

Abstract
Doravirine (DOR, formerly known as MK-1439) is a human immunodeficiency type 1 virus (HIV-1) nonnucleoside reverse transcriptase inhibitor (NNRTI) that is currently in phase 2b clinical trials. In vitro resistance selection of subtype B virus (MT4-green fluorescent protein [GFP] cells), as well as subtype A and C viruses (MT4-GFP/CCR5 cells) was conducted with DOR, rilpivirine (RPV), and efavirine (EFV) under low-multiplicity-of-infection conditions in a 96-well format. Resistance selection was performed with escalating concentrations of the NNRTIs ranging from the 95% effective concentration (1× EC95) to 1,000× EC95 in the presence of 10% fetal bovine serum. In the resistance selection of subtype B virus with DOR, a V106A mutant virus led to two mutation pathways, followed by the emergence separately of either F227L or L234I. In the resistance selection of subtype A and C viruses, similar mutation development pathways were detected, in which a V106A or V106M mutant was also the starting virus in the pathways. Mutations that are commonly associated with RPV and EFV in clinical settings were also identified in subtype B viruses such as the E138K and K103N mutants, respectively, in this in vitro resistance selection study. The susceptibility of subtype B mutant viruses selected by DOR, RPV, and EFV to NNRTIs was evaluated. Results suggest that mutant viruses selected by DOR are susceptible to RPV and EFV and mutants selected by RPV and EFV are susceptible to DOR. When the replication capacity of the V106A mutant was compared with that of the wild-type (WT) virus, the mutant virus was 4-fold less fit than the WT virus.
INTRODUCTION
Human immunodeficiency type 1 virus (HIV-1) infection has become a global epidemic, as there are more than 35 million people worldwide who are infected with HIV-1 and approximately 2.3 million people were newly infected in 2012 on the basis of the Joint United Nations Programme on HIV/AIDS 2013 report (1). HIV-1-infected patients have been successfully treated with highly active antiretroviral therapy (HAART). HAART regimens generally comprise three agents from at least two different mechanistic classes. Although HAART effectively controls the progress of disease and restores the immune function of patients, the treatment does not eradiate the virus. As a result, lifelong treatment must be maintained, which may lead to therapy fatigue and to medication noncompliance because of adverse effects. Under these circumstances, resistant viruses often emerge and lead to treatment failure. Consequently, mutant viruses with reduced susceptibility to the licensed agents remain a constant threat to HAART.
Reverse transcriptase (RT) plays an essential role in the HIV-1 replication cycle, converting single-stranded viral RNA into double-stranded DNA that is then integrated into the host cell genome by HIV-1 integrase for subsequent transcription and translation processes to generate infectious viral particles (2). As a result, inhibition of RT is one of the primary strategies used to suppress HIV-1 replication (3). Two different classes of RT inhibitors are available for treating HIV-1 infection. The first class is nucleoside RT inhibitors (NRTIs) which are active-site inhibitors and act as chain terminators of DNA polymerization catalyzed by RT because of the lack of a 3′ hydroxyl group for chain extension. The other class is non-NRTIs (NNRTIs), which bind to an exo site of RT, causing significant conformational changes within RT and leading to inhibition of the enzyme. Current HARRT for treating HIV-1-infected patients often contains an NNRTI and two NRTIs, such as Atripla (efavirenz [EFV]-tenofovir disproxil fumarate [TDF]-emtricitabine [FTC]) and Complera (rilpivirine [RPV]-TDF-FTC).
To date, there are five NNRTIs available for use in combination with other classes of antiviral agents to effectively suppress HIV-1 replication (4, 5). Nevirapine (NVP) was approved in 1996, and it has to be taken twice a day with food. In 2011, an extended-release version of NVP with once-a-day dosing was approved by the FDA. NVP has been associated with hepatotoxicity, substantial skin reactions (including Stevens-Johnson syndrome), hypersensitivity reactions, and, in some studies, excess rates of early discontinuation or inferior efficacy (6). Delavirdine (DLV) was approved in 1997, and it must be taken three times daily and is no longer among the recommended agents for initial therapy. EFV was approved in 1998, and it remains the preferred NNRTI for treatment initiation according to multiple guidelines. However, EFV is associated with substantial central nervous system intolerance and skin rash, as well as lipid abnormalities (7, 8). In addition, it can be a perpetrator of drug-drug interactions as it is a mixed inducer and inhibitor of CYP3A and CYP2B6 enzymes. Etravirine (ETR) was approved in 2008. It must be given twice daily and is not recommended as part of an initial treatment regimen. RPV was approved in 2011 and has been recommended by these guidelines as an alternative NNRTI agent for treatment initiation. RPV was shown to have suboptimal efficacy in patients with viral loads of >100,000 copies/ml or CD4+ cell counts below 200 copies/ml at the baseline (9, 10). In addition, the E138K substitution, accounting for the majority of mutants in patients who have experienced RPV-containing regimen failure, was found to be associated with the most prevalent NRTI mutants containing M184V/I substitutions (11–13).
A single mutation was often found to confer high-level resistance to the antiviral agents in the same class. For example, a virus with the K103N substitution exhibits a high level of resistance to EFV, NVP, and DLV. A Y181C mutant is highly resistant to NVP and DLV and displays a low level (∼4-fold) of resistance to RPV and ETR (11). In the clinical setting, a Y181C mutant has also been associated with virologic failures of regimens containing NVP, RPV, or ETR (10, 14). Moreover, both K103N and Y181C mutants are the two most prevalent NNRTI-associated, as well as transmitted, viruses (15, 16). Therefore, new NNRTIs that offer improved potency against the two mutants, a distinct resistance profile, dosing convenience, and a favorable safety and tolerability profile would provide HIV-1 patients with an additional choice for effective treatment. To this end, a novel NNRTI, doravirine (DOR), was identified as a potential new antiviral agent. DOR displayed efficacy against the wild-type (WT) virus, as well as the most common NNRTI-resistant variants such as the K103N and Y181C mutant viruses (11). The mutant profile of DOR is superior to that of EFV and comparable to that of RPV (11). In preclinical toxicity studies, no article-related findings were observed in either antemodem or postmodem analyses. Clinical pharmacology studies indicate that DOR can be taken once daily with or without food, which offers dosing convenience to patients (17). In addition, DOR is not a metabolic inducer or inhibitor, reducing the likelihood of causing significant drug-drug interactions (17).
