infect Dis 40:e113–e116. doi: 10.1086/430441. [DOI] [PubMed] [Google Scholar]
- 37.Garrabou G, Soriano A, Lopez S, Guallar JP, Giralt M, Villarroya F, Martinez JA, Casademont J, Cardellach F, Mensa J, Miro O. 2007. Reversible inhibition of mitochondrial protein synthesis during linezolid-related hyperlactatemia. Antimicrob Agents Chemother 51:962–967. doi: 10.1128/AAC.01190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gerson SL, Kaplan SL, Bruss JB, Le V, Arellano FM, Hafkin B, Kuter DJ. 2002. Hematologic effects of linezolid: summary of clinical experience. Antimicrob Agents Chemother 46:2723–2726. doi: 10.1128/AAC.46.8.2723-2726.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joshi L, Taylor SR, Large O, Yacoub S, Lightman S. 2009. A case of optic neuropathy after short-term linezolid use in a patient with acute lymphocytic leukemia. Clin Infect Dis 48:e73–e74. doi: 10.1086/597298. [DOI] [PubMed] [Google Scholar]
- 40.Metaxas EI, Falagas ME. 2009. Update on the safety of linezolid. Expert Opin Drug Saf 8:485–491. doi: 10.1517/14740330903049706. [DOI] [PubMed] [Google Scholar]
- 41.Apodaca AA, Rakita RM. 2003. Linezolid-induced lactic acidosis. N Engl J Med 348:86–87. doi: 10.1056/NEJM200301023480123. [DOI] [PubMed] [Google Scholar]
- 42.De Vriese AS, Coster RV, Smet J, Seneca S, Lovering A, Van Haute LL, Vanopdenbosch LJ, Martin JJ, Groote CC, Vandecasteele S, Boelaert JR. 2006. Linezolid-induced inhibition of mitochondrial protein synthesis. Clin Infect Dis 42:1111–1117. doi: 10.1086/501356. [DOI] [PubMed] [Google Scholar]
- 43.FDA. 2000. FDA briefing package, anti-infective drugs advisory committee, 68th meeting, March 24, 2000. New drug applications 21-130, 21-131, 21-132. Zyvox™ (linezolid) FDA, Washington, DC http://www.fda.gov/ohrms/dockets/ac/00/backgrd/3597b1.htm. [Google Scholar]
- 44.Cohen BH, Saneto RP. 2012. Mitochondrial translational inhibitors in the pharmacopeia. Biochim Biophys Acta 1819:1067–1074. doi: 10.1016/j.bbagrm.2012.02.023. [DOI] [PubMed] [Google Scholar]
- 45.Bernstein WB, Trotta RF, Rector JT, Tjaden JA, Barile AJ. 2003. Mechanisms for linezolid-induced anemia and thrombocytopenia. Ann Pharmacother 37:517–520. doi: 10.1345/aph.1C361. [DOI] [PubMed] [Google Scholar]