

Abstract
The pathogenic yeast Candida albicans can develop resistance to azole antifungal drugs by overexpressing ERG11, which encodes the drug target, or the multidrug efflux pumps MDR1 and CDR1/CDR2. The constitutive upregulation of these genes is usually caused by gain-of-function mutations in the zinc cluster transcription factors Upc2, Mrr1, and Tac1, respectively. These transcription factors are also required for the induction of their target genes in drug-susceptible strains in the presence of specific stimuli. By swapping the DNA-binding domains of Mrr1, Tac1, and Upc2 we investigated if the hybrid transcription factors could activate their new target genes in response to the same signals. When Tac1 was targeted to the MDR1 and ERG11 promoters, the expression of these genes became inducible by fluphenazine. Similarly, MDR1 and CDR2 were strongly upregulated by fluconazole when Upc2 was fused to the DNA-binding domains of Mrr1 and Tac1, respectively. In contrast, Mrr1 was unable to promote gene expression in response to benomyl when it was targeted to the CDR2 and ERG11 promoters instead of the MDR1 promoter. These results suggest that Tac1 and Upc2 themselves are activated by the inducers fluphenazine and fluconazole, respectively, whereas benomyl does not activate Mrr1 itself but a coregulatory factor that is present at the promoters of Mrr1 target genes. Strains in which the expression levels of Mrr1 and Tac1 target genes were controlled by Upc2 exhibited increased fluconazole resistance, demonstrating that the ability to efficiently upregulate the expression of efflux pumps in the presence of the drug results in enhanced intrinsic fluconazole resistance.
INTRODUCTION
Infections by the pathogenic yeast Candida albicans are commonly treated with azole antifungal drugs, which inhibit ergosterol biosynthesis. C. albicans can develop resistance to azole drugs by various mechanisms. In addition to mutations in the target enzyme, encoded by ERG11, the overexpression of ERG11 and overexpression of genes encoding multidrug efflux pumps are frequent causes of azole resistance in clinical C. albicans strains (1, 2). The constitutive upregulation of these genes is usually due to gain-of-function mutations in zinc cluster transcription factors, a family of transcriptional regulators that is specific for fungi. Activating mutations in Upc2 result in increased ERG11 expression (3–6), while gain-of-function mutations in Mrr1 are responsible for the overexpression of the multidrug efflux pump MDR1 (7–9) and hyperactive forms of Tac1 mediate the overexpression of the ABC transporters CDR1 and CDR2 (10–14).
The expression of MDR1, CDR1/CDR2, and ERG11 is also upregulated in drug-susceptible C. albicans strains in the presence of certain chemicals. For example, benomyl is a strong inducer of MDR1 expression (15, 16), fluphenazine efficiently stimulates the expression of CDR1 and CDR2 (16, 17), and ERG11 expression is upregulated by ergosterol biosynthesis inhibitors, such as fluconazole (18, 19). In each case, the respective zinc cluster transcription factor is required for the induction of gene expression (8, 13, 20). How Mrr1, Tac1, and Upc2 are activated in the presence of inducing chemicals is not known. It is possible that the inducers directly bind to the transcription factors and effect a conformational change that enables the activation domains to recruit the transcriptional machinery, as has been reported for the drug resistance regulators Pdr1/Pdr3 of Saccharomyces cerevisiae and Candida glabrata (21). On the other hand, the transcription factors may also be activated by alterations within the cell that are caused by a drug. For example, the depletion of ergosterol and/or the accumulation of precursor molecules may be a signal that induces ERG11 upregulation when ergosterol biosynthesis is inhibited by azoles (22). It is also possible that Mrr1, Tac1, and Upc2 are not the direct sensors of inducing signals, which may act on upstream regulators or on coregulatory transcription factors. For instance, MDR1 expression is also upregulated by oxidative stress, e.g., in the presence of H2O2 (23). Oxidative stress activates the bZIP transcription factor Cap1 (24, 25), and loss of Cap1 abolishes MDR1 induction by H2O2 (26, 27). Although Mrr1 is also required for H2O2-induced MDR1 upregulation (8, 27), it is likely that Cap1, and not Mrr1, is the sensor of this stimulus.
Most zinc cluster transcription factors contain an N-terminal DNA-binding domain, a C-terminal transcription activation domain, and a large central regulatory domain (28). This is also the case for Mrr1, Tac1, and Upc2, which contain the typical Zn2Cys6 DNA-binding motif in the N terminus. Fusion proteins comprising the N-terminal 129 amino acids of Tac1 or the N-terminal 148 amino acids of Upc2 bound to Tac1 and Upc2 target genes, respectively (13, 29), and the N-terminal 128 amino acids of Mrr1 were sufficient for targeting the transcription factor to the MDR1 promoter (30). Activating mutations are located in the (putative) regulatory or activation domains of the three transcription factors (1, 2). However, it is not known whether the regulatory/activation domains of Mrr, Tac1, and Upc2 contain the necessary information to respond to a specific stimulus. If this is the case, targeting the transcription factors to different promoters by exchanging their DNA-binding domains should result in the activation of the new target gene in response to the same signal. In the present study, we have addressed this question by swapping the DNA-binding domains of Mrr1, Tac1, and Upc2 and investigating the effect on the inducibility of drug resistance genes by specific stimuli.
MATERIALS AND METHODS
Strains and growth conditions.
The C. albicans strains used in this study are listed in Table 1. All strains were stored as frozen stocks with 15% glycerol at −80°C and subcultured on yeast extract-peptone-dextrose (YPD) agar plates (10 g yeast extract, 20 g peptone, 20 g glucose, and 15 g agar per liter) at 30°C. The strains were routinely grown in YPD liquid medium at 30°C in a shaking incubator. The selection of nourseothricin-resistant transformants and recycling of the SAT1 flipper cassette were performed as described previously (31).
TABLE 1.
C. albicans strains used in this studya
| Strain(s) | Parent | Relevant characteristic or genotype | Reference or source |
|---|---|---|---|
| SC5314 | Wild-type reference | 50 | |
| SCMG3A and -B | SC5314 | MDR1/mdr1::PMDR1-GFP-caSAT1 | 32 |
| SCCG3A and -B | SC5314 | CDR2/cdr2::PCDR2-GFP-caSAT1 | 32 |
| SCEG2A and -B | SC5314 | ERG11/erg11::PERG11-GFP-caSAT1 | 5 |
| SCMRR1M4A and -B | SC5314 | mrr1Δ::FRT/mrr1Δ::FRT | 8 |
| SCTAC1M4A and -B | SC5314 | tac1Δ::FRT/tac1Δ::FRT | 32 |
| UPC2M4A and -B | SC5314 | upc2Δ::FRT/upc2Δ::FRT | 3 |
| SCΔmrr1MDR1G42A | SCMRR1M4A | mrr1Δ::FRT/mrr1Δ::FRT | This study |
| MDR1/mdr1::PMDR1-GFP-FRT | |||
| SCΔmrr1MDR1G42B | SCMRR1M4B | mrr1Δ::FRT/mrr1Δ::FRT | This study |
| MDR1/mdr1::PMDR1-GFP-FRT | |||
| SCΔtac1CDR2G52A | SCTAC1M4A | tac1Δ::FRT/tac1Δ::FRT | This study |
| CDR2/cdr2::PCDR2-GFP-FRT | |||
| SCΔtac1CDR2G52B | SCTAC1M4B | tac1Δ::FRT/tac1Δ::FRT | This study |
| CDR2/cdr2::PCDR2-GFP-FRT | |||
| SCΔupc2ERG11G42A | UPC2M4A | upc2Δ::FRT/upc2Δ::FRT | This study |
| ERG11/erg11::PERG11-GFP-FRT | |||
| SCΔupc2ERG11G42B | UPC2M4B | upc2Δ::FRT/upc2Δ::FRT | This study |
| ERG11/erg11::PERG11-GFP-FRT |
Derivatives containing the various hybrid transcription factors are not listed.