To further characterize DOR, in vitro resistance selection was conducted to understand the potential mutation development pathways of the virus in the presence of DOR. There are at least nine different HIV-1 subtypes in the M group (A, B, C, D, F, G, H, J, and K), and subtypes A and C are the most prevalent HIV globally (18). Subtype B virus, on the other hand, is predominant in North America and Europe. Therefore, DOR resistance selection was performed with HIV-1 subtypes A, B, and C under low-multiplicity-of-infection (MOI) conditions in the presence of 10% fetal bovine serum (FBS). In addition, two licensed NNRTIs, EFV and RPV, were also included in these studies as comparators. Resistance selection with subtype B virus was also conducted under high-MOI conditions at fixed concentrations of the inhibitors in the presence of 10% FBS (19). Mutants selected under high-MOI conditions are consistent with the results obtained under low-MOI conditions. The susceptibility of the selected mutant viruses to NNRTIs was evaluated under single replication cycle conditions. The results suggest that the mutants selected by DOR were susceptible to RPV and EFV and the mutants selected by RPV and EFV were susceptible to DOR.
MATERIALS AND METHODS
The cells used (293T) were from the ATCC (Manassas, VA). Fugene HD transfection reagent was bought from Promega (Madison, WI). Dulbecco's modified Eagle medium was purchased from Life Technology (Grand Island, NY). Fetal bovine serum was obtained from HyClone (Logan, UT). G418 and hygromycin were from Life Technology (Grand Island, NY). pQBILTRGagGEPNeo was purchased from MP Biomedicals (Santa Ana, CA). The Bravo liquid-handling station used came from Agilent Technologies (Santa Clara, CA). The Acumen eX3 cytometer was bought from TTP Labtech (Cambridge, MA). MagMAX Express 96, the MagMAX 96 viral RNA isolation kit, RPMI medium, the Superscript III one-step RT-PCR system, and the TOPO TA cloning system for sequencing were obtained from Life Technologies (Grand Island, NY). Black and clear 96-well poly-d-lysine-coated or noncoated plates were purchased from BD Biosciences (San Jose, CA). The SeqScape software V3 and ABI 3100 analyzer used were from Applied Biosystems (Foster City, CA). The WT and K103N and Y181C mutant HIV-1 R8 strains were produced by Advanced Biotechnologies Inc. (Columbia, MD). RPMI 1640 medium were purchased from Invitrogen (Carlsbad, CA).
Generation of mutant viruses.
Each mutant was created by the site-directed mutagenesis method via gene synthesis and subcloning into plasmid RT112, which contained full-length R8 provirus DNA. The resulting clones were subjected to fluorescence sequencing to verify the presence of the desired mutation(s) and the absence of extraneous mutations.
Cells and viruses.
Cells (293T) were grown in Dulbecco's modified Eagle medium containing 10% FBS. MT4-green fluorescent protein (GFP) cells were grown in RPMI 1640 medium containing 10% FBS and 0.4 mg/ml G418; MT4-GFP/CCR5 cells were grown in RPMI 1640 medium containing 10% FBS, 0.4 mg/ml G418, and 0.4 mg/ml hygromycin.
293T cells were seeded at 2.5 × 106 per well of a 10-cm-diameter dish. After incubation for 24 h, the cells were transfected with 18 µg of provirus plasmid DNA using Fugene HD transfection reagent. The supernatant was harvested at 48 h posttransfection. Each mutant virus was evaluated for of MT4-GFP reporter cell infectivity.
Development of MT4-GFP/CCR5 reporter cells.
MT4 cells were transfected with pQBILTRGagGEPNeo, in which Gag-fused GFP was expressed under the control of the long terminal repeat (LTR) promoter with G418 as a selectable marker. A stably expressing cell clone was obtained by G418 drug selection and subsequent cell sorting to isolate single Gag-GFP-expressing cells (20). MT4-GFP cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 1% Pen-Strep, and 0.4 mg/ml G418 and incubated in a humidified incubator with 5% CO2 at 37°C.
MT4-GFP/CCR5 cells were generated by introducing stably expressed CCR5 genes into MT4-GFP cells. A single MT4-GFP/CCR5 cell clone was selected by the hygromycin and subsequent cell sorting method. The best final cell clone was determined on the basis of the robustness of the fluorescent signal after infection with both M- and T-tropic virus assays and was further validated by assessing compound potency with the selected cell clones in a single replication cycle assay. MT4-GFP/CCR5 cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 1% Pen-Strep, 0.4 mg/ml G418, and 0.4 mg/ml hygromycin and then incubated in a humidified incubator with 5% CO2 at 37°C.
In vitro resistance selection with HIV-1 subtype A, B, and C viruses under low-MOI conditions.
In vitro resistance selection was performed in a 96-well format with each row containing a series of concentrations of NNRTIs in 1:2 serial dilutions, with the highest concentration (1,000 times the 95% effective concentration [1,000× EC95]) in column 1 and the lowest (1× EC95) in column 11. Column 12 served as a virus control with no compound. Therefore, each row represented an independent resistance selection experiment. Cells (MT4-GFP/CCR5 for A and C viruses and MT4-GFP for subtype B virus; cell density, 75,000/well) and viruses (subtype A [92W026], B [R8], or C [93MW959], each at an MOI of 0.01) were added to 96-well plates containing serial dilutions of NNRTIs.
After 3 to 4 days of culturing, a new selection cycle was initiated. The procedure used was as follows. A preprepared compound plate containing NNRTI concentrations as described above was thawed; this was followed by the addition of fresh MT4-GFP cells (with or without a CCR5 coreceptor, depending on the virus subtype). Supernatant (30 μl) was removed from each well of the plate from the previous selection cycle and split into three 10-μl portions. These three portions were used to infect the freshly prepared cells in the wells at NNRTI concentrations 1, 2, and 4 times the concentration in the original well from which the supernatant was removed. This procedure was repeated every 3 to 4 days on a Bravo liquid-handling station. For every passage, the plates were scanned with an Acumen eX3 cytometer to monitor the level of viral replication. When evidence of virus replication was observed via detection of green fluorescence in the wells of column 1 (with the highest concentration of NNRTIs), the supernatant was removed from the plate and used for genotyping analysis.
Resistance selection with HIV-1 subtype B virus using MT4-GFP cells under high-MOI conditions.
The resistance selection protocol used is similar to that employed for selection under low-MOI conditions, except that the selection pressure was fixed at 3, 10, or 30 times the EC95 of the NNRTI with no distribution of supernatant from the low-concentration wells to the higher-concentration wells during each passage. Viral breakthrough was judged by the appearance of green fluorescence in wells upon scanning with the Acumen eX3 cytometer.
Analysis of RT mutation(s) in the breakthrough viruses from the resistance selection studies.
Viral RNA was extracted with the MagMAX 96 viral RNA isolation kit from culture supernatant of breakthrough virus stock from the resistance studies described above. The RT-encoding region was amplified by the one-step RT-PCR method. PCR products were genotyped by an automated population-based full-length sequencing method (covering amino acids 1 to 440 of the RT region). The primers used for PCR amplification of subtype B virus were AAGCAGGAGCCGATAGACAA (forward) and TAATCCCGAATCCTGCAAAGCTAGA (reverse). The sequencing primers were 5′-CCCTGTGGAAGCAC and 5′-GGATGTGGGCGATGC. The amplification primers used for subtype A sequencing were AGGCTATAGGTACAGTATTAGTAGGACCTAC (forward) and TGTTCAGCTTGATCCCTTACCTG (reverse). Primers GACCTACACCTGTCAACATAATT, TGTCTTCCTCTGTCAGTAACATAC, GGAAAGGATCACCGGCAATA, and TTGCTCTATGCTGCCCTATTT were used for sequencing analysis. The primers used in PCR amplification for subtype C sequencing were GACACAGGAGCAGATGATACAG (forward) and AGCACTTTCCTGATTCCACTAC (reverse). Primers ACCTGTCAACATAATTGGAAGAAAT, TTCTGCCTTCCTTTGTCAGTAA, GATGGAAAGGATCACCAGCA, and GCTCTATGTTGCCCTATTTCTAAG were used for sequence analysis. Sequencing results were reported as amino acid changes compared with WT HIV-1 92RW026 (subtype A), R8 (subtype B), and 93MW959 (subtype C) reference sequences.
Relative replication capacity of the V106A mutant.
The relative replication capacity of the V106A mutant was measured in a growth competition experiment. MT4-GFP cells (2.5 × 106) were infected with either WT or V106A mutant virus at an estimated MOI of 0.001 in a final volume of 5 ml at 37°C in 5% CO2 for 1 h. Cells were then washed, counted, and resuspended in complete RPMI medium, and various ratios of WT and V106A mutant virus-infected cells were mixed and added to a 12-well cell culture plate at a cell density of 0.2 × 106 per well. After incubation for 3 to 4 days, 10 μl of culture supernatant was used to infect 2 × 106 fresh MT4-GFP cells in the plates. At each passage, 50 μl of supernatant from each well was harvested for population or clonal sequence analysis. For clonal sequence analysis, RT-PCR products were cloned into the TOPO TA vector and then transformed into Top10 competent cells; 30 bacterial colonies from each sample were analyzed. The relative prevalence of WT and mutant viruses at each passage was quantitated on the basis of the clonal sequence proportion of the viruses. The experiments were performed in triplicate.
RESULTS
DOR displays efficacy against the WT virus, as well as the two most prevalent NNRTI-associated mutant viruses containing K103N and Y181C substitutions, with EC95s of 20, 42, and 27 nM in the presence of 50% NHS, respectively (11). These potencies are more than 30-fold better than that of EFV against the K103N mutant virus and more than 9- and 4-fold better than those of ETR and RPV versus the Y181C mutant virus, respectively (11).
To evaluate potential mutations that would be selected by DOR in vitro with HIV-1 subtype A, B, and C viruses, resistance selection experiments were conducted in a 96-well format using MT4-GFP cells (for subtype B virus) with the CCR5 coreceptor (for subtype A and C viruses), which is controlled by transcription of the LTR to express GFP. Each well of the plate contained different concentrations of the compounds under investigation, ranging from a low compound concentration (column 11) to a high compound concentration (column 1). Under low-MOI conditions, as described in Materials and Methods, supernatant from wells with a lower concentration of DOR was distributed to wells containing a higher concentration of the compound during each passage. As a result, a small portion of the mutant viruses that broke through at the lower concentration of DOR would be transferred to wells containing higher concentrations of the compound, facilitating viral breakthrough even under higher selective pressure. By this strategy, viral breakthrough was observed with compound concentrations as high as 1,000× EC95. The plates were scanned with the Acumen eX3 cytometer after each passage during the resistance selection process. Detection of green fluorescence was indicative of viral breakthrough and was found to progress gradually from column 11 to column 1 as viral breakthrough started from column 11, which contained the lowest concentration of NNRTI (1× EC95), as shown in Fig. 1. Breakthrough viruses were harvested and subjected to population sequence analyses after green fluorescence was observed on the entire plate, as shown in Fig. 1 at day 81. Mutation data were collected from two independent experiments with a total of 16 repeats (two plates, eight rows each).
FIG 1.

The progress of viral breakthrough from low to high concentrations of compounds. Compound concentrations escalate from column 11 (lowest) to column 1 (highest). Panels: A, EFV; B, RPV; C, DOR. Gray suggests that there is no viral breakthrough. Black indicates that there may be a minor portion of breakthrough viruses but the green fluorescence is too weak to be detected. Dark green indicates a high level of viral breakthrough. Light green indicates a medium level of viral breakthrough or high level of viral breakthrough causing some extent of cell death.
Resistance selection in subtype B virus.
The results of resistance selection with subtype B virus are shown in Table 1. Two major mutation development pathways were associated with DOR exposure in this study. V106A was the first mutant identified in breakthrough viruses at a lower concentration of DOR. As the compound concentration increased, an additional amino acid substitution emerged, such as F227L or L234I, to give a V106A/F227L or V106A/L234I double mutant. As the concentration increased further, an additional F227L substitution evolved in the V106A/L234I mutant to give a triple mutant that overcame the selection pressure. Other minor mutation development pathways were also observed in the selection with DOR, such as starting with a V108I mutant, followed by the emergence of V106A and L234I substitutions sequentially, or starting with an L234I mutant virus, followed by the sequential addition of V108I and V106A substitutions.
TABLE 1.