Plasmid constructions.
Plasmids pMDR1G3 and pERG11G2, in which the GFP reporter gene is fused to the MDR1 and ERG11 promoters, respectively, have been described previously (5, 32). To allow recycling of the nourseothricin resistance marker after genomic insertion of the reporter fusions, the caSAT1 marker in these plasmids was replaced by the SAT1 flipper cassette. An ApaI-SalI fragment from pMDR1G3 containing the MDR1 promoter was ligated together with a SalI-BamHI fragment from pOP4G4 (33) containing GFP, the TEF3 transcription termination sequence, and an FLP recombination target sequence (FRT) into the ApaI/BamHI-digested pMDR1M2 (27), which contains the remainder of the SAT1 flipper cassette and MDR1 downstream sequences. The resulting plasmid pMDR1G4 contains a PMDR1-GFP reporter fusion that can be targeted to the endogenous MDR1 locus in the C. albicans genome, followed by recycling of the SAT1 flipper cassette. To obtain an analogous cassette with a PERG11-GFP reporter fusion, an ApaI-BglII fragment from pERG11G2 containing the PERG11-GFP fusion was first substituted for the POP4-GFP fragment in pOP4G4, generating pERG11G3. ERG11 downstream sequences were then amplified by PCR from genomic DNA of strain SC5314 with the primers ERG17 (5′-AATACCGCGGCAACTTTCTTTCGATTCAGTG-3′) and ERG16 (5′-CTAAGAGCTCGAATCCTGGTCCTATATTAGC-3′), digested at the introduced SacII and SacI sites (underlined), and substituted for the OP4 downstream sequences in pERG11G3 to yield pERG11G4. The analogous plasmid pCDR2G5 with a PCDR2-GFP reporter fusion was described in a previous report (34).
Hybrids between MRR1, TAC1, and UPC2 were generated in the following way. A part of the MRR1 open reading frame (ORF) encoding amino acids 129 to 1108 was amplified from plasmid pZCF36K2 (8) with the primer ZCF36-95 (5′-ATATGGCGCCAATAATCAACAAACAGCTTCTG-3′) and a primer binding within the TACT1 sequence. The PCR product was digested at the introduced KasI site and at the BglII site behind the MRR1 stop codon and ligated together with an ApaI-KasI fragment from plasmid pZCF36DB1 (30) containing ADH1 upstream sequences and the 5′ part of the MRR1 ORF (encoding amino acids 1 to 128) in the ApaI/BglII-digested pZCF36E2 (9). The resulting plasmid pMRR1DB1-MRR1 encodes a protein in which the DNA-binding domain and the remainder of Mrr1 are fused via a Gly-Ala linker and which is expressed from the ADH1 promoter after integration into the genomic ADH1 locus. To construct an MRR1DB-TAC1 fusion, the 3′ part of the TAC1 ORF encoding amino acids 130 to 981 was amplified from plasmid pTAC1E2 (34) with the primers TAC1-16 (5′-ATATGGCGCCGATCTAGAATCGAGATTGAGTCG-3′) and TAC1-9 (5′-TTTTGGATCCTTAAATCCCCAAATTATTGTCAAAG-3′). The PCR product was digested at the introduced KasI and BamHI sites and ligated behind the MRR1 DNA-binding domain in the KasI/BglII-digested pMRR1DB1-MRR1, generating pMRR1DB1-TAC1. Similarly, the 3′ part of the UPC2 ORF encoding amino acids 150 to 712 was amplified from plasmid pUPC2E2 (34) with the primers UPC2-9 (5′-ATATGGCGCCCCAACTAATCCACTTAGTGCTTTG-3′) and UPC2-2 (5′-ATATAGATCTATTTCATATTCATAAACCCATTATC-3′). The PCR product was digested at the introduced KasI and BglII sites and cloned in the KasI/BglII-digested pMRR1DB1-MRR1 to yield pMRR1DB1-UPC2. Fusions of the 3′ parts of MRR1, TAC1, and UPC2 to the TAC1 DNA-binding domain (encoding amino acids 1 to 129) were obtained by ligating the KasI-SacI fragments from pMRR1DB1-MRR1, pMRR1DB1-TAC1, and pMRR1DB1-UPC2 (containing the respective MRR1, TAC1, and UPC2 sequences, the ACT1 transcription termination sequence, the caSAT1 marker, and 3′ ADH1 sequences) with the KasI/SacI-digested plasmid pTAC1DBH1 (34). Similarly, fusions of the 3′ parts of MRR1, TAC1, and UPC2 to the UPC2 DNA-binding domain (encoding amino acids 1 to 148) were obtained by ligating the KasI-SacI fragments from pMRR1DB1-MRR1, pMRR1DB1-TAC1, and pMRR1DB1-UPC2 with the KasI/SacI-digested plasmid pUPC2DBH1 (34).
Fusions of the TAC1 and UPC2 DNA-binding domains with hyperactive forms of MRR1 were also generated. An EcoRI-BglII fragment from plasmid pZCF36E3 (9) containing the 3′ part of the MRR1P683S allele was substituted for the corresponding wild-type MRR1 sequences in pTAC1DB1-MRR1 and pUPC2DB1-MRR1 to produce plasmids pTAC1DB1-MRR1-4 and pUPC2DB1-MRR1-4, respectively. An EcoRI-SalI fragment from plasmid pZCF36EH2 (27) containing the 3′ part of a hemagglutinin (HA)-tagged MRR1 allele, the ACT1 transcription termination sequence, and part of the caSAT1 marker was substituted for the corresponding sequences in pTAC1DB1-MRR1 and pUPC2DB1-MRR1 to generate plasmids pTAC1DB1-MRR1-5 and pUPC2DB1-MRR1-5, respectively. To obtain hybrids of the TAC1 or UPC2 DNA-binding domains and MRR1 with a C-terminal fusion to the GAL4 activation domain, an EcoRI-SalI fragment from plasmid pMRR1-GAD1 (34) was inserted in the EcoRI/SalI-digested pTAC1DB1-MRR1, yielding pTAC1DB1-MRR1-6. Substitution of the UPC2 DNA-binding domain for the TAC1 DNA-binding domain in the latter plasmid resulted in pUPC2DB1-MRR1-6.
Strain constructions.
C. albicans strains were transformed by electroporation (35) with the following gel-purified linear DNA fragments. The inserts from pMDR1G4, pCDR2G5, and pERG11G4 were used to integrate PMDR1-GFP, PCDR2-GFP, and PERG11-GFP reporter fusions at the corresponding endogenous loci in the mrr1Δ mutants SCMRR1M4A and -B, the tac1Δ mutants SCTAC1M4A and -B, and the upc2Δ mutants UPC2M4A and -B, respectively. Subsequent excision of the SAT1 flipper cassette resulted in the reporter strains SCΔmrr1MDR1G42A and -B, SCΔtac1CDR2G52A and -B, and SCΔupc2ERG11G42A and -B, respectively. The inserts from pMRR1DB1-MRR1, pMRR1DB1-TAC1, pMRR1DB1-UPC2, pTAC1DB1-MRR1, pTAC1DB1-TAC1, pTAC1DB1-UPC2, pUPC2DB1-MRR1, pUPC2DB1-TAC1, pUPC2DB1-UPC2, pTAC1DB1-MRR1-4, pTAC1DB1-MRR1-5, pTAC1DB1-MRR1-6, pUPC2DB1-MRR1-4, pUPC2DB1-MRR1-5, and pUPC2DB1-MRR1-6 were used to integrate genes encoding hybrid transcription factors and control constructs under the control of the ADH1 promoter at the ADH1 locus of the reporter strains described above and of their parental mrr1Δ, tac1Δ, and upc2Δ mutants. The correct integration of each construct and the excision of the SAT1 flipper cassette were confirmed by Southern hybridization using the flanking sequences as probes.
Isolation of genomic DNA and Southern hybridization.
Genomic DNA from C. albicans strains was isolated as described previously (31). The DNA was digested with appropriate restriction enzymes, separated on a 1% agarose gel, transferred by vacuum blotting onto a nylon membrane, and fixed by UV cross-linking. Southern hybridization with enhanced chemiluminescence-labeled probes was performed with the Amersham ECL direct nucleic acid labeling and detection system (GE Healthcare UK Limited, Little Chalfont, Buckinghamshire, United Kingdom) according to the instructions of the manufacturer.
Flow cytometry.
Overnight cultures of the GFP reporter and control strains were diluted 10−2 in fresh YPD medium and grown for 3 h at 30°C. After addition of 50 μg/ml benomyl, 10 μg/ml fluphenazine, 50 μg/ml fluconazole, or no drug, the cultures were incubated for an additional hour. The cell suspensions were 10-fold diluted in 1 ml cold phosphate-buffered saline (PBS), and flow cytometry was performed using a MACSQuantAnalyzer (Miltenyi Biotec, Bergisch Gladbach, Germany) equipped with an argon laser emitting at 488 nm. Fluorescence was detected using the B1 fluorescence channel equipped with a 525-nm band-pass filter (bandwidth of 50 nm). A total of 20,000 cells were analyzed per sample and counted at a flow rate of approximately 500 cells/s. Fluorescence data were collected by using logarithmic amplifiers. The mean fluorescence (arbitrary values) was determined with MACSQuantify (version 2.4; Miltenyi Biotec) software.
Fluconazole susceptibility testing.
To determine the fluconazole susceptibilities of the strains, a 2-fold dilution series of fluconazole (Sigma GmbH, Deisenhofen, Germany) was prepared in the assay medium, starting from an initial concentration of 256 μg/ml. Susceptibility tests were carried out using a previously described microdilution method (36), except that the assays were performed in SD medium (6.7 g of yeast nitrogen base without amino acids [YNB; BIO 101, Vista, CA], 20 g of glucose, and 0.77 g of complete supplement medium [CSM; BIO101]) instead of high-resolution (HR) medium (32). MICs of fluconazole were determined after 24 h of growth at 37°C, because the growth inhibition of the tested strains was more clearly observed at this time point than after longer incubation times.
RESULTS
Induction of MDR1, CDR2, and ERG11 expression by specific stimuli.
To investigate the upregulation of Mrr1, Tac1, and Upc2 target genes by specific inducers, we used reporter strains expressing GFP under the control of the MDR1, CDR2, or ERG11 promoter in a wild-type background. CDR2 was chosen as a Tac1 target gene because it is not significantly expressed in the absence of inducers, whereas CDR1 exhibits relatively high, Tac1-independent basal expression levels (13, 34). The reporter strains were grown in the presence or absence of the inducers benomyl, fluphenazine, or fluconazole, and the induction of each promoter was assessed by quantifying the fluorescence of the cells (Fig. 1). In accordance with previous results (8, 32, 37–39), MDR1 was not detectably expressed under uninduced conditions and was strongly induced by benomyl but not by fluphenazine or fluconazole (Fig. 1A). CDR2 was also not detectably expressed in the absence of inducers but was strongly upregulated by fluphenazine, whereas benomyl and fluconazole had no effect (Fig. 1B). Basal ERG11 expression levels were well detected with GFP as a reporter, and the ERG11 promoter was further upregulated in the presence of fluconazole but not by benomyl and fluphenazine (Fig. 1C). These results confirmed the specific induction of MDR1, CDR2, and ERG11 expression by the inducers benomyl, fluphenazine, and fluconazole, respectively, under the experimental conditions used. The experiments also demonstrated the suitability of the GFP reporter fusions for the analysis of the differential induction of the three genes.
FIG 1.
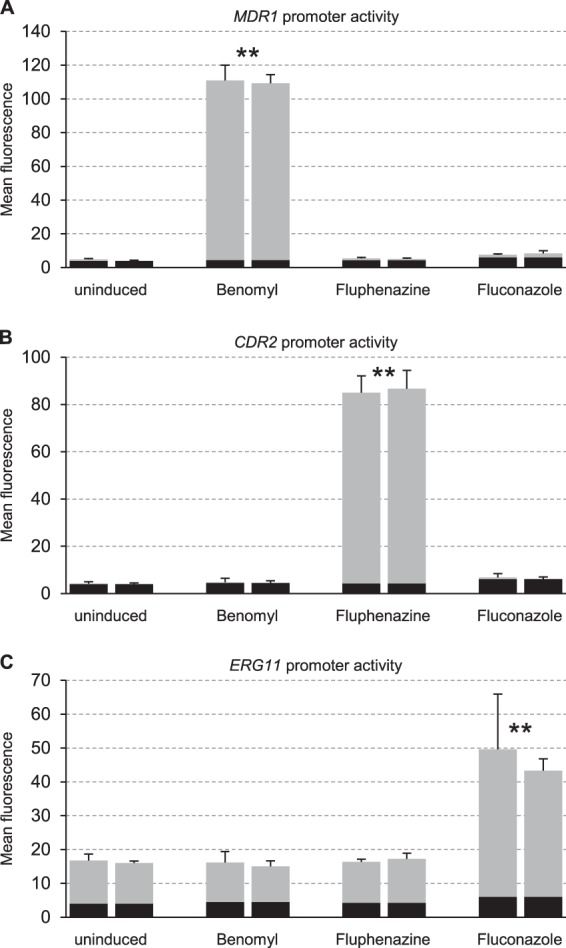
Inducibility of the MDR1, CDR2, and ERG11 promoters by benomyl, fluphenazine, and fluconazole. Transformants of the wild-type strain SC5314 expressing the GFP reporter gene from the MDR1 (A), CDR2 (B), or ERG11 (C) promoter were grown in the absence or presence of inducers as indicated. The mean fluorescence of the cells was determined by flow cytometry. The results obtained with two independently generated reporter strains are shown in each case (means and standard deviations from three experiments). The following strains were used: SCMG3A and -B (MDR1), SCCG3A and -B (CDR2), and SCEG2A and -B (ERG11). The background fluorescence of the parental strain SC5314 is indicated by the black part of each column. Fluorescence values that are at least 2-fold higher than those of the uninduced controls (combined data of the two independently generated strains) are marked by asterisks (**, P < 0.01, two-tailed t test).
Construction of hybrid transcription factors.