Resistance selection with DOR, EFV, and RPV in subtype B virus using MT4-GFP cells in the presence 10% FBS
| NNRTI | Pathway | Evolution of mutants as NNRTI concn increasesa |
||
|---|---|---|---|---|
| DOR | 1 | V106A → V106A/F227L | ||
| (9.6 ± 1.8) (>2,000) | ||||
| 2 | V106A → V106A/L234I → V106A/L234I/F227L | |||
| (9.6 ± 1.8) (173 ± 17.5) (>2,000) | ||||
| EFV | 1 | L100I → L100I/K103N | ||
| (16 ± 0.6) (>2,000) | ||||
| 2 | L100I → L100I/V179D → L100I/V179D/P225H or M230L | |||
| (16 ± 0.6) (76 ± 15) (465 ± 127) | ||||
| 3 | K103N → K103N/L100I | |||
| (58 ± 13) (>2,000) | ||||
| RPV | 1 | E138K → E138K/L100I → E138K/L100I/V179I | ||
| (3.8 ± 0.6) (2.0 ± 0.01) (8.7 ± 3.8) | ||||
| 2 | E138K → E138K/V106A | |||
| (3.8 ± 0.6) (8.1 ± 2.1) | ||||
| 3 | K101P → K101P/V179I | |||
| (40 ± 18) (199 ± 40) | ||||
Experiments were conducted at least three times. Values in parentheses indicate fold resistance (means ± standard deviation) of the mutants.
As discussed above, the V106A mutant is the major variant responsible for viral breakthrough in resistance selection with DOR in subtype B viruses. Therefore, the replication capability of the V106A mutant was compared with that of the WT virus to evaluate the potential for V106A to revert to the WT in the absence of drug pressure. Several methods can be used to assess viral replication capability (21). In this study, the approach of coculturing mutant and WT viruses, following by clonal sequencing of the supernatant at each passage, was used. The replication capacity of the V106A mutant was assessed by culturing a fixed ratio of WT and V106A mutant viruses on the same plate (22). The supernatant was removed for clonal sequencing to determine the percentage of each virus at different time points. As shown in Fig. 2, the percentage of V106A mutant versus WT virus continued to decrease as the passage number increased. With this trend, the V106A mutant will eventually be replaced by the WT virus. The replication capacity of the V106A virus was calculated according to the equation 1 + s = 1 + ln[(Mt/M0)/(Rt/R0)] [1] (22).
FIG 2.
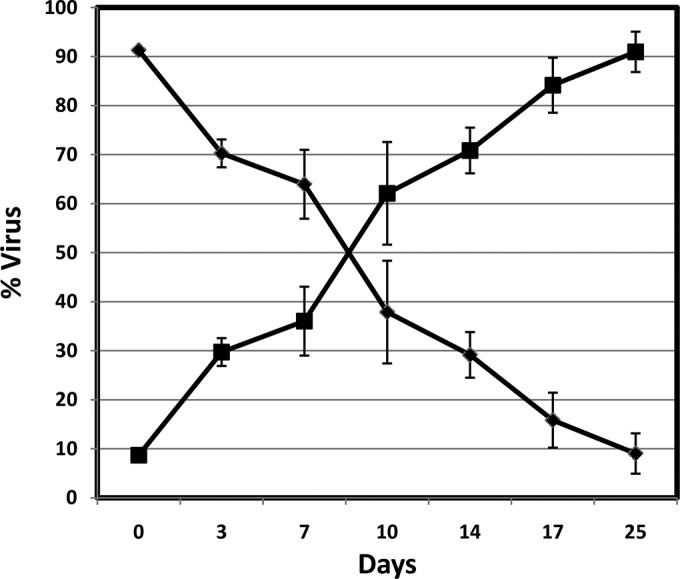
Relative prevalence of WT and V106A mutant viruses in cultures from the replication capacity study. Solid diamonds represent the V106A mutant virus, and solid squares represent the WT virus. The experiment was conducted in triplicate, and the error bars represent standard deviations.
The term 1 + s is the relative fitness or difference (n-fold) between the growth constants of the WT and mutant strains. Rt is the proportion of the WT strain at time t, Mt is the proportion of the V106A mutant at time t, R0 is the proportion of the WT strain at time zero, and M0 is the proportion of the V106A mutant at time zero. The results suggest that the replication capacity of the V106A mutant is 25% of that of the WT virus, which is consistent with previous reports indicating that the V106A mutant's fitness is significantly compromised compared with that of the WT virus (23). As a result, the V106A mutant may not be able to survive under the high selective pressure of DOR.
Resistance selection with the benchmark compound EFV was also conducted under the same conditions for comparison. As shown in Table 1, three mutation development pathways were identified. Two of them started with the L100I mutant, followed by an additional K103N or V179D substitution. The L100I/K103N-containing virus was highly resistant to EFV, with a fold change (FC) in potency relative to the WT virus of >200-fold. On the other hand, the L100I/V179D mutant, with an FC of 75 to EFV, requires an additional mutation to overcome the selective pressure as the DOR concentration is increased to 160× EC95, rendering the emergence of the P225H or M230L substitution to give a triple mutant (L100I/V179D/P225H or L100I/V179D/M230L) to circumvent the selection pressure. The third mutation pathway was initiated by a K103N mutant followed by the addition of an L100I substitution to give the K103N/L100I double mutant, which is similar to the first mutation development pathway.
Three mutation development pathways were also identified in the resistance selection with another licensed NNRTI, RPV. The first pathway was initiated with the E138K substitution, followed by the emergence of an L100I substitution. Upon a further increase in the RPV concentration, a triple mutant (E138K/L100I/V179I) breakthrough virus was identified. The second pathway was also started with an E138K mutant, followed by the addition of the V106A substitution under higher selective pressure. K101P mutant was the first mutant observed in the third mutation pathway, and a V179I substitution was found to evolve subsequently at higher concentrations of RPV.
Resistance selection in subtype B virus with MT4-GFP cells was also conducted under high-MOI conditions at DOR and EFV concentrations of 3, 10, and 30 times the EC95. With DOR, most of the viral breakthroughs (n = 24) were observed at only 3 times the EC95. Population sequencing analysis indicated that the mutations selected under these conditions are similar to those observed under low-MOI conditions. In other words, the most frequently identified mutant viruses contained mutations that gave the V106A, L234I, F227L, and V108I substitutions. Although the V108I mutation was not listed as the major mutation pathway under low-MOI conditions, it was involved in several minor mutation pathways. Similarly, the majority of the viral breakthroughs occurred at 3 times the EC95 in the selection with EFV. The mutations selected under these conditions are also consistent with the results obtained under low-MOI conditions, in which K103N, L100I, and V179D were the major mutants identified in the breakthrough viruses. The same conditions were employed for resistance selection with RPV. Only a few viral breakthroughs were observed at 3 times the EC95. The amino acid substitutions were found to be K101E, E138K, and Y181C, and they are commonly associated with viruses found in patients who have experienced virologic failure while on a regimen containing RPV (24).