To investigate whether the induction of the MDR1, CDR2, and ERG11 promoters by benomyl, fluphenazine, and fluconazole, respectively, could be transferred from one to the other genes by retargeting the corresponding transcription factor, we swapped the DNA-binding domains of Mrr1, Tac1, and Upc2. The resulting hybrid transcription factors, in which the two parts of each protein were fused via a Gly-Ala linker (encoded by a KasI site used for cloning), are illustrated in Fig. 2. As a control, the DNA-binding domain of each transcription factor was also fused in the same way to the remainder of its own protein to exclude a possible negative effect of the inserted Gly-Ala linker. The hybrid transcription factors and control constructs were introduced into reporter strains lacking the relevant wild-type transcription factor and expressed from the ADH1 promoter.
FIG 2.

Expression of hybrid zinc cluster transcription factors in C. albicans. (A) General structure of the cassettes used for expression of hybrid transcription factors. The DNA-binding domains of the transcription factors are represented by the light gray box. PADH1, ADH1 promoter; TACT1, transcription termination sequence of the ACT1 gene; caSAT1, Candida-adapted nourseothricin resistance marker; 3′ADH1, sequence from the 3′ part of the ADH1 gene. The flanking ADH1 sequences (PADH1 and 3′ADH1) served for integration into the ADH1 locus by homologous recombination. (B) Schematic diagrams of the hybrid transcription factors constructed in this study. Mrr1, Tac1, and Upc2 sequences are represented in blue, red, and green, respectively. The extent of the DNA-binding domain (DB) and the remainder of the proteins are indicated (amino acid positions correspond to those in the wild-type transcription factors). The Gly-Ala linker that was inserted between the two parts of each protein is also shown.
Inducibility of the MDR1 promoter by hybrid transcription factors.
Figure 3 shows the activity of the MDR1 promoter in strains from which the endogenous MRR1 alleles were deleted and which contained hybrid transcription factors consisting of the Mrr1 DNA-binding domain and the regulatory/activation domains of Tac1 or Upc2. As previously reported (8, 27, 32), no induction of MDR1 expression by benomyl was observed in the absence of Mrr1, and the inducibility by benomyl was restored when the MRR1DB-MRR1 control construct was expressed in the mutants (Fig. 3A). When the regulatory/activation domain of Mrr1 was replaced by that from Tac1, MDR1 induction by benomyl was largely, but not completely, abolished (Fig. 3A). However, MDR1 expression in these strains could be efficiently stimulated by fluphenazine (Fig. 3B), demonstrating that the presence of Tac1 at the MDR1 promoter rendered it responsive to this inducer. A slight induction of the MDR1 promoter by the hybrid Mrr1DB-Tac1 protein was also observed in the presence of fluconazole (Fig. 3C; see also Discussion). Interestingly, MDR1 was constitutively expressed even in the absence of inducers when the Mrr1 DNA-binding domain was fused to the regulatory/activation domain of Upc2. MDR1 promoter activity was further increased in the presence of fluconazole (Fig. 3C), but not by the original inducer benomyl (Fig. 3A) or by fluphenazine (Fig. 3B). Therefore, Upc2 could upregulate MDR1 expression in the presence of fluconazole when it was fused to the Mrr1 DNA-binding domain, similar to the fluphenazine-induced MDR1 expression by Mrr1DB-Tac1 (see also Discussion). In additional control experiments, we tested MDR1 expression in strains in which the Mrr1 DNA-binding domain was replaced by those of Tac1 or Upc2. No induction of the MDR1 promoter was observed in these strains, except for a slight upregulation by the hybrid Upc2DB-Mrr1 protein in the presence of benomyl (Fig. 3A to C).
FIG 3.

Inducibility of the MDR1 promoter in mrr1Δ mutants carrying a PMDR1-GFP reporter fusion and the indicated hybrid transcription factors by benomyl (A), fluphenazine (B), and fluconazole (C). Strains were grown in the absence (−) or presence (+) of inducers, and the mean fluorescence of the cells was determined by flow cytometry. The results obtained with two independently generated reporter strains are shown in each case (means and standard deviations from three experiments). The background fluorescence of otherwise identical strains without GFP is indicated by the black part of each column. Values of the uninduced cultures are from the same experiments in panels A to C and are included for comparison with each inducer. Fluorescence values that are at least 2-fold higher than those of the controls (combined data of the two independently generated strains) are marked by asterisks (**, P < 0.01; *P, < 0.05, two-tailed t test).
Inducibility of the CDR2 promoter by hybrid transcription factors.
Figure 4 shows the activity of the CDR2 promoter in strains lacking the endogenous TAC1 alleles and expressing TAC1DB-MRR1 or TAC1DB-UPC2 fusions. The inability of the tac1Δ mutants to upregulate CDR2 expression in the presence of fluphenazine was complemented by the TAC1DB-TAC1 control construct (Fig. 4A). When the Tac1 DNA-binding domain was fused to Upc2, CDR2 expression could be induced by fluconazole instead of fluphenazine (Fig. 4A and C). Of note, no constitutive CDR2 expression was observed when Upc2 was targeted to the CDR2 promoter, which is in contrast to the activation of the MDR1 promoter in the absence of inducers by the Mrr1DB-Upc2 hybrid transcription factor (see above). Therefore, Upc2 could promote CDR2 expression when it was targeted to the CDR2 promoter, but it had to be activated by the inducer fluconazole. When the Tac1 DNA-binding domain was fused to Mrr1, no CDR2 expression was observed in the presence of any of the inducers, indicating that Mrr1 was unable to effect benomyl-induced gene expression when targeted to the CDR2 promoter instead of the MDR1 promoter (Fig. 4B). Not surprisingly, no induction of CDR2 expression occurred when the DNA-binding domain of Tac1 was replaced by those of Mrr1 or Upc2, which do not bind to the CDR2 promoter (27, 40).
FIG 4.

Inducibility of the CDR2 promoter in tac1Δ mutants carrying a PCDR2-GFP reporter fusion and the indicated hybrid transcription factors by fluphenazine (A), benomyl (B), and fluconazole (C). Strains were grown in the absence (−) or presence (+) of inducers, and the mean fluorescence of the cells was determined by flow cytometry. The results obtained with two independently generated reporter strains are shown in each case (means and standard deviations from three experiments). The background fluorescence of otherwise identical strains without GFP is indicated by the black part of each column. Values of the uninduced cultures are from the same experiments in panels A to C and are included for comparison with each inducer. Fluorescence values that are at least 2-fold higher than those of the controls (combined data of the two independently generated strains) are marked by asterisks (**, P < 0.01, two-tailed t test).
Inducibility of the ERG11 promoter by hybrid transcription factors.
Figure 5 shows the activity of the ERG11 promoter in upc2Δ mutants expressing UPC2DB-MRR1 or UPC2DB-TAC1 fusions. As previously reported (20, 29), no induction of the ERG11 promoter by fluconazole occurred in the absence of Upc2; this deficiency was complemented after insertion of the UPC2DB-UPC2 control construct (Fig. 5A). The fluconazole inducibility was lost when the Upc2 DNA-binding domain was fused to Tac, but the hybrid Upc2DB-Tac1 transcription factor upregulated ERG11 expression in response to fluphenazine (Fig. 5C), demonstrating that Tac1 could activate both MDR1 and ERG11 expression in response to this normally noninducing stimulus when targeted to the respective promoter. Interestingly, basal ERG11 expression levels were reduced by Upc2DB-Tac1 (the fluorescence of the cells was 1.7-fold lower than that of the controls, P < 0.01), indicating that Tac1 bound to the ERG11 promoter acted as a repressor, unless it was activated by fluphenazine. When the Upc2 DNA-binding domain was fused to Mrr1, no upregulation of ERG11 expression by any of the inducers was observed. Therefore, Mrr1 could not induce gene expression in response to benomyl when targeted to the ERG11 (Fig. 5B) or CDR2 promoter (Fig. 4B). When the DNA-binding domain of Upc2 was replaced by those of Mrr1 or Tac1, which do not bind to the ERG11 promoter (27, 41), no upregulation of ERG11 expression occurred in the presence of any of the inducers (Fig. 5A to C).