Resistance selection in subtype A virus.
In the resistance selection of subtype A virus with DOR using MT4-GFP/CCR5 cells, the major mutation pathway started with the V106A mutant at a lower concentration of the compound (a small portion of the breakthrough viruses had the V106M substitution), followed by the development of the F227L mutant (a small portion of the breakthrough viruses had a C or V substitution). A minor mutation pathway also emerged in this selection study with V108I as the first mutant identified in the breakthrough virus. As the compound concentration increased, the L234I and V106A(I) substitutions sequentially developed to give a triple mutant [V108I/L234I/V106A(I)]. The results are shown in Table 2. In the resistance selection with EFV, there were also two mutation pathways identified, in which the L100I and V106M mutants led each of the pathways, followed by the emergence of Y188H(C) or V179D and Y188C or L100I substitutions, respectively. For resistance selection with RPV, the major mutation pathway was led by the E138K mutant with the emergence of an L100I substitution at a higher concentration of RPV. This pathway is similar to the pathway detected in the selection with subtype B virus. However, as the RPV concentration continued to increase, an additional substitution that was different from the pathway in subtype B virus emerged to give the triple mutant E138K/L100I/V108I, as opposed to E138K/L100I/V179I with subtype B virus. A minor pathway was started with the Y181C mutant with the emergence of a V108I substitution at a higher concentration of RPV.
TABLE 2.
Resistance selection with DOR, EFV, and RPV in subtype A virus using MT4-GFP/CCR5 cells in the presence 10% FBS
| NNRTI | Pathway | Evolution of mutants as NNRTI concn increasesa | Population |
|---|---|---|---|
| DOR | 1 | V106A(M) → V106A(M)/F227L(C/V) | Major |
| 2 | V108I → V108I/L234I → V108I/L234I/V106A(I) | Minor | |
| EFV | 1 | L100I → L100I/Y188H(C) or V179D | |
| 2 | L106 M → L106 M/Y188C or L100I | ||
| RPV | 1 | E138K → E138K/L100I → E138K/L100I/V108I | Major |
| 2 | Y188C → Y181C/V108I | Minor |
An amino acid in the parentheses represents a minor population.
Resistance selection in subtype C virus.
The resistance selection results obtained with subtype C virus are shown in Table 3. Two mutation pathways were identified in the selection with DOR. The V106A mutant again led the first selection pathway, followed by the emergence of an F227I substitution. The other pathway was led by the V106M mutant, followed by the addition of an F227C substitution. As for the results obtained with EFV, they were similar to those obtained from the resistance selection with subtype A virus, where mutants L100I and V106M led the two mutation pathways. The L100I mutant was followed by sequential emergence of V106M and Y188C (or F227C) substitutions at higher concentrations of EFV instead of the Y188H or V179D substitution obtained with subtype A virus. The V106M pathway was followed by a Y188C substitution as the compound concentration increased. Unique mutation development pathways were discovered in the resistance selection experiments with RPV in subtype C virus. Although the common mutation E138K was found to be associated with RPV, the subsequent mutation gave a unique double mutation, E138K/K101E. In spite of the E138K or K101E single mutation being generally associated with RPV, the double mutant has not been identified before. E138 and K101 form a salt bridge in the RT region of the WT virus, and either an E138K or a K101E substitution breaks the salt bridge, conferring resistance to RPV. The E138K/K101E substitutions, however, may restore the salt bridge formation and thus restore sensitivity to RPV. More studies are required to understand the impact of this double substitution on viral replication capacity and susceptibility to NNRTIs. In addition, the L100V/M230I double mutant was identified in the breakthrough viruses also representing a unique mutation development pathway because the L100I and M230L substitutions are more common at these positions. As the compound concentration increased, an additional amino acid change of F227C emerged to give a triple mutant. The third mutation pathway started with mutant E138Q, which is also not a usual substitution at this position, followed by the emergence of L100I.
TABLE 3.
Resistance selection with DOR, EFV, and RPV in subtype C virus using MT4-GFP/CCR5 cells in the presence 10% FBS
| NNRTI | Pathway | Evolution of mutants as NNRTI concn increases |
|---|---|---|
| DOR | 1 | V106A → V106A/F227I |
| 2 | V106 M → V106 M/F227C | |
| EFV | 1 | L100I → L100I/V106 M → L100I/V106 M/Y188C |
| or F227C | ||
| 2 | V106 M → V106 M/Y188C | |
| RPV | 1 | E138K → E138K/K101E |
| 2 | L100V/M230I → L100V/M230I/F227C | |
| 3 | E138Q → E138Q/L100I |
Susceptibility of mutants selected in subtype B virus to NNRTIs.
In the process of developing an antiviral agent, it is important to understand the cross-resistance profile of a novel compound with other licensed antiviral agents in the same class. Therefore, mutants selected by DOR, RPV, and EFV in this study were generated via site-directed mutagenesis and tested for their susceptibility to the three NNRTIs. As shown in Fig. 3A, the V106A mutant displayed an approximately 10-fold resistance to DOR. In addition, the double substitutions V106A/F227L and V106A/L234I and the triple substitution V106A/L234I/V108I conferred >150-fold resistance to DOR. However, the mutants were susceptible to RPV and EFV, with an FC of <6, except for theV106A/F227L mutant, which showed a 22-fold resistance to EFV. The susceptibilities of mutants selected by EFV to NNRTIs are shown in Fig. 3B. The L100I and K103N mutants displayed 10- and 56-fold resistance to EFV, respectively. The L100I/V179D double mutant exhibited a >75-fold resistance to EFV, and the level of resistance of the L100I/K103N and L100I/V179D/P225H mutants to EFV was >200-fold. However, those mutants are highly susceptible to DOR, with an FC of ≤2. They also had a low level of resistance to RPV, except for the L100I/K103N mutant, which displayed a 16-fold resistance to RPV. Figure 3C shows the susceptibilities of mutants selected by RPV to NNRTIs. Interestingly, although they were selected by RPV, most of the mutants were <10-fold resistant to RPV. The K101P and K101P/V179I mutants, on the other hand, were highly resistant to RPV, with FCs of 40 and 199, respectively. The mutant viruses also showed 32- and 76-fold resistance to EFV. In contrast, all of the mutants selected by RPV were susceptible to DOR with an FC of <10. In summary, mutants selected by DOR are susceptible to RPV and EFV and mutants selected by RPV and EFV are susceptible to DOR.
FIG 3.

Susceptibilities of selected mutant viruses to NNRTIs. (A) Mutants selected by DOR. (B) Mutants selected by EFV. (C) Mutants selected by RPV. The x axis shows the sequential appearance of mutant viruses selected by the NNRTIs.
DISCUSSION
Among NNRTIs, DOR displays the best potency against the two most prevalent NNRTI-associated mutants containing K103N and Y181C substitutions (11). On the basis of the X-ray structure of the RT-DOR complex, the nitrogen atoms in the methyl-triazolone ring interact with the backbone, but not with the side chain, of K103 via two hydrogen bonds, as shown in Fig. 4. As a result, the impact of the K103N substitution on the interactions between the residue and DOR is minimal, as the interactions between N103 and the DOR triazole ring remain intact. Y181 does not play an important role in the binding of DOR to RT, given the long distance between the cyanochlorophenol group of DOR and Y181 based on the X-ray structure. Therefore, it is not surprising that the Y181C mutation does not confer high resistance to DOR, with an FC of <2, likely explaining the lack of selection of this mutant in resistance selection experiments. In contrast, the cyanochlorophenol moiety forms π-π stacking with Y188, and the Y188L substitution eliminates the π-π interactions and creates a clash with DOR; thus, the Y188L substitution confers significant resistance to DOR, suggesting that the π-π interactions between the residues are crucial for the binding of DOR to RT.
FIG 4.
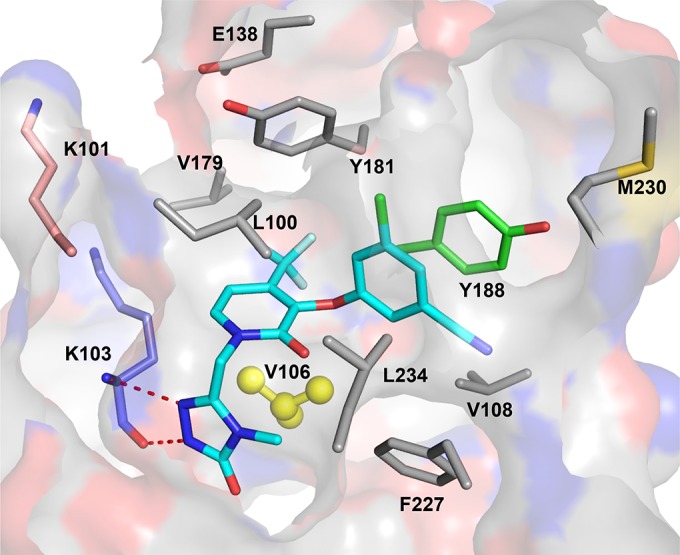
X-ray structure of the RT-DOR complex. Residues that are selected by DOR during resistance selection are shown. A red dashed line indicates the formation of a hydrogen bond between DOR and residue K103. Highlighted in yellow is V106, which is the major residue involved in the development of resistance to DOR.
As mentioned above, the V106 substitution accounts for the majority of the mutants selected by DOR in the in vitro resistance selection study. The side chain of V106 is close to the central ring of DOR, with a distance of 3.5 Å based on the X-ray structure shown in Fig. 4. It appears that the isopropyl group on the Val side chain makes van der Waals interactions with DOR in the NNRTI binding pocket. Replacement of the isopropyl group with a methyl group (V106A) weakens the interactions between RT and DOR, rendering 10-fold resistance of the virus to DOR. Interestingly, substitutions at position V106 were often found to be associated with substitution at F227 as the concentration of DOR increased in the resistance selection with subtypes A, B, and C. Although the F227 residue has a limited interactions with the triazole ring of DOR, it is in close proximity to V106, at a distance of 3.5 Å based on the X-ray structure. Therefore, residue substitution at position 227 may, in turn, alter the interactions between the residue at 227 and V106A, causing conformation changes of the latter, which may further reduce the interactions between the V106A mutant and DOR, causing significant resistance to DOR.
The amino acid at position 106 is a valine in the WT virus. GTA represents the most common codon at the position (>95%) in subtype A and B viruses, and GTG is the major codon in subtype C virus at the position (97%) (25). The V106M mutant confers a high level of resistance to EFV, and the substitution is a signature mutation associated with EFV in subtype C virus in the clinical setting (26). However, although the V106M mutant confers a high level of resistance to EFV, the mutant was not associated with EFV in subtype B virus because two base changes are required for the substitution (from GTA to ATG) (27). In contrast, only a single base change is needed for the V106M substitution in subtype C virus (GTG to ATG). Interestingly, the codon at position 106 was found to be a mixture of GTA and GTG in the WT subtype A virus (92RW026) that we used in this study. As a result, in the resistance selection with EFV, V106M was selected in the subtype A virus because the V106M mutant displays a high level of resistance to EFV and required only a single base change for the conversion (from GTG to ATG) (22). The V106A mutant was not selected, as the virus is susceptible to EFV although it also requires only a single base change (GTG to GCG) (22). On the other hand, both the V106A and V106M mutants were identified in the breakthrough viruses from the resistance selection with DOR in subtype A and C viruses. As mentioned earlier, the V106A mutant of subtype B virus exhibits an approximately 10-fold resistance to DOR. It is reasonable to hypothesize that the V106A and V106M mutations in subtype A and C viruses also confer a similar level of resistance to DOR, enabling the virus to survive under the high selective pressure of DOR.
In the in vitro resistance selection study with EFV, the K103N substitution, which represents the most prevalent mutant associated with the NNRTI, was selected in one of the three mutation pathways. In addition, L100I was the lead mutant in two out of three mutation pathways followed by the emergence of a K103N or V179D substitution at a higher concentration of EFV. The L100I/K103N mutant is one of the frequent double mutants associated with EFV (28). Although two out of three mutation pathways started with the L100I mutant, that substitution is rarely identified as the only change in samples from patients treated with an EFV-containing regimen who experience virologic failure (29). It always appears along with K103N substitution. Part of the reason why the L100I mutant is not seen in the clinical setting may be the high trough concentration of EFV (∼5 μM) in plasma at 24 h after dosing, which is more than enough to suppress an L100I mutant, thereby requiring an additional mutation that gives a L100I/K103N double mutant to overcome the selective pressure. In addition, although the V179D mutant is not listed as the prevalent NNRTI-associated virus, it has often emerged with other mutants under the selective pressure of EFV (30). Moreover, in the resistance selection with RPV, the E138K substitution represented the first substitution in the two out of three mutation pathways, which is consistent with the finding that the E138K mutant was the most frequently selected mutant in a clinical setting, although it was often associated with M184I/V (9, 10). In addition, the K101P mutant, which is also often associated with RPV in clinical findings, was also identified leading the third mutation pathway in breakthrough viruses (31). Therefore, results from in vitro resistance selection, to some extent, can predict the potential mutations that may develop in the clinical setting.