FIG 5.

Inducibility of the ERG11 promoter in upc2Δ mutants carrying a PERG11-GFP reporter fusion and the indicated hybrid transcription factors by fluconazole (A), benomyl (B), and fluphenazine (C). Strains were grown in the absence (−) or presence (+) of inducers, and the mean fluorescence of the cells was determined by flow cytometry. The results obtained with two independently generated reporter strains are shown in each case (means and standard deviations from three experiments). The background fluorescence of otherwise identical strains without GFP is indicated by the black part of each column. Values of the uninduced cultures are from the same experiments in panels A to C and are included for comparison with each inducer. Fluorescence values that are at least 2-fold higher than those of the controls (combined data of the two independently generated strains) are marked by asterisks (**, P < 0.01, two-tailed t test).
Hyperactive forms of Mrr1 can activate gene expression when fused to heterologous DNA-binding domains.
The inability of Tac1DB-Mrr1 and Upc2DB-Mrr1 fusion proteins to activate the CDR2 and ERG11 promoters suggested that Mrr1 itself is unable to react to the presence of the inducer benomyl. However, it was also possible that these fusion proteins were simply unstable or incorrectly folded and therefore nonfunctional. To address this possibility, we constructed analogous fusions with constitutively active forms of Mrr1 that do not require the inducer benomyl. The tested fusion proteins contained the P683S gain-of-function mutation, which was previously found in a fluconazole-resistant clinical C. albicans isolate (8), a C-terminal 3×HA tag, which acts like a gain-of-function mutation (27), or a C-terminal Gal4 activation domain (34). When expressed in tac1Δ and upc2Δ mutants carrying PCDR2-GFP and PERG11-GFP reporter fusions, respectively, the target genes were constitutively activated by all three respective hybrid transcription factors, albeit with different efficiencies (Fig. 6). These results argued that the hybrid Tac1DB-Mrr1 and Upc2DB-Mrr1 transcription factors were not intrinsically unstable. Their failure to induce gene expression in response to benomyl suggests that benomyl acts on a coregulatory factor, which is present at the MDR1 but not the CDR2 and ERG11 promoters, rather than on Mrr1 itself.
FIG 6.

Activation of the CDR2 and ERG11 promoters by hybrid transcription factors in which the Tac1 (A) or Upc2 (B) DNA-binding domains were fused to mutated forms of Mrr1. The Tac1DB-Mrr1 and Upc2DB-Mrr1 fusions correspond to those shown in Fig. 2 (amino acids 129 to 1108 of Mrr1) but contain the P683S gain-of-function mutation (MRR1P683S), a C-terminal 3×HA epitope tag (MRR1-HA), or a C-terminal Gal4 activation domain with a 3×HA tag (MRR1-GAD). The hybrid transcription factors were expressed in tac1Δ mutants carrying a PCDR2-GFP reporter fusion (A) or in upc2Δ mutants carrying a PERG11-GFP reporter fusion (B). The mean fluorescence of cells grown in the absence of inducers was determined by flow cytometry. The results obtained with two independently generated reporter strains are shown in each case (means and standard deviations from three experiments). The background fluorescence of otherwise identical strains without GFP is indicated by the black part of each column. Fluorescence values that are at least 2-fold higher than those of the controls (combined data of the two independently generated strains) are marked by asterisks (**, P < 0.01, two-tailed t test).
Fluconazole susceptibilities of strains containing hybrid transcription factors.
Mrr1 and Tac1 are not active under standard growth conditions and also do not induce their target genes in the presence of fluconazole. Deletion of MRR1 and TAC1 from a drug-susceptible wild-type strain, like SC5314, therefore does not result in fluconazole hypersusceptibility (8, 34). Upc2, when targeted to the MDR1 or CDR2 promoters by fusion with the Mrr1 and Tac1 DNA-binding domains, respectively, constitutively activated MDR1 expression (Fig. 3) and upregulated MDR1 and CDR2 expression in the presence of fluconazole (Fig. 3C and 4C). This observation suggested that strains containing Mrr1DB-Upc2 or Tac1DB-Upc2 proteins should exhibit increased fluconazole resistance. We therefore tested the drug susceptibilities of mrr1Δ, tac1Δ, and upc2Δ mutants expressing hybrid transcription factors or control constructs (Fig. 7). Unlike Mrr1 itself, the Mrr1DB-Upc2 hybrid transcription factor conferred a 32-fold increased fluconazole resistance in the mrr1Δ mutants, reflecting the strong upregulation of MDR1 (Fig. 3C) and presumably other Mrr1 target genes that contribute to fluconazole resistance (27) by this protein. Similarly, the Tac1DB-Upc2 hybrid protein caused a 4-fold increase in fluconazole resistance in the tac1Δ mutants, as expected from its ability to upregulate CDR2 (Fig. 4C) and possibly other Tac1 target genes in the presence of fluconazole. The Mrr1DB-Tac1 hybrid also caused a minor (2-fold) increase in fluconazole resistance in the mrr1Δ mutants, in accordance with its ability to slightly activate the MDR1 promoter in the presence of fluconazole (Fig. 3C). However, this hybrid transcription factor did not confer increased resistance in tac1Δ mutants, presumably because it had to compete with (inactive) wild-type Mrr1 for binding to the MDR1 promoter in this strain background. Overexpression of UPC2 from the ADH1 promoter rescued the fluconazole hypersusceptibility of the upc2Δ mutants, and the strains exhibited slightly higher resistance (2-fold) than the wild type. The Mrr1DB-Upc2 and Tac1DB-Upc2 hybrid transcription factors also increased the resistance of the upc2Δ mutants, presumably because these strains now overexpressed Mrr1 and Tac1 target genes, respectively, in the presence of fluconazole.
FIG 7.
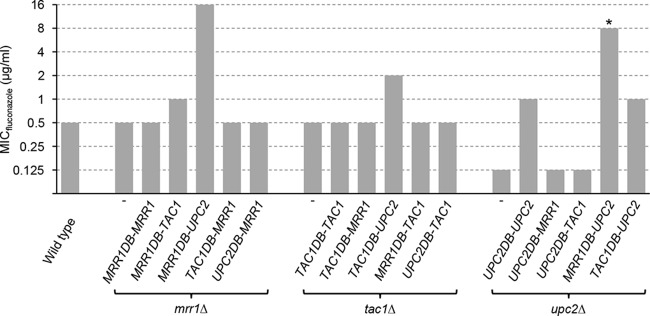
Fluconazole susceptibilities of the wild-type strain SC5314 and of mrr1Δ, tac1Δ, and upc2Δ mutants expressing the indicated hybrid transcription factors. *The MIC for one of the two independently constructed upc2Δ mutants expressing the MRR1DB-UPC2 fusion was slightly lower (4 μg/ml). In all other cases, the two independently generated strains behaved identically.
DISCUSSION
The zinc cluster proteins Mrr1, Tac1, and Upc2 mediate drug resistance in many azole-resistant, clinical C. albicans strains that have acquired gain-of-function mutations in these transcription factors, which result in their constitutive activation. Mrr1, Tac1, and Upc2 are also required for the upregulation of their target genes in response to specific stimuli in drug-susceptible strains. However, it has not been established if the ability to respond to the presence of an inducer is intrinsic to these transcription factors or if the stimuli result in the activation of other transcription factors that bind to the same promoters and help Mrr1, Tac1, and Upc2 to promote gene expression. Here, we have addressed this question by exchanging the DNA-binding domains of Mrr1, Tac1, and Upc2 and assessing the inducibility of their target genes by specific stimuli.
Fluphenazine induces the expression of CDR2 and other Tac1 target genes in a Tac1-dependent fashion. When Tac1 was targeted to the MDR1 or ERG11 promoters, which are not normally activated by this inducer, these promoters now became responsive to fluphenazine. Similarly, Upc2 could upregulate the expression of MDR1 and CDR2 in the presence of fluconazole when its DNA-binding domain was replaced by those of Mrr1 and Tac1, respectively. These results support the idea that Tac1 and Upc2 themselves are the targets for activation by the inducers fluphenazine and fluconazole. The drugs might directly bind to the transcription factors to promote a conformational transition to an activated state, but it is also possible that Tac1 and Upc2 are modified (e.g., by phosphorylation or some other protein modification) via a signaling pathway that is activated in the presence of the drugs.
In contrast, when the DNA-binding domain of Mrr1 was replaced by those of Tac1 and Upc2, the hybrid transcription factors failed to induce the expression of the new target genes CDR2 and ERG11, respectively, in response to benomyl. The fact that analogous fusion proteins with constitutively activated forms of Mrr1 mediated CDR2 and ERG11 overexpression argues that the Tac1DB-Mrr1 and Upc2DB-Mrr1 hybrid transcription factors were correctly targeted to the CDR2 and ERG11 promoters, respectively. It is therefore possible that another transcription factor that binds to the MDR1 but not the CDR2 and ERG11 promoters is activated by benomyl and cooperates with Mrr1 to induce MDR1 expression. Benomyl also causes oxidative stress (42), and benomyl-induced MDR1 expression is strongly reduced in the absence of the oxidative stress response regulator Cap1 (27, 43). Cap1 does not bind to the CDR2 and ERG11 promoters (43) and can therefore facilitate MDR1 but not CDR2 and ERG11 upregulation in the presence of benomyl. Nevertheless, a significant induction of MDR1 expression by benomyl still occurs in the absence of Cap1 (26, 27, 43), demonstrating that benomyl or benomyl-induced effects on the cells are also sensed in a different way. Like Cap1, the MADS box transcription factor Mcm1 binds to the MDR1 but not to the CDR2 and ERG11 promoters and contributes to benomyl-induced MDR1 expression (37, 44–46). Therefore, if Mrr1 is not itself activated by benomyl, the absence of its coregulators may prevent it from upregulating CDR2 and ERG11 expression when it is targeted to these promoters by fusion with the Tac1 and Upc2 DNA-binding domains. It should be noted that a weak but significant induction by benomyl was observed when Mrr1 was targeted to an artificial promoter by fusion with the Tet repressor TetR (30). Therefore, the possibility that some coactivator-independent activation of Mrr1 itself may occur in the presence of benomyl cannot be excluded. It is also worth mentioning that the hyperactive Mrr1 containing the P683S mutation can activate MDR1 expression in the absence of Cap1 but requires Mcm1 to do so (27, 37). It is possible that this hyperactive form of Mrr1 may cooperate with other transcriptional regulators to promote the (comparatively weak) constitutive CDR2 and ERG11 expression that was observed when it was targeted to these promoters via the Tac1 and Upc2 DNA-binding domains (Fig. 6).
Interestingly, the Mrr1DB-Tac1 hybrid transcription factor induced MDR1 expression not only in response to fluphenazine but also, albeit very weakly, in the presence of benomyl and fluconazole (Fig. 3). The induction by benomyl can be explained if activated Cap1 also interacts with Tac1 that is present instead of Mrr1 at the MDR1 promoter, resulting in a slight MDR1 upregulation. The weak but significant induction of the MDR1 promoter by fluconazole in strains containing the Mrr1DB-Tac1 hybrid transcription factor instead of Mrr1 is somewhat surprising, as neither Mrr1 nor Tac1 is activated by fluconazole. It must be mentioned that the very low basal MDR1 expression levels in drug-susceptible strains like SC5314 are indeed elevated in the presence of fluconazole (47). The increase in basal MDR1 transcription can be detected by reverse transcription (RT)-PCR (47) but did not significantly enhance the fluorescence of cells containing a PMDR1-GFP reporter fusion over the background fluorescence (Fig. 1A). Importantly, the level of MDR1 upregulation in the presence of fluconazole is insufficient to confer increased drug resistance, because deletion of MDR1 in fluconazole-susceptible C. albicans strains does not result in hypersusceptibility to the drug (27, 48, 49). The induction of MDR1 expression by fluconazole in wild-type strains is most likely also mediated by Upc2 binding to the MDR1 promoter (40). Upc2 may upregulate MDR1 expression in the presence of fluconazole more efficiently when Tac1 is present instead of Mrr1 at the MDR1 promoter. This is also reflected by the slightly increased (2-fold) fluconazole resistance of the strains with the Mrr1DB-Tac1 hybrid protein compared with that of the wild-type strain (Fig. 7).
As Upc2 does bind to the MDR1 promoter, replacement of the Mrr1 DNA-binding domain by the Upc2 DNA-binding domain would still allow the hybrid transcription factor to bind to the MDR1 promoter. Nevertheless, MDR1 expression was only slightly induced by benomyl in strains containing the Upc2DB-Mrr1 hybrid protein (Fig. 3A). Apparently, shifting the binding site of Mrr1 in the MDR1 promoter to the Upc2 binding site prevented the hybrid transcription factor from productively interacting with coregulatory transcription factors, like Cap1 and Mcm1, resulting in a failure to efficiently induce MDR1 expression in the presence of benomyl.
Remarkably, when fused to the Mrr1 DNA-binding domain, Upc2 caused a constitutive activation of the MDR1 promoter (Fig. 3). This observation is compatible with a model in which Mrr1 is constitutively localized to the MDR1 promoter but needs to be activated in order to induce gene expression. Upc2, in contrast, might bind to its target promoters only under inducing conditions. In that case, constitutive binding of the Mrr1DB-Upc2 hybrid protein to the MDR1 promoter could result in MDR1 expression even in the absence of an inducer and further upregulation in the presence of fluconazole. However, Tac1 also binds to its target genes in the absence of inducers (41), but CDR2 expression was induced by the Tac1DB-Upc2 fusion protein only in the presence of fluconazole (Fig. 4C). Upc2 may therefore be in a partially activated state when fused to the Mrr1 DNA-binding domain but not when fused to the Tac1 DNA-binding domain. Yet, both hybrid transcription factors could be stimulated in the presence of fluconazole.
Mrr1 and Tac1 do not induce the expression of efflux pumps in the presence of fluconazole, which may explain the high susceptibility of the majority of C. albicans strains to this drug. If Mrr1 and Tac1 acquired the ability to be activated by azole drugs, like the artificially created Mrr1DB-Upc2 and Tac1DB-Upc2 hybrid proteins, this would result in efflux pump overexpression in the presence of azoles and, consequently, increased drug resistance (Fig. 7). In C. glabrata, the zinc cluster transcription factor Pdr1, which regulates the expression of several drug efflux pumps, has this ability and is activated by azoles and other toxic compounds, resulting in the high intrinsic azole resistance of this pathogenic yeast (21).