As aforementioned, the mutants selected by DOR were susceptible to RPV and EFV and mutants selected by RPV, as well as EFV, were susceptible to DOR. These results suggest that DOR can be used to treat naive patients because other NNRTIs are still capable of suppressing the mutant viruses that cause treatment failure under a DOR regimen. Furthermore, DOR may be used to treat experienced patients because DOR is highly active against mutants selected by RPV and EFV. As a result, DOR can be a valuable addition to the currently available NNRTI antiviral agents.
REFERENCES
- 1.Joint United Nations Programme on HIV/AIDS. 2013. Global report: UNAIDS report on the global AIDS epidemic 2013. World Health Organization, Geneva, Switzerland: http://www.unaids.org/sites/default/files/en/media/unaids/contentassets/documents/epidemiology/2013/gr2013/UNAIDS_Global_Report_2013_en.pdf. [Google Scholar]
- 2.Götte M, Li X, Wainberg MA. 1999. HIV-1 reverse transcription: a brief overview focused on structure-function relationships among molecules involved in initiation of the reaction. Arch Biochem Biophys 365:199–210. doi: 10.1006/abbi.1999.1209. [DOI] [PubMed] [Google Scholar]
- 3.Castro HC, Loureiro NI, Pujol-Luz M, Souza AM, Albuquerque MG, Santos DO, Cabral LM, Frugulhetti IC, Rodrigues CR. 2006. HIV-1 reverse transcriptase: a therapeutical target in the spotlight. Curr Med Chem 13:313–324. doi: 10.2174/092986706775476089. [DOI] [PubMed] [Google Scholar]
- 4.Jayaweera D, Dilanchian P. 2012. New therapeutic landscape of NNRTIs for treatment of HIV: a look at recent data. Expert Opin Pharmacother 13:2601–2612. doi: 10.1517/14656566.2012.742506. [DOI] [PubMed] [Google Scholar]
- 5.de Béthune MP. 2010. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: a review of the last 20 years (1989-2009). Antiviral Res 85:75–90. doi: 10.1016/j.antiviral.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Taiwo BO. 2006. Nevirapine toxicity. Int J STD AIDS 17:364–369. doi: 10.1258/095646206777323346. [DOI] [PubMed] [Google Scholar]
- 7.Maggiolo F. 2009. Efavirenz: a decade of clinical experience in the treatment of HIV. J Antimicrob Chemother 64:910–928. doi: 10.1093/jac/dkp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vrouenraets SM, Wit FW, van Tongeren J, Lange JM. 2007. Efavirenz: a review. Expert Opin Pharmacother 8:851–871. doi: 10.1517/14656566.8.6.851. [DOI] [PubMed] [Google Scholar]
- 9.Cohen CJ, Andrade-Villanueva J, Clotet B, Fourie J, Johnson MA, Ruxrungtham K, Wu H, Zorrilla C, Crauwels H, Rimsky LT, Vanveggel S, Boven K. 2011. Rilpivirine versus efavirenz with two background nucleoside or nucleotide reverse transcriptase inhibitors in treatment-naive adults infected with HIV-1 (THRIVE): a phase 3, randomised, non-inferiority trial. Lancet 378:229–237. doi: 10.1016/S0140-6736(11)60983-5. [DOI] [PubMed] [Google Scholar]
- 10.Molina JM, Cahn P, Grinsztejn B, Lazzarin A, Mills A, Saag M, Supparatpinyo K, Walmsley S, Crauwels H, Rimsky LT, Vanveggel S, Boven K. 2011. Rilpivirine versus efavirenz with tenofovir and emtricitabine in treatment-naive adults infected with HIV-1 (ECHO): a phase 3 randomised double-blind active-controlled trial. Lancet 378:238–246. doi: 10.1016/S0140-6736(11)60936-7. [DOI] [PubMed] [Google Scholar]
- 11.Lai MT, Feng M, Falgueyret J P, Tawa P, Witmer M, DiStefano D, Li Y, Burch J, Sachs N, Lu M, Cauchon E, Campeau LC, Grobler J, Yan Y, Ducharme Y, Cote B, Asante-Appiah E, Hazuda DJ, Miller MD. 2014. In vitro characterization of MK-1439, a novel HIV-1 nonnucleoside reverse transcriptase inhibitor. Antimicrob Agents Chemother 58:1652–1663. doi: 10.1128/AAC.02403-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu HT, Asahchop EL, Oliveira M, Quashie PK, Quan Y, Brenner BG, Wainberg MA. 2011. Compensation by the E138K mutation in HIV-1 reverse transcriptase for deficits in viral replication capacity and enzyme processivity associated with the M184I/V mutations. J. Virol 85:11300–11308. doi: 10.1128/JVI.05584-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu HT, Colby-Germinario SP, Asahchop EL, Oliveira M, McCallum M, Schader SM, Han Y, Quan Y, Sarafianos SG, Wainberg MA. 2013. Effect of mutations at position E138 in HIV-1 reverse transcriptase and their interactions with the M184I mutation on defining patterns of resistance to nonnucleoside reverse transcriptase inhibitors rilpivirine and etravirine. Antimicrob Agents Chemother 57:3100–3109. doi: 10.1128/AAC.00348-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alcaro S, Alteri C, Artese A, Ceccherini-Silberstein F, Costa G, Ortuso F, Bertoli A, Forbici F, Santoro MM, Parrotta L, Flandre P, Masquelier B, Descamps D, Calvez V, Marcelin AG, Perno CF, Sing T, Svicher V. 2011. Docking analysis and resistance evaluation of clinically relevant mutations associated with the HIV-1 non-nucleoside reverse transcriptase inhibitors nevirapine, efavirenz and etravirine. ChemMedChem 6:2203–2213. doi: 10.1002/cmdc.201100362. [DOI] [PubMed] [Google Scholar]
- 15.D'Aquila RT, Schapiro JM, Brun-Vezinet F, Clotet B, Conway B, Demeter LM, Grant RM, Johnson VA, Kuritzkes DR, Loveday C, Shafer RW, Richman DD. 2003. Drug resistance mutations in HIV-1. Top HIV Med 11:92–96. [PubMed] [Google Scholar]
- 16.Johnson VA, Brun-Vezinet F, Clotet B, Conway B, D'Aquila RT, Demeter LM, Kuritzkes DR, Pillay D, Schapiro J M, Telenti A, Richman DD, International AIDS Society-USA Drug Resistance Mutations Group . 2003. Drug resistance mutations in HIV-1. Top HIV Med 11:215–221. [PubMed] [Google Scholar]
- 17.Morales-Ramirez JO, Gatell JM, Hagins DP, Thompson M, Arasteh K, Hoffmann C, Harvey C, Teppler H. Safety and antiviral effect of MK-1439, a novel NNRTI, (+Truvada) in ART-naive HIV infected patients.21st Conference on Retroviruses and Opportunistic Infections, Boston, MA 3 to 6 March 2014 National AIDS Treatment Advocacy Project, New York, NY http://www.natap.org/2014/CROI/croi_28.htm. [Google Scholar]
- 18.Takebe Y, Kusagawa YS, Motomura K. 2004. Molecular epidemiology of HIV: tracking AIDS pandemic. Pediatr Int 46:236–244. doi: 10.1046/j.1442-200x.2004.01869.x. [DOI] [PubMed] [Google Scholar]
- 19.Vingerhoets J, Azijn H, Fransen E, De Baere I, Smeulders L, Jochmans D, Andries K, Pauwels R, de Béthune MP. 2005. TMC125 displays a high genetic barrier to the development of resistance: evidence from in vitro selection experiments. J Virol 79:12773–12782. doi: 10.1128/JVI.79.20.12773-12782.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang YJ, McKenna PM, Hrin R, Felock P, Lu M, Jones KG, Coburn CA, Grobler J A, Hazuda DJ, Miller MD, Lai MT. 2010. Assessment of the susceptibility of mutant HIV-1 to antiviral agents. J Virol Methods 165:230–237. doi: 10.1016/j.jviromet.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Quiñones-Mateu ME, Arts EJ. 2002. Fitness of drug resistant HIV-1: methodology and clinical implications. Drug Resist Updat 5:224–233. doi: 10.1016/S1368-7646(02)00123-1. [DOI] [PubMed] [Google Scholar]
- 22.Holland JJ, de la Torre JC, Clarke DK, Duarte E. 1991. Quantitation of relative fitness and great adaptability of clonal populations of RNA viruses. J Virol 65:2960–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Picado J, Martinez MA. 2008. HIV-1 reverse transcriptase inhibitor resistance mutations and fitness: a view from the clinic and ex vivo. Virus Res 134:104–123. doi: 10.1016/j.virusres.2007.12.021. [DOI] [PubMed] [Google Scholar]
- 24.Rimsky L, Vingerhoets J, Van Eygen V, Eron J, Clotet B, Hoogstoel A, Boven K, Picchio G. 2012. Genotypic and phenotypic characterization of HIV-1 isolates obtained from patients on rilpivirine therapy experiencing virologic failure in the phase 3 ECHO and THRIVE studies: 48-week analysis. J Acquir Immune Defic Syndr 59:39–46. doi: 10.1097/QAI.0b013e31823df4da. [DOI] [PubMed] [Google Scholar]
- 25.Turner D, Brenner B, Moisi D, Detorio M, Cesaire R, Kurimura T, Mori H, Essex M, Maayan S, Wainberg MA. 2004. Nucleotide and amino acid polymorphisms at drug resistance sites in non-B-subtype variants of human immunodeficiency virus type 1. Antimicrob Agents Chemother 48:2993–2998. doi: 10.1128/AAC.48.8.2993-2998.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brenner B, Turner D, Oliveira M, Moisi D, Detorio M, Carobene M, Marlink RG, Schapiro J, Roger M, Wainberg MA. 2003. A V106M mutation in HIV-1 clade C viruses exposed to efavirenz confers cross-resistance to non-nucleoside reverse transcriptase inhibitors. AIDS 17:F1–F5. doi: 10.1097/00002030-200301030-00001. [DOI] [PubMed] [Google Scholar]
- 27.Lai MT, Lu M, Felock PJ, Hrin RC, Wang YJ, Yan Y, Munshi S, McGaughey GB, Tynebor RM, Tucker TJ, Williams TM, Grobler J A, Hazuda DJ, McKenna PM, Miller MD. 2010. Distinct mutation pathways of non-subtype B HIV-1 during in vitro resistance selection with nonnucleoside reverse transcriptase inhibitors. Antimicrob Agents Chemother 54:4812–4824. doi: 10.1128/AAC.00829-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koval CE, Dykes C, Wang J, Demeter LM. 2006. Relative replication fitness of efavirenz-resistant mutants of HIV-1: correlation with frequency during clinical therapy and evidence of compensation for the reduced fitness of K103N + L100I by the nucleoside resistance mutation L74V. Virology 353:184–192. doi: 10.1016/j.virol.2006.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bacheler LT, Anton ED, Kudish P, Baker D, Bunville J, Krakowski K, Bolling L, Aujay M, Wang XV, Ellis D, Becker MF, Lasut AL, George HJ, Spalding DR, Hollis G, Abremski K. 2000. Human immunodeficiency virus type 1 mutations selected in patients failing efavirenz combination therapy. Antimicrob Agents Chemother 44:2475–2484. doi: 10.1128/AAC.44.9.2475-2484.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gatanaga H, Ode H, Hachiya A, Hayashida T, Sato H, Oka S. 2010. Combination of V106I and V179D polymorphic mutations in human immunodeficiency virus type 1 reverse transcriptase confers resistance to efavirenz and nevirapine but not etravirine. Antimicrob Agents Chemother 54:1596–1602. doi: 10.1128/AAC.01480-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Picchio GR, Rimsky LT, Van Eygen V, Haddad M, Napolitano LA, Vingerhoets J. 4 April 2014. Prevalence in the USA of rilpivirine resistance-associated mutations in clinical samples and effects on phenotypic susceptibility to rilpivirine and etravirine. Antivir Ther doi: 10.3851/IMP2771. [DOI] [PubMed] [Google Scholar]