ACKNOWLEDGMENTS
This study was supported by the Deutsche Forschungsgemeinschaft (DFG grant MO 846/7 and SFB 630).
Nico Dunkel, Dagmar Sedding, and Anna-Lena Geiselhöringer are acknowledged for their help in the construction of some plasmids and strains.
REFERENCES
- 1.Morschhäuser J. 2010. Regulation of multidrug resistance in pathogenic fungi. Fungal Genet Biol 47:94–106. doi: 10.1016/j.fgb.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Sanglard D, Coste A, Ferrari S. 2009. Antifungal drug resistance mechanisms in fungal pathogens from the perspective of transcriptional gene regulation. FEMS Yeast Res 9:1029–1050. doi: 10.1111/j.1567-1364.2009.00578.x. [DOI] [PubMed] [Google Scholar]
- 3.Dunkel N, Liu TT, Barker KS, Homayouni R, Morschhäuser J, Rogers PD. 2008. A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryot Cell 7:1180–1190. doi: 10.1128/EC.00103-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flowers SA, Barker KS, Berkow EL, Toner G, Chadwick SG, Gygax SE, Morschhäuser J, Rogers PD. 2012. Gain-of-function mutations in UPC2 are a frequent cause of ERG11 upregulation in azole-resistant clinical isolates of Candida albicans. Eukaryot Cell 11:1289–1299. doi: 10.1128/EC.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heilmann CJ, Schneider S, Barker KS, Rogers PD, Morschhäuser J. 2010. An A643T mutation in the transcription factor Upc2p causes constitutive ERG11 upregulation and increased fluconazole resistance in Candida albicans. Antimicrob Agents Chemother 54:353–359. doi: 10.1128/AAC.01102-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoot SJ, Smith AR, Brown RP, White TC. 2011. An A643V amino acid substitution in Upc2p contributes to azole resistance in well-characterized clinical isolates of Candida albicans. Antimicrob Agents Chemother 55:940–942. doi: 10.1128/AAC.00995-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunkel N, Blaß J, Rogers PD, Morschhäuser J. 2008. Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Mol Microbiol 69:827–840. doi: 10.1111/j.1365-2958.2008.06309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morschhäuser J, Barker KS, Liu TT, Blaß-Warmuth J, Homayouni R, Rogers PD. 2007. The transcription factor Mrr1p controls expression of the MDR1 efflux pump and mediates multidrug resistance in Candida albicans. PLoS Pathog 3:e164. doi: 10.1371/journal.ppat.0030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schubert S, Rogers PD, Morschhäuser J. 2008. Gain-of-function mutations in the transcription factor MRR1 are responsible for overexpression of the MDR1 efflux pump in fluconazole-resistant Candida dubliniensis strains. Antimicrob Agents Chemother 52:4274–4280. doi: 10.1128/AAC.00740-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coste A, Selmecki A, Forche A, Diogo D, Bougnoux ME, d'Enfert C, Berman J, Sanglard D. 2007. Genotypic evolution of azole resistance mechanisms in sequential Candida albicans isolates. Eukaryot Cell 6:1889–1904. doi: 10.1128/EC.00151-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coste A, Turner V, Ischer F, Morschhäuser J, Forche A, Selmecki A, Berman J, Bille J, Sanglard D. 2006. A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in Candida albicans. Genetics 172:2139–2156. doi: 10.1534/genetics.105.054767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coste AT, Crittin J, Bauser C, Rohde B, Sanglard D. 2009. Functional analysis of cis- and trans-acting elements of the Candida albicans CDR2 promoter with a novel promoter reporter system. Eukaryot Cell 8:1250–1267. doi: 10.1128/EC.00069-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coste AT, Karababa M, Ischer F, Bille J, Sanglard D. 2004. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot Cell 3:1639–1652. doi: 10.1128/EC.3.6.1639-1652.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Znaidi S, De Deken X, Weber S, Rigby T, Nantel A, Raymond M. 2007. The zinc cluster transcription factor Tac1p regulates PDR16 expression in Candida albicans. Mol Microbiol 66:440–452. doi: 10.1111/j.1365-2958.2007.05931.x. [DOI] [PubMed] [Google Scholar]
- 15.Gupta V, Kohli A, Krishnamurthy S, Puri N, Aalamgeer SA, Panwar S, Prasad R. 1998. Identification of polymorphic mutant alleles of CaMDR1, a major facilitator of Candida albicans which confers multidrug resistance, and its in vitro transcriptional activation. Curr Genet 34:192–199. doi: 10.1007/s002940050385. [DOI] [PubMed] [Google Scholar]
- 16.Karababa M, Coste AT, Rognon B, Bille J, Sanglard D. 2004. Comparison of gene expression profiles of Candida albicans azole-resistant clinical isolates and laboratory strains exposed to drugs inducing multidrug transporters. Antimicrob Agents Chemother 48:3064–3079. doi: 10.1128/AAC.48.8.3064-3079.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Micheli M, Bille J, Schueller C, Sanglard D. 2002. A common drug-responsive element mediates the upregulation of the Candida albicans ABC transporters CDR1 and CDR2, two genes involved in antifungal drug resistance. Mol Microbiol 43:1197–1214. doi: 10.1046/j.1365-2958.2002.02814.x. [DOI] [PubMed] [Google Scholar]
- 18.Franz R, Kelly SL, Lamb DC, Kelly DE, Ruhnke M, Morschhäuser J. 1998. Multiple molecular mechanisms contribute to a stepwise development of fluconazole resistance in clinical Candida albicans strains. Antimicrob Agents Chemother 42:3065–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henry KW, Nickels JT, Edlind TD. 2000. Upregulation of ERG genes in Candida species by azoles and other sterol biosynthesis inhibitors. Antimicrob Agents Chemother 44:2693–2700. doi: 10.1128/AAC.44.10.2693-2700.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silver PM, Oliver BG, White TC. 2004. Role of Candida albicans transcription factor Upc2p in drug resistance and sterol metabolism. Eukaryot Cell 3:1391–1397. doi: 10.1128/EC.3.6.1391-1397.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thakur JK, Arthanari H, Yang F, Pan SJ, Fan X, Breger J, Frueh DP, Gulshan K, Li DK, Mylonakis E, Struhl K, Moye-Rowley WS, Cormack BP, Wagner G, Naar AM. 2008. A nuclear receptor-like pathway regulating multidrug resistance in fungi. Nature 452:604–609. doi: 10.1038/nature06836. [DOI] [PubMed] [Google Scholar]
- 22.Song JL, Harry JB, Eastman RT, Oliver BG, White TC. 2004. The Candida albicans lanosterol 14-α-demethylase (ERG11) gene promoter is maximally induced after prolonged growth with antifungal drugs. Antimicrob Agents Chemother 48:1136–1144. doi: 10.1128/AAC.48.4.1136-1144.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harry JB, Oliver BG, Song JL, Silver PM, Little JT, Choiniere J, White TC. 2005. Drug-induced regulation of the MDR1 promoter in Candida albicans. Antimicrob Agents Chemother 49:2785–2792. doi: 10.1128/AAC.49.7.2785-2792.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alarco AM, Raymond M. 1999. The bZip transcription factor Cap1p is involved in multidrug resistance and oxidative stress response in Candida albicans. J Bacteriol 181:700–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, De Micheli M, Coleman ST, Sanglard D, Moye-Rowley WS. 2000. Analysis of the oxidative stress regulation of the Candida albicans transcription factor, Cap1p. Mol Microbiol 36:618–629. doi: 10.1046/j.1365-2958.2000.01877.x. [DOI] [PubMed] [Google Scholar]
- 26.Rognon B, Kozovska Z, Coste AT, Pardini G, Sanglard D. 2006. Identification of promoter elements responsible for the regulation of MDR1 from Candida albicans, a major facilitator transporter involved in azole resistance. Microbiology 152:3701–3722. doi: 10.1099/mic.0.29277-0. [DOI] [PubMed] [Google Scholar]
- 27.Schubert S, Barker KS, Znaidi S, Schneider S, Dierolf F, Dunkel N, Aid M, Boucher G, Rogers PD, Raymond M, Morschhäuser J. 2011. Regulation of efflux pump expression and drug resistance by the transcription factors Mrr1, Upc2, and Cap1 in Candida albicans. Antimicrob Agents Chemother 55:2212–2223. doi: 10.1128/AAC.01343-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacPherson S, Larochelle M, Turcotte B. 2006. A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol Mol Biol Rev 70:583–604. doi: 10.1128/MMBR.00015-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacPherson S, Akache B, Weber S, De Deken X, Raymond M, Turcotte B. 2005. Candida albicans zinc cluster protein Upc2p confers resistance to antifungal drugs and is an activator of ergosterol biosynthetic genes. Antimicrob Agents Chemother 49:1745–1752. doi: 10.1128/AAC.49.5.1745-1752.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schubert S, Popp C, Rogers PD, Morschhäuser J. 2011. Functional dissection of a Candida albicans zinc cluster transcription factor, the multidrug resistance regulator Mrr1. Eukaryot Cell 10:1110–1121. doi: 10.1128/EC.05100-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reuß O, Vik Å Kolter R, Morschhäuser J. 2004. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341:119–127. doi: 10.1016/j.gene.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 32.Sasse C, Schillig R, Dierolf F, Weyler M, Schneider S, Mogavero S, Rogers PD, Morschhäuser J. 2011. The transcription factor Ndt80 does not contribute to Mrr1-, Tac1-, and Upc2-mediated fluconazole resistance in Candida albicans. PLoS One 6:e25623. doi: 10.1371/journal.pone.0025623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sasse C, Hasenberg M, Weyler M, Gunzer M, Morschhäuser J. 2013. White-opaque switching of Candida albicans allows immune evasion in an environment-dependent fashion. Eukaryot Cell 12:50–58. doi: 10.1128/EC.00266-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schillig R, Morschhäuser J. 2013. Analysis of a fungus-specific transcription factor family, the Candida albicans zinc cluster proteins, by artificial activation. Mol Microbiol 89:1003–1017. doi: 10.1111/mmi.12327. [DOI] [PubMed] [Google Scholar]
- 35.Köhler GA, White TC, Agabian N. 1997. Overexpression of a cloned IMP dehydrogenase gene of Candida albicans confers resistance to the specific inhibitor mycophenolic acid. J Bacteriol 179:2331–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruhnke M, Eigler A, Tennagen I, Geiseler B, Engelmann E, Trautmann M. 1994. Emergence of fluconazole-resistant strains of Candida albicans in patients with recurrent oropharyngeal candidosis and human immunodeficiency virus infection. J Clin Microbiol 32:2092–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mogavero S, Tavanti A, Senesi S, Rogers PD, Morschhäuser J. 2011. Differential requirement of the transcription factor Mcm1 for activation of the Candida albicans multidrug efflux pump MDR1 by its regulators Mrr1 and Cap1. Antimicrob Agents Chemother 55:2061–2066. doi: 10.1128/AAC.01467-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramírez-Zavala B, Mogavero S, Schöller E, Sasse C, Rogers PD, Morschhäuser J. 2014. SAGA/ADA complex subunit Ada2 is required for Cap1-but not Mrr1-mediated upregulation of the Candida albicans multidrug efflux pump MDR1. Antimicrob Agents Chemother 58:5102–5110. doi: 10.1128/AAC.03065-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sasse C, Schillig R, Reimund A, Merk J, Morschhäuser J. 2012. Inducible and constitutive activation of two polymorphic promoter alleles of the Candida albicans multidrug efflux pump MDR1. Antimicrob Agents Chemother 56:4490–4494. doi: 10.1128/AAC.00264-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Znaidi S, Weber S, Al-Abdin OZ, Bomme P, Saidane S, Drouin S, Lemieux S, De Deken X, Robert F, Raymond M. 2008. Genomewide location analysis of Candida albicans Upc2p, a regulator of sterol metabolism and azole drug resistance. Eukaryot Cell 7:836–847. doi: 10.1128/EC.00070-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu TT, Znaidi S, Barker KS, Xu L, Homayouni R, Saidane S, Morschhäuser J, Nantel A, Raymond M, Rogers PD. 2007. Genome-wide expression and location analyses of the Candida albicans Tac1p regulon. Eukaryot Cell 6:2122–2138. doi: 10.1128/EC.00327-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lucau-Danila A, Lelandais G, Kozovska Z, Tanty V, Delaveau T, Devaux F, Jacq C. 2005. Early expression of yeast genes affected by chemical stress. Mol Cell Biol 25:1860–1868. doi: 10.1128/MCB.25.5.1860-1868.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Znaidi S, Barker KS, Weber S, Alarco AM, Liu TT, Boucher G, Rogers PD, Raymond M. 2009. Identification of the Candida albicans Cap1p regulon. Eukaryot Cell 8:806–820. doi: 10.1128/EC.00002-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lavoie H, Sellam A, Askew C, Nantel A, Whiteway M. 2008. A toolbox for epitope-tagging and genome-wide location analysis in Candida albicans. BMC Genomics 9:578. doi: 10.1186/1471-2164-9-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Riggle PJ, Kumamoto CA. 2006. Transcriptional regulation of MDR1, encoding a drug efflux determinant, in fluconazole-resistant Candida albicans strains through an Mcm1p binding site. Eukaryot Cell 5:1957–1968. doi: 10.1128/EC.00243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tuch BB, Galgoczy DJ, Hernday AD, Li H, Johnson AD. 2008. The evolution of combinatorial gene regulation in fungi. PLoS Biol 6:e38. doi: 10.1371/journal.pbio.0060038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sellam A, Askew C, Epp E, Lavoie H, Whiteway M, Nantel A. 2009. Genome-wide mapping of the coactivator Ada2p yields insight into the functional roles of SAGA/ADA complex in Candida albicans. Mol Biol Cell 20:2389–2400. doi: 10.1091/mbc.E08-11-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morschhäuser J, Michel S, Staib P. 1999. Sequential gene disruption in Candida albicans by FLP-mediated site-specific recombination. Mol Microbiol 32:547–556. doi: 10.1046/j.1365-2958.1999.01393.x. [DOI] [PubMed] [Google Scholar]
- 49.Sanglard D, Ischer F, Monod M, Bille J. 1996. Susceptibilities of Candida albicans multidrug transporter mutants to various antifungal agents and other metabolic inhibitors. Antimicrob Agents Chemother 40:2300–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gillum AM, Tsay EYH, Kirsch DR. 1984. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and Escherichia coli pyrF mutations. Mol Gen Genet 198:179–182. doi: 10.1007/BF00328721. [DOI] [PubMed] [Google Scholar]